Introduction To Autonomic Pharmacology
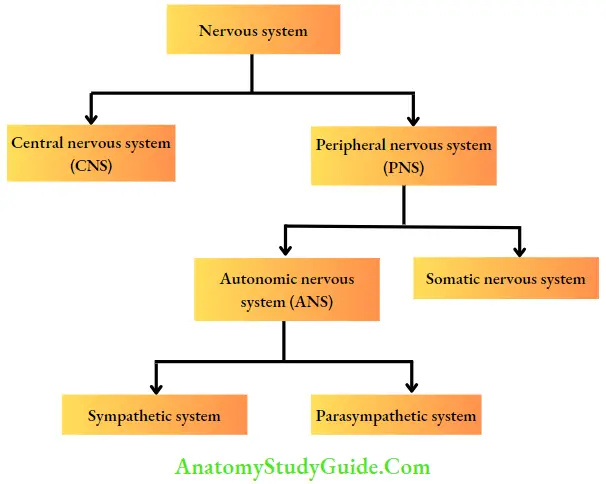
The nervous system is divided into the central nervous system (CNS: brain and spinal cord) and the peripheral nervous system (PNS). PNS can be further divided into the somatic nervous system and the autonomic nervous system (ANS). The differences between these two systems are given.
Table of Contents
Read And Learn More: Pharmacology for Dentistry Notes
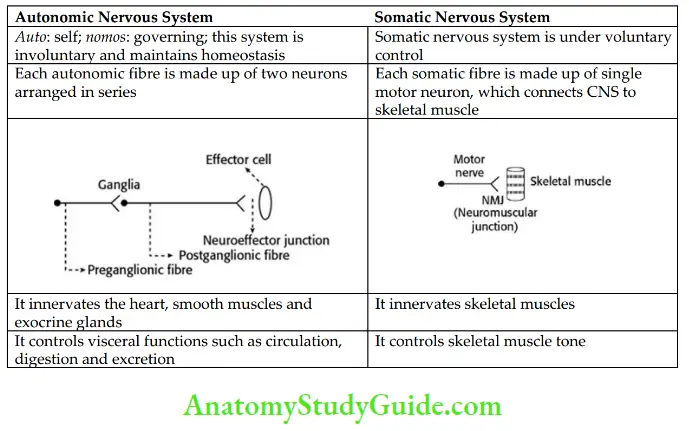
Differences Between ANS and Somatic Nervous System
The ANS has two divisions – sympathetic and parasympathetic. The sympathetic division arises from the thoracolumbar region (T1–L3, thoracolumbar outflow) and the parasympathetic division arises from two separate regions in the CNS.
The cranial outflow arises from cranial nerves (3, 7, 9, and 10) and sacral outflow from S2, S3, and S4 spinal roots.
In the sympathetic system, the preganglionic fibres are short and the postganglionic fibres are long. On the contrary, the parasympathetic preganglionic fibres are long and postganglionic fibres are short.
Most of the visceral organs have dual nerve supply, i.e. they are supplied by both divisions of the ANS, but the effects of one system predominate.
The ciliary muscle, pancreatic and gastric glands receive only parasympathetic supply; sweat glands, hair follicles, spleen, and most of the blood vessels have only sympathetic supply. Their stimulation usually produces the opposite effect on the innervating organ.


Cholinergic System
Cholinergic Transmission
Acetylcholine (ACh) is the main neurotransmitter in the cholinergic system. The neurons that synthesize, store, and release ACh are called cholinergic neurons.
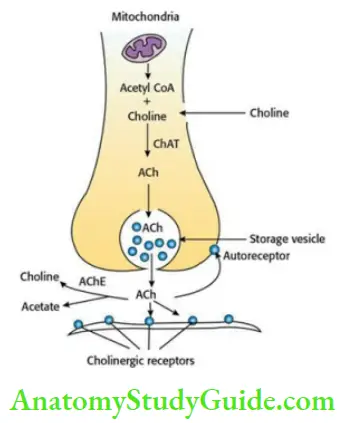
- Synthesis Of Acetylcholine Choline enters the cholinergic neuron by carrier-mediated transport, where it reacts with acetyl-CoA with the help of choline acetyltransferase (ChAT) to form ACh. The ACh is then stored in storage vesicles. It is released into the synaptic cleft when an action potential reaches the nerve terminals. The released ACh interacts with cholinergic receptors on effector cell and activates them. In the synaptic cleft, ACh is rapidly hydrolysed by the acetylcholinesterase (AChE) enzyme.
- Cholinesterase ACh is rapidly hydrolysed to choline and acetic acid by enzyme, cholinesterase. There are two types of cholinesterase:
- True cholinesterase or AChE: It is found in cholinergic neurons, ganglia, RBCs, and neuromuscular junction (NMJ). It rapidly hydrolyses ACh and methacholine.
- Pseudocholinesterase or butyrylcholinesterase: It is found in plasma, liver, and glial cells. Pseudocholinesterase can act on a wide variety of esters including ACh but does not hydrolyse methacholine.
- Cholinergic receptors They are divided broadly into two types – muscarinic and nicotinic. Muscarinic receptors are further divided into five different subtypes: M1–M5. Only M1, M2, and M3 are functionally recognized; M4 and M5 subtypes are found in CNS. All muscarinic receptors are G-protein-coupled receptors and regulate the production of intracellular second messengers.

- Nicotinic receptors are divided into two subtypes – NM and NN. Activation of these receptors directly opens the ion channels and causes depolarization of the membrane. The characteristics of muscarinic and nicotinic receptors are shown in Table.

Characteristics of Muscarinic and Nicotinic Receptor Subtypes

Cholinergic Agents (Cholinomimetics, Parasympathomimetics)
ACh is a quaternary ammonium compound and is rapidly hydrolysed by cholinesterases. Hence, it has no therapeutic application. It has to be given intravenously to study its pharmacological actions. Even when given intravenously, a large amount of the drug is destroyed by pseudocholinesterase in blood.
- Classification

Choline Esters
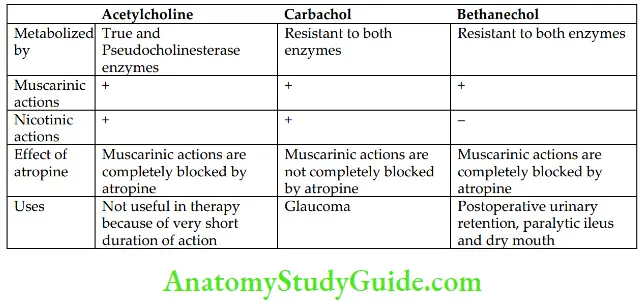
Choline esters include ACh, carbachol, and bethanechol.
1. Acetylcholine ACh produces muscarinic and nicotinic effects by interacting with respective receptors on the effector cells
Pharmacological Properties and Uses of Choline Esters
- Muscarinic actions
- Cardiovascular system
- Heart: The effects of ACh are similar to those following vagal stimulation. ACh, by stimulating M2 receptors of the heart, opens K+ channels resulting in hyperpolarization. Therefore, S–A and A–V nodal activity is reduced.

- Blood vessels: ACh stimulates M3 receptors of vascular endothelial cells, which release endothelium-dependent relaxing factor (EDRF; NO), leading to vasodilatation and a fall in blood pressure (BP).

- Heart: The effects of ACh are similar to those following vagal stimulation. ACh, by stimulating M2 receptors of the heart, opens K+ channels resulting in hyperpolarization. Therefore, S–A and A–V nodal activity is reduced.
- Smooth muscles
- Gastrointestinal tract

- Urinary bladder

- Bronchi

- Gastrointestinal tract
- Exocrine glands: All parasympathomimetic agents stimulate salivary secretion. They also increase lacrimal, sweat, bronchial, and other gastrointestinal (GI) secretions.
- Eye: ACh does not produce any effect on topical administration because of its poor penetration through tissues.
- Cardiovascular system

-
- The action of muscarinic agonists on eye can be depicted as follows:
- Nicotinic actions To elicit nicotinic actions, larger doses of ACh are required.
- Autonomic ganglia: Higher doses of ACh produce dangerous muscarinic effects, especially on the heart. Hence, prior administration of atropine is necessary to elicit nicotinic actions. Higher doses of ACh stimulate both sympathetic as well as parasympathetic ganglia, causing tachycardia and a rise in BP.

-
- Skeletal muscles: At high concentrations, ACh initially produces twitching, and fasciculations followed by prolonged depolarization of NMJ and paralysis.
- CNS: Intravenously administered ACh does not cause any central effects because of its poor penetration through BBB.
2. Bethanechol It has selective muscarinic actions on the gastrointestinal tract (GIT) and urinary bladder. It is preferred in postoperative urinary retention and paralytic ileus.

Cholinomimetic Alkaloids
They mimic the actions of ACh; examples are pilocarpine, muscarine, and arecoline.
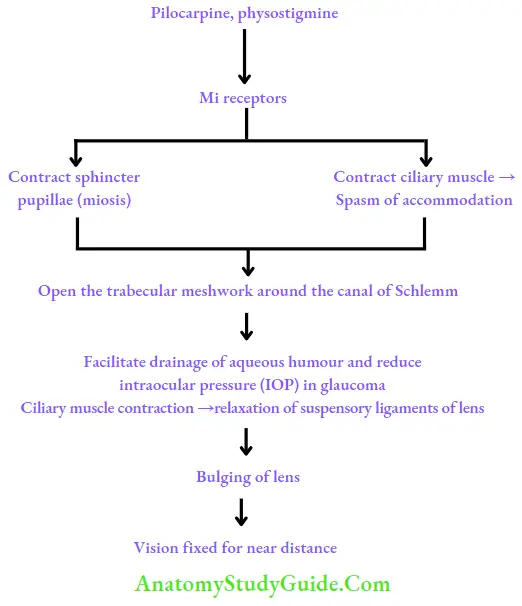
- Pilocarpine is a cholinomimetic alkaloid obtained from Pilocarpus plant. It is a tertiary amine. It produces muscarinic and nicotinic effects by directly interacting with the receptors. It has predominant muscarinic actions especially on secretory activity.
- Uses
- Pilocarpine is used as a sialagogue (a drug used to augment salivary secretion). It can be used cautiously by oral route in xerostomia (dry mouth) that follows head and neck radiation treatment. Other sialagogues are cevimeline and bethanechol. Cevimeline (30 mg TDS), an M3 agonist can be used to treat xerostomia and dry eyes. It is long acting and has fewer side effects compared to pilocarpine. Bethanechol can also be used in xerostomia and has lesser diaphoretic (sweating) effect.
- Uses
- Sjögren syndrome: It is an autoimmune disorder characterized by dryness of all mucosae (dry eyes, dry mouth, etc.). Pilocarpine can be used orally 5 mg three times daily with food.
- Pilocarpine 0.5%–4% solution is used topically in the treatment of open-angle and acute congestive glaucoma. It increases the tone of ciliary muscle and causes miosis by contracting sphincter pupillae, opens the trabecular meshwork around the canal of Schlemm, facilitates drainage of aqueous humour, and reduces intraocular pressure (IOP). It acts rapidly but has a shorter duration of action. Pilocarpine Ocusert that releases the drug slowly over 7 days is available.
- Pilocarpine is used alternatively with mydriatics to break adhesions between the iris and lens.
- Pilocarpine is used to reverse the pupillary dilatation after refraction testing.
- Adverse effects They are salivation, sweating, bradycardia, diarrhea, bronchospasm; pulmonary oedema can occur following systemic therapy.
- Muscarine It is an active ingredient of poisonous mushroom, Amanita muscaria. It has no therapeutic application.
- Arecoline It is an alkaloid obtained from areca nut. It has muscarinic and nicotinic actions similar to choline esters.
- Adverse effects They are salivation, sweating, bradycardia, diarrhea, bronchospasm; pulmonary oedema can occur following systemic therapy.
Anticholinesterases
They inhibit the enzyme cholinesterases, which is responsible for hydrolysis of ACh. Thus, ACh is not metabolized, gets accumulated at muscarinic and nicotinic sites, and produces cholinergic effects. Hence, anticholinesterases are called indirectly acting cholinergic drugs.
- Reversible anticholinesterases
- Physostigmine
- Neostigmine
- Pyridostigmine
- Edrophonium
- Rivastigmine
- Donepezil
Anticholinesterases Mechanism of action: ACh is rapidly hydrolysed by both true and pseudocholinesterase. Reversible anticholinesterases inhibit both true and pseudocholinesterase reversibly – thus, ACh gets accumulated and produces cholinergic effects.

- Physostigmine (eserine): It is an alkaloid obtained from Physostigma venenosum. It is a tertiary amine and has good penetration through tissues. Its actions are similar to those of other cholinergic agents.
- Uses
- Glaucoma: Physostigmine reduces IOP by producing miosis, thus it facilitates the drainage of aqueous humor. On chronic use, it accelerates cataract formation; hence it is rarely used in glaucoma.
- Atropine poisoning: Intravenous physostigmine is used for severe atropine and other antimuscarinic drug poisoning because it has both central and peripheral actions. It competitively reverses the effects of atropine poisoning; but it should be used cautiously by slow i.v. injection as it may cause bradycardia.
- Uses
- Neostigmine: Neostigmine is a synthetic anticholinesterase agent. Its actions are pronounced on NMJ, gastrointestinal system (GIT), and urinary bladder than on cardiovascular system (CVS) or eye. On skeletal muscle, it has both direct and indirect actions.Comparative Features of Physostigmine and Neostigmine

- Indirect actions: By inhibiting cholinesterases, neostigmine increases ACh concentration at NMJ.
- Direct action: Because of structural similarity with ACh (i.e. quaternary ammonium compound), neostigmine also directly stimulates NM receptors at NMJ. Thus, it improves muscle power in patients with myasthenia gravis. Neostigmine does not cross BBB and has no central side effects. Therefore, neostigmine is preferred over physostigmine in myasthenia gravis. It is available for oral, subcutaneous, i.m., and i.v. administration.
- Pyridostigmine: All features are the same as neostigmine. Pyridostigmine has a longer duration of action and can be given twice daily in sustained-release form; hence, it is preferred over neostigmine in myasthenia gravis. Even though pyridostigmine is less potent than neostigmine, it is better tolerated by myasthenic patients.
- Edrophonium: It is a quaternary ammonium compound. On i.v. administration, it has a rapid onset but short duration of action (8–10 min).
- Uses
- Edrophonium is used in the diagnosis of myasthenia gravis.
- It is used to differentiate a myasthenic crisis from a cholinergic crisis.
- In curare poisoning, edrophonium is preferred because of its rapid onset of action.
- Uses
- Adverse effects of anticholinesterases They are due to overstimulation of both muscarinic and nicotinic receptors – increased sweating, salivation, nausea, vomiting, abdominal cramps, bradycardia, diarrhea, tremors, and hypotension.
- Therapeutic uses of reversible anticholinesterases
- Eye.
- Glaucoma.
- To reverse pupillary dilatation after refraction testing.
- Miotics are used alternatively with mydriatics to break adhesions between the iris and lens.
- Myasthenia gravis.
- Postoperative urinary retention and paralytic ileus.
- Curare poisoning and reversal of nondepolarizing neuromuscular blockade.
- Belladonna poisoning.
- Alzheimer’s disease.
- Eye.
- Glaucoma The aqueous humour formed by the ciliary process is drained mainly through trabecular meshwork. Glaucoma is optic nerve damage with loss of visual function, frequently associated with raised IOP. Normal IOP varies between 10 and 20 mm Hg. Management of this disorder is almost always directed at lowering the existing IOP either by improving drainage or decreasing the formation of aqueous humor.
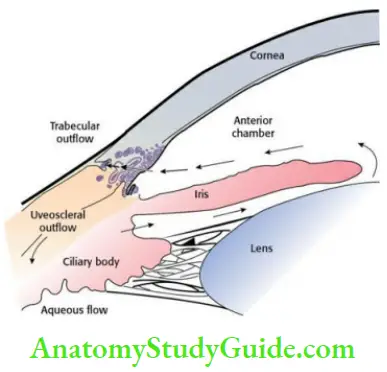
-
- Acute congestive glaucoma: It is usually precipitated by mydriatics in people with narrow iridocorneal angles and shallow anterior chambers. Acute congestive glaucoma is always a medical emergency. Once the attack is controlled, treatment is surgical or laser iridotomy.
- Chronic simple glaucoma: It is a genetically predisposed condition affecting the patency of trabecular meshwork. The IOP rises gradually. Pharmacotherapy is the definitive treatment in a majority of cases.
- Drugs for glaucoma
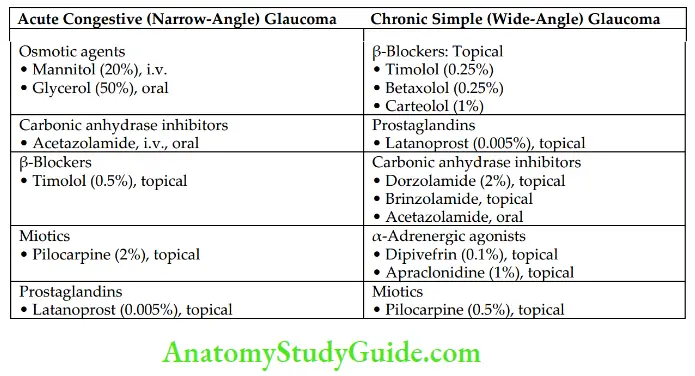
- Osmotic agents: Mannitol (20%) i.v. infusion (1.5 g/kg body weight) and 50% glycerol oral (1.5 g/kg) are used. They draw fluid from the eye into the circulation by osmotic effect and reduce IOP in acute congestive glaucoma.
- Carbonic anhydrase inhibitors: Acetazolamide (oral, i.v.), dorzolamide (topical), and brinzolamide (topical) are carbonic anhydrase inhibitors. They inhibit carbonic anhydrase enzyme and lower IOP by decreasing the formation of aqueous humor. Topical carbonic anhydrase inhibitors, which have a much lower risk of systemic side effects, are preferred over systemic carbonic anhydrase inhibitors in chronic simple glaucoma. In acute congestive glaucoma, acetazolamide is administered orally.
- β-Adrenergic blockers: They decrease the production of aqueous humor. Topical nonselective β-blockers are timolol, betaxolol, levobunolol, and carteolol. Timolol is widely used in glaucoma because
-
-
- It lacks local anesthetic properties,
- It does not affect pupil size or accommodation,
- It has a longer duration of action,
- It is well tolerated,
- It is less expensive and
- Topical timolol is safer and highly effective. Betaxolol is a selective β1-blocker used in glaucoma, but it is less effective than nonselective agents. Betaxolol is protective of retinal neurons. Levobunolol, a nonselective β-blocker, is long-acting. β- Blockers should be cautiously used in bronchial asthma and heart failure.
- Prostaglandins (PGs): Topical PGs such as latanoprost and bimatoprost (PGF2α analogs) are the preferred agents for initial therapy in open-angle glaucoma because of their longer duration of action, high efficacy, and low incidence of systemic toxicity. They are also useful in acute congestive glaucoma. They reduce IOP probably by facilitating uveoscleral outflow. They usually do not cause systemic side effects but may cause ocular irritation and iris pigmentation.
- Miotics: Pilocarpine is a tertiary amine and is well absorbed through the cornea. It is used topically in the treatment of open-angle and acute congestive glaucoma. It facilitates drainage of aqueous humor and reduces IOP.
- α-Adrenergic agonists:
- Apraclonidine is used topically as an adjunct in glaucoma. It does not cross BBB and hence has no hypotensive effect like clonidine. They act on α2– receptors on the ciliary epithelium.
- Apraclonidine → α2-Agonist → Reduces formation of aqueous humour → Decreases IOP
- Dipivefrin is a prodrug of adrenaline. It penetrates the cornea and gets converted into adrenaline with the help of esterases. Drugs Used for Treating Glaucoma
-
- Myasthenia gravis Myasthenia gravis is an autoimmune disorder where antibodies are produced against N M receptors of NMJ resulting in a decrease in the number of NM receptors. There is an increased incidence of myasthenia gravis in patients with thymoma. Thymectomy can induce remission in most of the cases. In myasthenia, there is marked muscular weakness varying in degree at different times. Myasthenia gravis is diagnosed by
-
- Typical signs and symptoms— weakness and easy fatigability. Edrophonium test— edrophonium (2–10 mg) given slow i.v. shows dramatic improvement of symptoms in patients with myasthenia gravis but not in other muscular dystrophies.
- Demonstration of circulating antibodies to NM receptors.
- Treatment Anticholinesterases (neostigmine and pyridostigmine) are effective in providing symptomatic relief. They inhibit the metabolism of ACh, thus prolonging its action at the receptor.
- Neostigmine also directly activates the NM receptors. Pyridostigmine is commonly used. Long-term use or overdose of anticholinesterases leads to cholinergic crisis (severe muscular weakness and neuromuscular paralysis due to prolonged depolarization).
- This may be differentiated from the myasthenic crisis (severe weakness due to exacerbation of myasthenia) by injecting a small dose of edrophonium (2 mg, i.v.). If the patient shows improvement in muscle power → myasthenic crisis.
- If the muscular weakness deteriorates → cholinergic crisis. Ventilators should be kept ready before injecting edrophonium as it may aggravate the cholinergic crisis, which is dangerous.
- Corticosteroids and other immunosuppressants like azathioprine or cyclophosphamide are useful for the induction and maintenance of remission in myasthenia gravis. Plasmapheresis and immune therapy may be useful in resistant cases.
- Postoperative urinary retention and paralytic ileus Neostigmine is used because it increases the tone of the smooth muscle and relax the sphincters.

- Curare poisoning and reversal of nondepolarizing neuromuscular blockade Edrophonium or neostigmine is used. They antagonize neuromuscular blockade by increasing the concentration of ACh at the NMJ. Prior administration of atropine is a must to block the muscarinic side effects.
- Belladonna poisoning Physostigmine is preferred because it reverses both the central and peripheral effects of atropine poisoning.
- Alzheimer’s disease is a degenerative disease of the cerebral cortex. Donepezil, galantamine, and rivastigmine are cerebroselective anticholinesterases. They increase cerebral levels of ACh and have been shown to produce some benefits in these patients.
Mnemonic for therapeutic uses of reversible anticholinesterases:
GLAUCOMA
- GI atony (Paralytic ileus)
- L—
- Alzheimer’s disease
- Urinary retention (postoperative urinary retention)
- Curare poisoning
- Ocular conditions – glaucoma and others
- Myasthenia gravis
- Atropine poisoning (Belladonna poisoning)
- Irreversible anticholinesterases
- Organophosphorus insecticides All organophosphorus (OP) compounds except echothiophate have no therapeutic applications. Echothiophate is rarely used in resistant cases of glaucoma. OP compounds have only toxicological importance. OP poisoning is one of the most common poisoning all over the world. Common OP compounds are parathion, malathion, floss, etc. They irreversibly inhibit cholinesterases and cause accumulation of ACh at muscarinic and nicotinic sites.
- Signs and symptoms
- Muscarinic effects: Profuse sweating, salivation, lacrimation, increased tracheobronchial secretions, bronchospasm, vomiting, abdominal cramps, miosis, bradycardia, hypotension, involuntary urination, and defecation.
- Nicotinic effects: Twitching, fasciculation, muscle weakness, and paralysis are due to prolonged depolarization.
- Central effects: Headache, restlessness, confusion, convulsions, coma, and death usually due to respiratory failure
- Diagnosis OP poisoning can be diagnosed by:
- History of exposure.
- Characteristic signs and symptoms.
- Estimating the cholinesterase activity in the blood, which is decreased.
- Treatment General Measures
- Remove the contaminated clothes; wash the skin with soap and water.
- Gastric lavage should be continued till the returning fluid is clear.
- The airway should be maintained.
- Artificial respiration is given, if necessary.
- Diazepam should be used cautiously by slow i.v. injection to control convulsions.
- Specific measures
- Atropine: Atropine is the first drug to be given in organophosphorus poisoning. Inject atropine 2 mg i.v. stat and it should be repeated every 5–10 min doubling the dose, if required, till the patient is fully atropinized (fully dilated, nonreactive pupils, flushed skin, tachycardia, etc.). Atropine should be continued for 7–10 days.
- Signs and symptoms
- Organophosphorus insecticides All organophosphorus (OP) compounds except echothiophate have no therapeutic applications. Echothiophate is rarely used in resistant cases of glaucoma. OP compounds have only toxicological importance. OP poisoning is one of the most common poisoning all over the world. Common OP compounds are parathion, malathion, floss, etc. They irreversibly inhibit cholinesterases and cause accumulation of ACh at muscarinic and nicotinic sites.

-
-
-
- Animation 2: Treatment of OP poisoning. Atropine competitively blocks the muscarinic effects of organophosphorus compounds (competitive antagonism).
- Oximes: Atropine is not effective for reversal of neuromuscular paralysis. Neuromuscular transmission can be improved by giving cholinesterase reactivators such as pralidoxime and obidoxime. Pralidoxime is administered i.v. slowly in a dose of 1–2 g. As shown above, OP compounds inactivate cholinesterases by phosphorylating the esteratic site of the enzyme. Oximes bind with high affinity to anionic site and dephosphorylate the enzyme and reactivate it. Early administration of oximes is necessary before the phosphorylated enzyme undergoes ‘aging’ and becomes resistant to reactivation. Oximes are not effective in carbamate poisoning; they also have mild antiChE activity.
-
-

- Delayed toxicity of organophosphates:
Prolonged exposure to OP compounds can cause neurotoxicity.
Anticholinergic Agents (Cholinergic Receptor Blockers)
Various anticholinergic agents are shown below.
Anticholinergic Agents Classification

Antimuscarinic Agents (Muscarinic Receptor Antagonists)
These drugs block muscarinic-receptor-mediated actions of ACh on the heart, CNS, smooth muscles, and exocrine glands. Atropine and scopolamine are belladonna alkaloids. Atropine is obtained from Atropa belladonna and scopolamine from Hyoscyamus niger.
- Anticholinergic Agents Mechanism of action Both natural and synthetic drugs competitively block the muscarinic effects of ACh (competitive antagonism).

- Classification of antimuscarinic agents
- Natural alkaloids (belladonna alkaloids): Atropine, scopolamine (hyoscine).
- Semisynthetic derivatives:
- Hyoscine butylbromide
- Homatropine (mydriatic)
- Ipratropium bromide, tiotropium bromide (used in bronchial asthma)
- Synthetic antimuscarinic agents:
- Used as mydriatic – cyclopentolate, tropicamide.
- Used in peptic ulcer – pirenzepine, telenzepine.
- Used as an antispasmodic – dicyclomine, flavoxate, oxybutynin, tolterodine.
- Used as a pre-anesthetic agent – glycopyrrolate.
- Used in parkinsonism – benzhexol (trihexyphenidyl), benztropine, biperiden.
- Used in sialorrhoea – glycopyrrolate, propantheline bromide. (Other drugs useful in sialorrhoea are atropine, scopolamine, and botulinum toxin A.)
Atropine
Atropine is the prototype drug and the chief alkaloid of belladonna. It is a tertiary amine. It blocks the action of ACh on all muscarinic receptors. Atropine is administered by topical (eye), oral, and parenteral routes.
- Pharmacological Actions of Atropine

-
- CNS: In therapeutic doses, atropine has a mild CNS-stimulant effect. It produces an anti-parkinsonian effect by reducing cholinergic overactivity in basal ganglia. It suppresses vestibular disturbances and produces an anti-motion-sickness effect. Large doses can produce excitement, restlessness, agitation, hallucinations, medullary paralysis, coma, and death.
- CVS: At low doses, atropine causes initial bradycardia due to the blockade of presynaptic muscarinic autoreceptors (M1) on vagal nerve endings. In therapeutic doses, tachycardia is seen due to the blockade of M2 receptors of the heart; it also improves A–V conduction. In high doses, flushing of the face and hypotension may occur due to cutaneous vasodilatation.
- Glands: All secretions under the cholinergic influence are reduced due to the blockade of M3 receptors, i.e. sweat, salivary, nasal, throat, bronchial, gastric, lacrimal, etc. Milk and bile secretions are not affected. The skin and mucous membranes become dry.
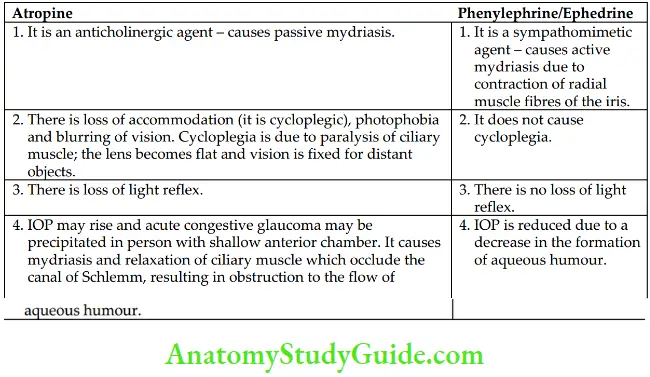
- Eye: Effects of atropine on the eye are depicted below Effects on the eye last for 7–10 days following topical administration of atropine.
- Smooth muscles:
- GIT: Atropine decreases tone and motility of the gut but increases sphincter tone and may cause constipation. It also relaxes the smooth muscle of the gall bladder.
- Urinary bladder: Atropine relaxes the detrusor muscle of the bladder, but increases the tone of the trigone and sphincter – may cause urinary retention, especially in elderly men with an enlarged prostate.
- Bronchi: Atropine relaxes the bronchial smooth muscle. It also reduces secretion and mucociliary clearance resulting in mucus plugs that may block the airway. Effects of Atropine and Phenylephrine/Ephedrine on Eye
- Pharmacokinetics Atropine, scopolamine, and most of the synthetic tertiary amines are well absorbed from the conjunctiva and GI tract; widely distributed all over the body; cross BBB; partly metabolized in the liver; and partly excreted unchanged in urine.
- Atropine substitutes Atropine acts on all subtypes of muscarinic receptors. Atropine substitutes have a selective or relatively selective action on a particular organ, hence producing fewer adverse effects than atropine.
- For the eye (as mydriatic)
- Homatropine
- Semisynthetic atropine derivative.
- Less potent than atropine.
- Duration of action (mydriasis and cycloplegia) is 1–3 days.
- Cyclopentolate and tropicamide
- Synthetic atropine derivatives with rapid onset (tropicamide is the fastest acting) and shorter duration of action than atropine.
- The action of cyclopentolate lasts for 24 h; tropicamide is the shortest acting and the action lasts for 6 h.
- Homatropine
- Antispasmodics
- Dicyclomine
- Tertiary amine.
- Has antispasmodic and antiemetic properties.
- Useful in dysmenorrhoea and abdominal colic.
- Oxybutynin
- Has selective action at M1 and M3 receptors in the urinary bladder and salivary gland.
- Has regioselective action – useful for relief of spasms after urologic surgeries, for increasing bladder capacity in paraplegics, and in nocturnal enuresis.
- Tolterodine
- More selective for the urinary bladder than salivary glands; hence dryness of mouth is less.
- Used to decrease frequency and urgency in detrusor overactivity.
- Flavoxate
- Similar to oxybutynin.
- Used to relieve urgency and frequency due to cystitis, prostatitis, or urethritis.
- Oxybutynin, flavoxate, and tolterodine are regioselective anticholinergics.
- Dicyclomine
- Ipratropium bromide and tiotropium bromide
- Synthetic atropine derivatives are administered by the inhalation route.
- Have a selective action on bronchial smooth muscle – bronchodilatation (mainly in the larger airways).
- Do not affect mucociliary clearance.
- Tiotropium (24 h) is longer acting than ipratropium (6 h).
- Dryness of mouth is the main side effect of these agents.
- Pirenzepine
- Has selective action on gastric acid secretion (M1) – useful in peptic ulcer.
- Anticholinergic side effects – dryness of mouth, constipation, tachycardia, and urinary retention are rare.
- Benzhexol and benztropine
- They are centrally-acting anticholinergic agents used in Parkinsonism.
- Glycopyrrolate
- Quaternary ammonium compound – central side effects are rare.
- Used for pre-anesthetic medication.
- For the eye (as mydriatic)
- Therapeutic uses of atropine and its substitutes
- As mydriatic and cycloplegic – Tropicamide is used topically for producing mydriasis and cycloplegia during refraction testing. Tropicamide is the preferred mydriatic, as it has a quick onset and short duration of action. In iridocyclitis – atropine mydriatics are used alternatively with miotics to break or prevent adhesions between the iris and lens.
- As pre-anesthetic medication: Atropine or glycopyrrolate is used. They are used prior to the administration of general anesthetics:
- To prevent vagal bradycardia during anesthesia.
- To prevent laryngospasm by decreasing respiratory secretions.
- Glycopyrrolate is a quaternary ammonium compound and has only peripheral anticholinergic effects.
- Sialorrhoea (hypersalivation): Synthetic derivatives (glycopyrrolate) are used to decrease salivary secretion, for example during dental procedures and in heavy-metal poisoning. They are also useful in drooling (saliva beyond the margins of the lips).
- COPD and bronchial asthma: Ipratropium bromide and tiotropium bromide are used in COPD and bronchial asthma. They are administered by a metered-dose inhaler or nebulizer. They produce bronchodilatation without affecting mucociliary clearance; hence are preferred over atropine.
- Anticholinergics are useful as antispasmodics in dysmenorrhoea, and intestinal and renal colic.
- Urinary disorders: Oxybutynin and flavoxate have a more prominent effect on bladder smooth muscle and, hence are used to relieve spasms after urologic surgery. Tolterodine has selective action on bladder smooth muscle (M3); hence, is used to relieve urinary incontinence.
- Poisoning:
- In OP poisoning, atropine is a life-saving drug
- In some types of mushroom poisoning (Inocybe species), atropine is the drug of choice.
- Atropine is used in curare poisoning with neostigmine to counteract the muscarinic effects of neostigmine.
- As vagolytic: Atropine is used to treat sinus bradycardia and partial heart block due to increased vagal activity. It improves A–V conduction by vagolytic effect.
- Parkinsonism: Centrally acting anticholinergic drugs such as benzhexol (trihexyphenidyl), benztropine, and biperiden are the preferred agents for the prevention and treatment of drug-induced parkinsonism. They are also useful in idiopathic parkinsonism, but less effective than levodopa. They control tremors and rigidity of parkinsonism.
- Adverse effects and contraindications Adverse effects are due to the extension of pharmacological actions.
- GIT: Dryness of mouth and throat, difficulty in swallowing, constipation, etc.
- Eye: Photophobia, headache, blurring of vision; in elderly persons with shallow anterior chamber, they may precipitate acute congestive glaucoma. Hence, anticholinergics are contraindicated in glaucoma.
- Urinary tract: Difficulty in micturition and urinary retention, especially in elderly men with enlarged prostate. So, they are contraindicated in these patients.
- CNS: Large doses produce restlessness, excitement, delirium, and hallucinations.
- CVS: Tachycardia, palpitation and hypotension.
- Acute belladonna poisoning: It is more common in children. The presenting features include fever, dry and flushed skin, photophobia, blurring of vision, difficulty in micturition, restlessness, excitement, confusion, disorientation, and hallucinations. Severe poisoning may cause respiratory depression, cardiovascular collapse, convulsions, coma, and death.
- Treatment of belladonna poisoning (atropine poisoning): It is mainly symptomatic.
- Hospitalization.
- Gastric lavage in case of ingested poison.
- Tepid sponging to control hyperpyrexia.
- Diazepam to control convulsions.
- The antidote for severe atropine poisoning is physostigmine (1–4 mg). It is injected intravenously slowly. It is a tertiary amine – that counteracts both peripheral as well as central effects of atropine poisoning. Hence, physostigmine is preferred over neostigmine.
- Treatment of belladonna poisoning (atropine poisoning): It is mainly symptomatic.
Scopolamine
Scopolamine (hyoscine), another belladonna alkaloid, produces all the actions of atropine. In therapeutic doses, it produces prominent CNS depression with sedation and amnesia. Scopolamine has a shorter duration of action than atropine. It has more prominent actions on the eye and secretory glands. By blocking cholinergic activity, scopolamine suppresses vestibular disturbances and prevents motion sickness.

It is the drug of choice for motion sickness – can be administered orally or as a transdermal patch. It is more effective for the prevention of motion sickness; hence should be given (0.2 mg oral) at least half an hour before a journey. The patch is placed behind the ear over the mastoid process. The patch should be used at least 4–5 hours before the journey, and its effect lasts 72 hours. Scopolamine may cause sedation and dryness of the mouth.
- Drug interactions
H1-blockers, tricyclic antidepressants (TCAs), phenothiazines, etc. have atropine-like actions; hence, they may potentiate anticholinergic side effects.
Atropine alters the absorption of some drugs by delaying gastric emptying – the bioavailability of levodopa is reduced, whereas the absorption of tetracyclines and digoxin is enhanced due to increased GI transit time.
Ganglionic Blockers
They act at NN receptors of the autonomic ganglia (block both parasympathetic and sympathetic ganglia) and produce widespread complex effects.

- The ganglionic blockers have ‘atropine-like’ action on the heart (palpitation and tachycardia), eye (mydriasis and cycloplegia), GIT (dryness of mouth and constipation), bladder (urinary retention), impotence in males and decreased sweat secretion.
- Blockade of sympathetic ganglia results in marked postural hypotension. No selective ganglion blockers are available till now. Hence, they are rarely used in
therapy. - Nicotine is obtained from tobacco leaves. It has an initial stimulating and later a prolonged blocking effect on the autonomic ganglia. Tobacco smoking and chewing is a serious risk factors for oral, lung, heart, and other diseases.
- Nicotine is of no value in clinical practice except in the form of transdermal patches and chewing gum for the treatment of tobacco addiction
Skeletal Muscle Relaxants
Skeletal muscle relaxants decrease skeletal muscle tone by peripheral or central action.
Physiology of skeletal muscle contraction
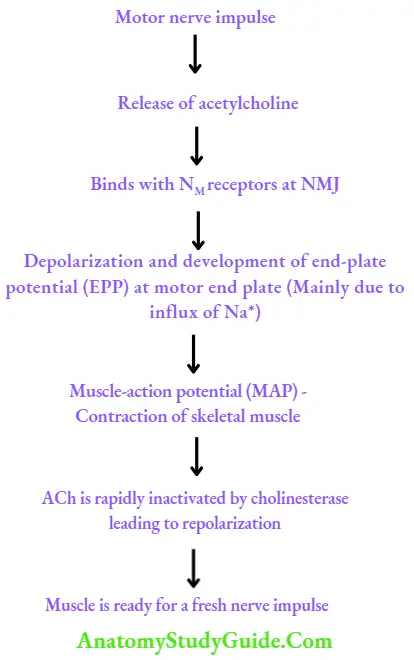
Skeletal muscle relaxants Classification
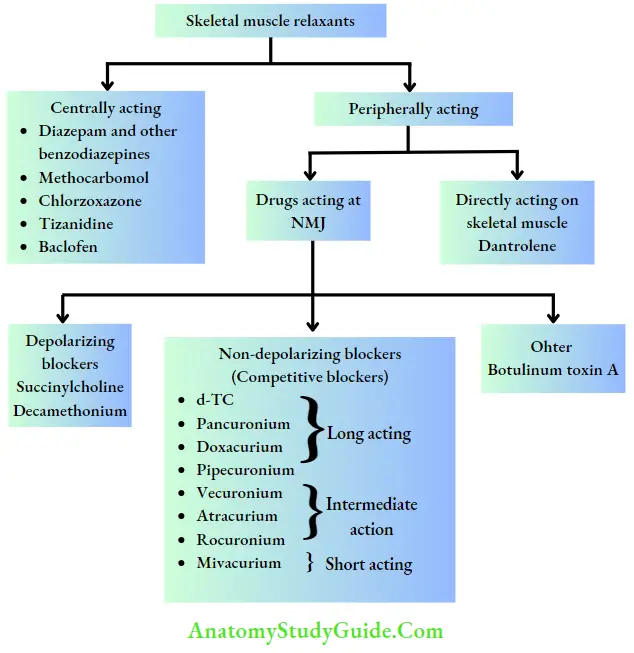
Centrally Acting Skeletal Muscle Relaxants
Most of the centrally acting skeletal muscle relaxants are available in combination with one or other NSAIDs. All of them cause a certain degree of sedation.
They are baclofen, diazepam and other benzodiazepines, tizanidine, chlorzoxazone, methocarbamol, etc. All are effective orally. Baclofen and benzodiazepines can also be administered parenterally. They are used to reduce spasms associated with temporomandibular joint pain, cerebral palsy, trauma, muscular strain, tetanus, etc.
Characteristics of Centrally Acting Skeletal Muscle Relaxants

Block release of excitatory transmitter in the spinal cord → depress polysynaptic reflexes.
Neuromuscular Blockers
Unlike centrally acting skeletal muscle relaxants, these drugs interfere with neuromuscular transmission and do not affect CNS. These are administered intravenously. These drugs include nondepolarizing (competitive) and depolarizing blockers.
- Depolarizing blockers: succinylcholine (suxamethonium) Succinylcholine (SCh) is a quaternary ammonium compound. The structure resembles two molecules of ACh linked together. It acts as a partial agonist at NM receptors, hence causing initial fasciculations and later flaccid paralysis due to prolonged depolarization (phase 1 block).
- With continued exposure to the drug, the membrane becomes desensitized, which leads to phase II block, which resembles the non-depolarizing block and is partially reversed by anticholinesterases. SCh is rapidly hydrolysed by pseudocholinesterase, hence has a very short duration of action (3–8 min).
- Transient apnoea is usually seen at the peak of its action. In people with liver disease or atypical pseudocholinesterase due to genetic defect, the metabolism of succinylcholine becomes slow, which results in severe neuromuscular blockade leading to respiratory paralysis with prolonged apnoea. This is referred to as ‘succinylcholine apnoea’. There is no antidote available; therefore:
- Fresh frozen plasma should be infused.
- Patient should be ventilated artificially until full recovery.
- Adverse effects
- Muscle pain is due to initial fasciculations (muscle soreness).
Increased IOP due to contraction of external ocular muscles and it lasts for few minutes. - Aspiration of gastric contents may occur due to increased intragastric pressure.
- Hyperkalaemia – fasciculations release K+ into the blood.
- Sinus bradycardia is due to vagal stimulation.
- Succinylcholine apnoea (prolonged apnoea).
- Malignant hyperthermia, especially when used with halothane in genetically susceptible individuals. This is treated with intravenous dantrolene, rapid cooling, inhalation of 100% oxygen, and control of acidosis.
- Competitive blockers (nondepolarizing blockers) Claude Bernard showed experimentally the site of action of curare. Curare is a mixture of alkaloids and was used as arrow poison. Among them, d-Tubocurarine (d-TC) is the most important alkaloid, which has NM-blocking activity. d-TC is the prototype drug of competitive blockers
Features of Nondepolarizing (Competitive) Blockers
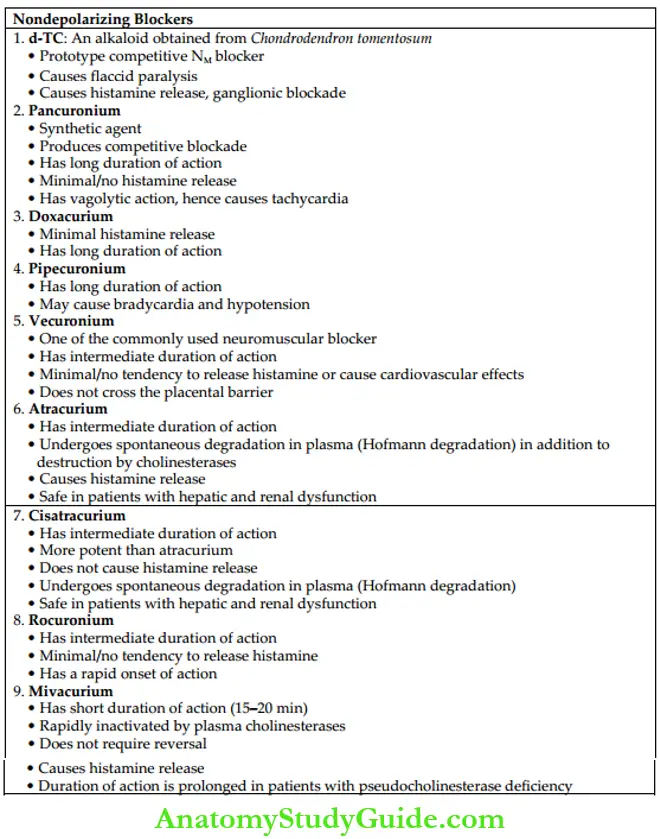
- Mechanism of action ACh is the agonist, whereas d-TC is the antagonist at NM receptors. Curariform drugs competitively antagonize the actions of ACh at the NM receptors of the NMJ. Anticholinesterases (neostigmine or edrophonium) are used to reverse the effects of competitive blockers by increasing the concentration of ACh.

- Actions Competitive blockers produce flaccid paralysis. The order of muscles affected is extrinsic eye muscles–neck (muscles of phonation and swallowing)–face–hands–feet– limbs–trunk and finally, the respiratory muscles (intercostal muscles and diaphragm). But recovery occurs in reverse order – the respiratory muscles are the first to recover. Consciousness and appreciation of pain are not affected.
- d-TC, mivacurium, and atracurium cause histamine release which can manifest as hypotension, bronchospasm, etc.
- Pancuronium, vecuronium, doxacurium, and rocuronium have minimal/no tendency to cause histamine release.
- Vecuronium, doxacurium, and rocuronium have a minimal tendency to cause cardiovascular effects like hypotension and cardiovascular collapse. These effects are prominent with d-TC, pancuronium, atracurium, and mivacurium.
- Among competitive neuromuscular blockers, rocuronium has a rapid onset of, hence it can be used for endotracheal intubation.
Comparative features of d-TC and SCh are shown in Table.

Comparative Features of d-TC and Succinylcholine
- Pharmacokinetics Neuromuscular blockers are quaternary ammonium compounds. They are highly ionized and, hence are poorly absorbed from the GI tract. They are administered intravenously. They are mainly confined to ECF space; and do not cross the placental and blood-brain barrier (BBB). They are metabolized in the liver and some are excreted unchanged in urine.
- Adverse effects The adverse effects of nondepolarizing drugs are hypotension, respiratory paralysis, bronchospasm, and aspiration of gastric contents.
Factors affecting the action of neuromuscular blockers
- pH changes: Metabolic acidosis and respiratory acidosis increase the duration of the block.
- Hypothermia: It potentiates neuromuscular block by delaying the metabolism and elimination of these drugs.
- Myasthenia gravis: Myasthenic patients are highly sensitive to competitive neuromuscular blockers.
- Aminoglycoside antibiotics: They potentiate the effect of both competitive as well as nondepolarizing blockers by inhibiting the presynaptic release of ACh.
- Inhalational anesthetics: Anaesthetics like halothane, isoflurane, and sevoflurane increase the effects of neuromuscular-blocking agents.
- Neuromuscular Blockers Drug interactions
1. Nondepolarizing blockers × antibiotics
Aminoglycosides inhibit the release of ACh from motor nerves and potentiate the effect of nondepolarizing blockers, hence requiring dose reduction in patients treated with aminoglycosides. Tetracyclines and clindamycin also potentiate the effect of nondepolarizing blockers.
2. Thiazides/loop diuretics × nondepolarizing blockers
Hypokalaemia caused by thiazides/loop diuretics may potentiate the effect of nondepolarizing blockers.
3. Succinylcholine × thiopentone
These drugs are chemically incompatible (in vitro; pharmaceutical interaction), hence resulting in precipitation when they are mixed in the same syringe.
4. General anesthetics × nondepolarizing blockers
Ether has curarimimetic effect on skeletal muscle, hence enhancing the effect of nondepolarizing blockers. Fluorinated anesthetics (isoflurane, desflurane, and sevoflurane) also produce similar effects but to a lesser extent.
- Neuromuscular Blockers Uses
- The main use of neuromuscular blockers is as an adjuvant to general anesthetics for producing satisfactory skeletal muscle relaxation during surgical procedures. Succinylcholine is preferred for short procedures, for example, diagnostic endoscopies, endotracheal intubation, and orthopedic manipulations. Vecuronium is commonly used in routine surgeries. Pancuronium and pipecuronium are used in surgeries of long duration.
- In dentistry: Muscle relaxants are useful to allow manipulation of bone fragments in fracture of the mandible and to facilitate opening of the mouth for diagnosis and treatment of trismus.
- Succinylcholine/mivacurium is used during electroconvulsive therapy (ECT) for psychiatric disorders to prevent trauma due to convulsions.
- Tetanus and status epilepticus when not controlled by other drugs.
- Competitive neuromuscular blockers can be used for ventilatory support in critically ill patients.
- Reversal of action of competitive neuromuscular blockers
Edrophonium or neostigmine by increasing the concentration of ACh reverses the effect of d-TC and other competitive blockers at NMJ. Prior atropine administration is necessary to block the muscarinic effects of anticholinesterases. Mivacurium (short-acting), atracurium (intermediate-acting), etc. do not require reversal.

- Sugammadex
It is administered intravenously for rapid reversal of the neuromuscular blocking action of rocuronium and vecuronium. It encapsulates the drugs, thus preventing their action.
Directly Acting Drug: Dantrolene
- Dantrolene is a directly acting skeletal muscle relaxant. It inhibits depolarization-induced Ca2+ release (by blocking ryanodine receptors) from the sarcoplasmic reticulum and produces skeletal muscle relaxation. Intravenous dantrolene is the life-saving drug in malignant hyperthermia.
- It is used orally to reduce spasms in multiple sclerosis, cerebral palsy, spinal injuries, etc. The side effects are drowsiness, diarrhea, dizziness, headache, fatigue, and rarely hepatotoxicity.
Botulinum Toxin A
- It is obtained from Clostridium botulinum, a gram-positive anaerobic bacterium. The toxin prevents the release of ACh into the synaptic cleft by inhibiting proteins necessary for the release of ACh. Thus, it normalizes the tone in hyperreactive or spastic muscles when given locally.
- It is given intradermally for antiwrinkle effect in cosmetic procedures and into the muscle in multiple doses for spasticity or dystonia. Botulinum toxin A is injected under ultrasound guidance into salivary glands in sialorrhoea and drooling.
- Adverse effects are pain at the site of injection, muscle paralysis, myalgia and occasionally rashes.
Adrenergic agonists (Sympathomimetic Agents)
Adrenergic agonists mimic the actions of sympathetic stimulation.
Adrenergic Transmission
The transmitter in the sympathetic system is noradrenaline (NA; norepinephrine). Nerves that synthesize, store, and release NA are called adrenergic (sympathetic) nerves.
- Synthesis of catecholamines begins with the amino acid tyrosine, which is transported into the adrenergic neuron by active transport. In the neuronal cytosol, tyrosine is converted to DOPA by tyrosine hydroxylase and DOPA to dopamine (DA) by DOPA decarboxylase.
- Dopamine enters the storage vesicles of nerve terminals by active transport, where it is converted to NA by the enzyme dopamine β-hydroxylase (this enzyme is present only in storage vesicles); NA formed gets stored in the vesicles.
- In the adrenal medulla, NA is further converted to adrenaline by N-methyltransferase. Small quantities of NA are released continuously into the synaptic cleft and large quantities during nerve stimulation.

Drugs Affecting Adrenergic Transmission and Their Uses
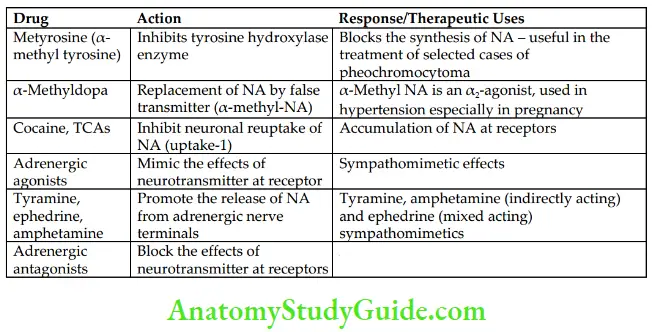
Three processes are involved in the termination of action of released NA in the synaptic cleft (the fate of released NA in the synaptic cleft):
- Most of the released NA is taken back into adrenergic nerve terminals (neuronal reuptake), which are either stored in vesicles or inactivated by mitochondrial monoamine oxidase (MAO) in the cytosol. Neuronal reuptake is the most important mechanism through which termination of action of NA takes place in the synaptic cleft.
- A small amount of NA from the synaptic cleft diffuses into circulation and gets inactivated in the liver by catechol-O-methyltransferase (COMT) and MAO.
- A small quantity of NA is transported into other tissues (extraneuronal uptake).
Metabolism Of Catecholamines
The main metabolite of catecholamines is vanillylmandelic acid (VMA). It is excreted in urine.
Types, Distribution, And Functions Of Adrenergic Receptors
Ahlquist divided adrenergic receptors into α and β types, which are located on the cell membrane. They are further divided into various subtypes, which are as follows:

The distribution of various adrenergic receptors is indicated.
- Effect of activation of α1-receptors
- Blood vessels: Constriction.
- GI sphincter (anal): Increase in tone.
- Urinary sphincter: Increase in tone.
- Radial muscle (iris): Contraction (mydriasis).
- Effect of activation of presynaptic α2-receptors
- Mediate negative feedback control on NA secretion (i.e. stimulation of α2– receptors decreases release of NA from sympathetic nerve endings).
- Effect of activation of postsynaptic vascular α2-receptors
- Mediate stimulatory effects: Vasoconstriction and vasoconstriction.
- Effect of activation of α1-receptors on various secretions
- Beta cells of islets of Langerhans in the pancreas: Decrease in insulin secretion.
- Ciliary epithelium: Reduction of aqueous humor secretion.
- Sympathetic nerve endings: Decrease in NA release.
- Effect of activation of β1-receptors
- Heart: Cardiac stimulation.
- Kidney: Promote renin release.
- Stimulatory effects due to activation of β2-receptors
- Liver: Stimulation of glycogenolysis.
- Skeletal muscle: Contraction.
- Ciliary epithelium: Increase in secretion of aqueous humor.
- Uptake of K+ into cells.
- Inhibitory effects due to activation of β2-receptors
- Bronchial, uterine (pregnant), vascular, and bladder smooth muscles: Relaxation.
- In GI smooth muscle, activation of both α- and β-receptors causes relaxation.
- Effect of activation of β3-receptors
- Adipose tissue: Lipolysis.

Adrenergic Drugs (Sympathomimetics)
The sympathomimetic drugs mimic the effects of sympathetic nerve stimulation. They are also referred to as adrenergic agonists.

- Classification of sympathomimetics
- On the basis of their chemical structure

- Catecholamines: Sympathomimetics with catechol nuclei are called catecholamines, for example, adrenaline, noradrenaline, dopamine, dobutamine, and isoprenaline.
- Noncatecholamines: Sympathomimetics that lack catechol nuclei are called noncatecholamines, for example, tyramine, ephedrine, amphetamine, phenylephrine, and salbutamol.
- On the basis of their mechanism of action:
- Direct acting: They act directly by stimulating adrenergic receptors.
- Indirect acting: They act by releasing NA from adrenergic nerve endings.
- Mixed acting: These drugs act both directly and indirectly.
- On the basis of their therapeutic use:
- To raise blood pressure in shock: Dopamine, noradrenaline, ephedrine, phenylephrine, methoxamine, and mephentermine.
- As bronchodilator: Salbutamol, terbutaline, salmeterol, formoterol.
- As cardiac stimulant: Adrenaline, isoprenaline, dobutamine.
- As CNS stimulant: Modafinil, amphetamine, dextroamphetamine.
- For local vasoconstrictor effect: Adrenaline.
- As nasal decongestant: Phenylephrine, xylometazoline, pseudoephedrine, oxymetazoline, naphazoline.
- As mydriatic: Ephedrine, phenylephrine.
- As anorexiant: Dextroamphetamine, mazindol, phentermine, sibutramine.
Summary of Sympathomimetic Agents

Sympathomimetics
- Adrenaline (epinephrine): α1-, α2-, β1-, β2– and β3-agonist It is a catecholamine, which is secreted mainly by the adrenal medulla. Adrenaline is a direct-acting nonselective adrenergic agonist.
- Pharmacological actions Adrenaline acts on α1-, α2-, β1-, β2– and β3-receptors.
- Cardiovascular system
- Heart: Adrenaline is a powerful cardiac stimulant. It acts mainly by interacting with β1-receptors and produces various effects. They are as follows:
- Increase in heart rate (positive chronotropic effect).
- Increase in myocardial contractility (positive inotropic effect).
- Increase in conduction velocity (positive dromotropic effect).
- Increase in cardiac output.
- Increase in automaticity.
- Cardiac work and oxygen requirement is markedly increased.
- Increase in the excitability and tendency to cause cardiac arrhythmias.
- Blood vessels and BP: Blood vessels of the skin and mucous membranes (α1– receptors) are constricted by adrenaline. It also constricts renal, mesenteric, pulmonary, and splanchnic vessels, but dilates blood vessels of skeletal muscle and coronary vessels (β2). Intravenous administration of adrenaline in moderate doses produces a biphasic effect. There is an initial rise in BP due to α1 (blood vessels) and β1 (heart) actions, followed by a fall in BP due to β2-mediated vasodilatation in skeletal muscle. Administration of adrenaline after α-blocker produces only a fall in BP (β2-action). This is referred to as the vasomotor reversal of Dale. If adrenaline is rapidly injected intravenously, there is an increase in both systolic and diastolic BP.

- Heart: Adrenaline is a powerful cardiac stimulant. It acts mainly by interacting with β1-receptors and produces various effects. They are as follows:
- Respiratory system: Adrenaline rapidly relaxes (β2) bronchial smooth muscle. It is a potent bronchodilator but has a short duration of action. It inhibits the release of inflammatory mediators from mast cells (β2). It also reduces secretions and relieves mucosal congestion by vasoconstrictor effect (α1).
- GIT: It relaxes the smooth muscle of the gut (α and β2). It reduces intestinal tone and peristaltic movements. But the effects are transient.
- Bladder: It relaxes the detrusor muscle (β2) and contracts the sphincter (β1). As a result, it may cause difficulty in urination.
- CNS: In therapeutic doses, adrenaline does not cross the BBB and hence CNS effects are very minimal. But in high doses, it may cause headaches, restlessness, and tremor.
- Eye: Adrenaline has poor penetration through the cornea when applied topically into the eye. Hence, it is administered as a prodrug.
- Metabolic effects: Adrenaline increases the blood glucose level by:
- Stimulating hepatic glycogenolysis (β2), which is the predominant effect.
- Reducing insulin secretion through α2-action.
- Decreasing the uptake of glucose by peripheral tissues.
- Other effects It reduces plasma K+ levels by promoting the uptake of K+ into cells, particularly into the skeletal muscle (β1).
- Cardiovascular system
- Pharmacokinetics Adrenaline is not suitable for oral administration because of its rapid inactivation in the GI mucosa and liver. Adrenaline can be given subcutaneously (s.c.). In anaphylactic shock, absorption of subcutaneous adrenaline is very poor, hence given intramuscularly. In cardiac arrest, it is given intravenously. It does not cross BBB, is rapidly metabolized by COMT and MAO, and the metabolites are excreted in urine.
- Adverse effects and contraindications The adverse effects of adrenaline are due to an extension of its pharmacological actions. They are tachycardia, palpitation, headache, restlessness, tremor, and a rise in BP.
- The serious side effects are cerebral hemorrhage and cardiac arrhythmias. In high concentrations, adrenaline may cause acute pulmonary edema due to the shift of blood from systemic to pulmonary circulation.
- Adrenaline is contraindicated in most cardiovascular diseases such as hypertension, angina, cardiac arrhythmias, and congestive cardiac failure (CCF). In patients on β-blockers, it may cause hypertensive crisis and cerebral hemorrhage due to unopposed action on vascular α1-receptors.
- Therapeutic uses of adrenaline (abcde)
- Anaphylactic shock: Adrenaline is the life-saving drug in anaphylactic shock. Adrenaline 0.3–0.5 mL of 1:1000 solution (1 mg/mL) is administered intramuscularly. It rapidly reverses the manifestations of severe allergic reactions. The beneficial effect of adrenaline in anaphylactic shock is shown below. Adrenaline produces the following effects:
- β1-mediated cardiac stimulation (↑ heart rate and ↑ force of contraction). +
- α1-mediated vasoconstriction ↑peripheral resistance. These actions help to ↑ BP
- α1-mediated vasoconstriction ↓mucosal oedema (↓ laryngeal oedema).
- β2-Stimulation bronchodilation, ↓ release of mediators from mast cells.
- It is a physiological antagonist of histamine.
- Cardiac resuscitation: In the treatment of cardiac arrest due to drowning or electrocution, adrenaline is injected intravenously in 1:10,000 (0.1 mg/mL) concentration along with other supportive measures such as external cardiac massage
- Prolong the Duration of local anesthesia: Adrenaline (1:100,000) with lignocaine. Adrenaline, by its vasoconstrictor effect (α1) delays absorption of local anesthetic and prolongs the duration of local anesthesia.
- Control Epistaxis and other capillary oozing: Adrenaline is used as a local hemostatic to control bleeding following tooth extraction and during surgical procedures in the nose, throat, larynx, etc. because of its vasoconstrictor effect.
- Bronchial asthma: Adrenaline is a powerful bronchodilator and has a rapid onset but a short duration of action. It is useful for acute attacks. Its use has declined because of its dangerous cardiac stimulant effect. The beneficial effects of adrenaline in bronchial asthma are shown. Adrenaline 0.3–0.5 mL of 1:1000 solution is given subcutaneously. It can be given by nebulization (as inhalation). Its use has declined because of its dangerous cardiac stimulant effect.
- Anaphylactic shock: Adrenaline is the life-saving drug in anaphylactic shock. Adrenaline 0.3–0.5 mL of 1:1000 solution (1 mg/mL) is administered intramuscularly. It rapidly reverses the manifestations of severe allergic reactions. The beneficial effect of adrenaline in anaphylactic shock is shown below. Adrenaline produces the following effects:
- Pharmacological actions Adrenaline acts on α1-, α2-, β1-, β2– and β3-receptors.

-
-
- Glaucoma: Adrenaline has poor penetration when applied locally into the eye; hence, it is administered as a prodrug.
-
- Noradrenaline: α1-, α2– and β1-agonist Noradrenaline is a catecholamine. It is the main neurotransmitter in the adrenergic system. It acts on α1-, α2– and β1-adrenergic receptors; has negligible β2 action. The main action of NA is on the cardiovascular system. It has a direct cardiac-stimulant effect (β1); and constricts all the blood vessels (α1) including those of skin, mucous membrane, renal, mesenteric, pulmonary, skeletal muscle, etc. The systolic, diastolic, and pulse pressures are increased. There is reflex bradycardia. Noradrenaline, like adrenaline, is not effective orally. It is not suitable for s.c., i.m. or direct i.v. injection because of necrosis and sloughing of the tissues at the site of injection. It is administered by i.v. infusion. It can be used to raise BP in hypotensive states, but it may decrease blood flow to vital organs by causing widespread vasoconstriction.
Comparative Features of Adrenaline and Noradrenaline
- Isoprenaline (isoproterenol): β1-, β2-, and β3-agonist It is a synthetic, nonselective β-receptor agonist with a catechol nucleus. It has potent β actions but no action at α-receptors. Isoprenaline is a powerful cardiac stimulant. It has positive inotropic, chronotropic, and dromotropic effects. It dilates renal, mesenteric, and skeletal muscle blood vessels. Systolic BP is minimally changed but the diastolic and mean arterial pressures are reduced. It relaxes bronchial and GI smooth muscles. Isoprenaline is not effective orally because of extensive first-pass metabolism. It can be given parenterally or as an aerosol. It is metabolized by COMT. Isoprenaline is used to increase the heart rate in heart block. In bronchial asthma, isoprenaline has been replaced by selective β2-agonists. Side effects are tachycardia, palpitation, cardiac arrhythmias, etc. due to its powerful cardiac-stimulant effect.
- Dobutamine: Relatively selective β1-agonist Dobutamine, a synthetic catecholamine, structurally resembles dopamine. It is a potent inotropic agent but causes only a slight increase in heart rate. It does not act on dopaminergic receptors. Total peripheral resistance is not significantly affected. It is administered by i.v. infusion in patients with acute heart failure. The side effects are tachycardia, rise in BP, etc.
- Salbutamol, terbutaline, salmeterol, formoterol: Selective β2-adrenergic agonists
Selective β2-agonists are the main drugs used in bronchial asthma, for example,n salbutamol, levalbuterol, terbutaline, salmeterol, and formoterol. Nonselective β-agonist like adrenaline is rarely used because of its cardiac side effects.- Pharmacological actions of selective β2-agonists are depicted. They cause bronchodilatation, relaxation of the pregnant uterus, dilatation of blood vessels supplying the skeletal muscles, promote hepatic glycogenolysis, and uptake of K+ into cells.

-
- Therapeutic uses
- Bronchial asthma: Selective β2-agonists are usually administered by aerosol. They produce prompt bronchodilatation (salbutamol, terbutaline, and formoterol) with minimal systemic side effects.
- Premature labor: On oral or parenteral administration, salbutamol and terbutaline relax the pregnant uterus by interacting with β2-receptors, hence are used to delay premature labor.
- Hyperkalemia: Selective β2-agonists are useful in hyperkalemia as they promote the uptake of potassium into cells, especially into skeletal muscles.
- Adverse effects of selective β2-agonists
- Tremor is due to the stimulation of β2-receptors of skeletal muscle. Tolerance develops to this effect on continued administration.
- Tachycardia and palpitation are due to stimulation of β1-receptors of the heart (β2– selectivity is not absolute – may cause cardiac side effects).
- Hyperglycaemia may occur in diabetics following parenteral administration of β2-agonists.
- Hypokalaemia is due to the shift of K+ into cells.
- Therapeutic uses
- Phenylephrine, methoxamine, mephentermine: Selective α1-adrenergic agonists Like ephedrine, mephentermine also has a cardiac-stimulant effect. They are used parenterally to raise the BP in hypotensive states. Phenylephrine is also used topically as a mydriatic and as a nasal decongestant.

- Nasal decongestants The commonly used α-agonists as nasal decongestants are naphazoline, oxymetazoline, xylometazoline (topical), pseudoephedrine (oral), and phenylephrine (oral, topical).
- They are used in allergic rhinitis, common cold, sinusitis, etc. These drugs stimulate α- receptors and cause vasoconstriction in the nasal mucous membrane, thus relieving nasal congestion. On prolonged use, they cause rebound congestion (after congestion).
- Atrophic rhinitis, anosmia, and local irritation are the other adverse effects seen with topical decongestants. If systemically absorbed, these drugs may aggravate hypertension.
- Pseudoephedrine and phenylephrine are the most commonly used oral preparations. These drugs cause less rebound phenomenon, but systemic side effects like hypertension and CNS stimulation are common.
- They should not be combined with MAO inhibitors because of the risk of hypertensive crisis, which could be fatal. Phenylpropanolamine was used as a nasal decongestant. It has been banned because of the increased incidence of stroke.
- Selective α2-adrenergic agonists
They include clonidine, α-methyldopa, and tizanidine. Apraclonidine and brimonidine, selective α2-agonists, are typically used in glaucoma.
Indirect-Acting Sympathomimetics
- Amphetamine is an indirect-acting sympathomimetic agent and has a potent CNS stimulant effect. It occurs in two isomers. The d-isomer has more potent CNS effects than the l-isomer on CVS. The side effects are restlessness, insomnia, confusion, fatigue, tremors, hallucinations, and suicidal tendencies. The cardiac side effects are tachycardia, palpitation, hypertension, angina, and cardiac arrhythmias.
- Treatment of acute intoxication
- Acidification of urine with ascorbic acid (vitamin C) promotes the excretion of amphetamine, which is a basic drug.
- Sedatives are effective in controlling CNS symptoms and sodium nitroprusside is for severe hypertension.
- Uses
- Narcolepsy: It is a sleep disorder characterized by recurrent episodes of uncontrollable desire for sleep. Amphetamine improves narcolepsy by its CNS stimulant effect.
- As an anorexiant: Amphetamine-like drugs reduce body weight by suppressing the hypothalamic feeding center. Tolerance to this effect develops rapidly.
- Attention-deficit hyperactivity disorder: Amphetamine acts paradoxically and controls the activity in children with hyperactivity disorder. The main adverse effects are loss of appetite and insomnia. Methylphenidate and dextroamphetamine are also useful in this disorder.
- Treatment of acute intoxication
Mixed-Acting Sympathomimetics
- Ephedrine: α- and β-agonist with NA release Ephedrine is a mixed-acting adrenergic agonist. It is an alkaloid, acts on α1-, α2-, β1– and β2-receptors and releases NA from sympathetic nerve endings.
- Pharmacological actions

-
- Intravenous ephedrine is the drug of choice to treat hypotension due to spinal anesthesia, as it increases peripheral vascular resistance, heart rate, cardiac output, and thus BP. The side effects are insomnia, hypertension, tachycardia, palpitation, and difficulty in urination; tachyphylaxis occurs on repeated administration.
- Dopamine: α1-, α2-, β1– and D1-agonist with NA release Dopamine (DA) is a catecholamine and the immediate metabolic precursor of noradrenaline (NA). It acts on dopaminergic D1 receptors as well as β1– and α1– adrenergic receptors. DA, like adrenaline and noradrenaline, is not effective orally. Dopamine is rapidly inactivated by COMT and MAO and is administered by i.v. infusion.
- Pharmacological actions
- At low doses, it selectively dilates renal, mesenteric, and coronary blood vessels by acting on D1 receptors increasing glomerular filtration rate (GFR) and urine output.
- At moderate doses, dopamine stimulates β1-receptors of the heart and increases myocardial contractility and cardiac output, but tachycardia is less prominent. It also stimulates dopaminergic receptors increasing GFR.
- At high doses (>10 mcg/kg/min), it stimulates vascular α1-adrenergic receptors and causes generalized vasoconstriction. This increases afterload and reduces blood flow to renal, mesenteric, and other vital organs. So, the beneficial effect seen with low-to-moderate doses of DA is lost at higher doses.
- Precautions and adverse effects During dopamine infusion, the dose, BP, heart rate, ECG, and urine output should be carefully monitored. The adverse effects seen are mainly due to sympathetic stimulation. They are nausea, vomiting, headache, hypertension, tachycardia, cardiac arrhythmias, and angina.
- Therapeutic uses
- Cardiogenic and septic shock: Dopamine can be used because it increases BP as well as selectively dilates renal, mesenteric, and coronary blood vessels, and improves blood flow to vital organs.
- Pharmacological actions
- Anorectics (anorexiants) Amphetamine-like drugs promote weight loss by acting on the hypothalamic feeding center.

The main adverse effects of these agents are addiction liability, rise in BP, palpitation, sleep disturbances, depression, and dry mouth.
Adrenergic Receptor Blockers
Adrenergic-receptor antagonists block the effects of sympathetic stimulation and adrenergic agonists mediated through α- and β-receptors.

Alpha-Adrenergic Blockers
Pharmacological effects of α-blockers
They block α-receptors, thus inhibiting the α-receptor-mediated responses of sympathetic stimulation and adrenergic drugs.

Alpha-Adrenergic Blockers Classification

Irreversible Nonselective Α-Blocker
- Phenoxybenzamine is a nonselective α-adrenergic blocker that blocks both α1– and α2– receptors. It binds covalently to α-receptors and causes irreversible blockade. It also inhibits the reuptake of NA into the adrenergic nerve endings.

-
- Pharmacological effects Peripheral vascular resistance is reduced due to the blockade of vascular α1– receptors and has a predominant vasodilating effect.
- Increased release of NA from the adrenergic nerve endings due to the blockade of presynaptic α2-receptors. This may cause cardiac stimulation and produce tachycardia, palpitation, cardiac arrhythmias, etc. Other effects are shown.
Phenoxybenzamine is given orally or through slow i.v. infusion. It has a slow onset but long duration of action because of the irreversible blockade of α-receptors. Its main use is in the treatment of pheochromocytoma. The side effects are postural hypotension, tachycardia, palpitation, diarrhea, nasal stuffiness, giddiness, and impotence.
Reversible Nonselective Α-Blockers
- Phentolamine Phentolamine is an imidazoline derivative. It competitively blocks the effects of NA at both α1– and α1-adrenergic receptors (competitive antagonism). Venodilatation is more than arteriolar dilatation. It can also block 5-HT receptors, K+ channels and cause histamine release from mast cells.

Phentolamine is given intravenously and has a rapid onset but a short duration of action.
-
- Adverse effects include tachycardia, palpitation, and arrhythmias; angina and MI may be precipitated.
- Tolazoline is similar to phentolamine and is rarely used.
Selective α1-Blockers
Prazosin is a potent and selective α1-adrenergic receptor blocker. It is given orally. It is well absorbed from the GI tract but undergoes extensive first-pass metabolism. The effects of α-blockade are depicted. Unlike nonselective α-blockers, selective α1-blockers (presynaptic α2-receptors are not blocked) produce minimal or no tachycardia. It causes both arteriolar and venodilatation; arteriolar dilatation is more prominent.
- Selective α1-Blockers Adverse effects
- First-dose phenomenon (a mechanism): Within 30–90 min of oral administration of the first dose of prazosin, postural hypotension, and syncopal attacks may be seen. Therefore, the initial dose should be small (1 mg). It is usually given at bedtime so that the patient remains in bed for several hours and the risk of syncopal attack is reduced. It may cause nasal stuffiness, tachycardia, impaired ejaculation, and impotence.
- Other selective α1-blockers
- Terazosin is similar to prazosin but less potent than prazosin. It is almost completely absorbed after oral administration and has a longer duration of action.
- Doxazosin is the longest-acting selective α1-blocker. The hemodynamic effects, bioavailability, and extent of metabolism are similar to prazosin.
- Alfuzosin blocks all subtypes of α1-receptors (α1A, α1B, and α1D). It is orally effective and used in benign prostatic hyperplasia (BPH).
- Tamsulosin is an uroselective α1-blocker (α1A). At low doses, it reduces the resistance to the flow of urine with little effect on BP. It is administered orally and is the preferred α1-blocker for the treatment of BPH in normotensive patients. It may cause retrograde ejaculation.
Therapeutic Uses Of α-Blockers
- Pheochromocytoma: It is a tumor of the adrenal medulla, which releases large amounts of adrenaline and NA. The signs and symptoms include a sudden and intermittent rise in BP with headache, palpitation, and excessive sweating.
- The diagnosis of pheochromocytoma is usually made by estimating catecholamines, vanillylmandelic acid (VMA), and other metabolites in blood and urine (normal VMA: 4–8 mg/24 h urine sample), computed tomography (CT), and magnetic resonance imaging (MRI) scans. The definitive treatment for pheochromocytoma is surgery.
- In the preoperative period, phenoxybenzamine is used to control hypertension. Blockade of vascular α1-receptors causes vasodilatation and a fall in BP. It is also used for inoperable cases.
- During surgery, handling of the tumor results in the sudden release of a large quantity of catecholamines, which may cause a marked rise in BP that can be controlled by i.v. phentolamine.
- Hypertensive emergencies: Intravenous phentolamine can be used in the following conditions because of its rapid onset of action:
- To control hypertensive episodes intraoperatively during surgery of pheochromocytoma.
- To control hypertensive crisis due to clonidine withdrawal.
- To control hypertensive crisis due to ‘cheese reaction’
- Essential hypertension: Among α-blockers, selective α1-antagonists are preferred to nonselective α-blockers in the treatment of mild-to-moderate hypertension. Selective α1-antagonists cause less tachycardia and have favorable effects on lipid profile.
- Benign prostatic hyperplasia (BPH): Selective α1-blockers are used in BPH; they decrease the tone of smooth muscle in the neck bladder and prostate resulting in a reduction in resistance to urinary flow. Prazosin, doxazosin, terazosin, and alfuzosin are particularly useful in patients who also have hypertension. Tamsulosin is preferred for BPH in normotensive patients.
- Selective α2-Adrenergic Blocker
- Yohimbine: Yohimbine is an alkaloid. It competitively blocks α2-receptors. It is an aphrodisiac but is rarely used therapeutically.
Beta-Adrenergic Blockers
β-Adrenergic antagonists block the β-receptor-mediated effects of sympathetic stimulation and adrenergic drugs.
Classification

Pindolol, acebutolol, and labetalol have partial agonistic activity (intrinsic sympathomimetic activity). They stimulate β-receptors partially in the absence of catecholamines.
Propranolol, acebutolol, labetalol, metoprolol, and pindolol have membrane-stabilizing activity (local anesthetic activity).
Mechanism of action
Propranolol is the prototype drug. β-Blockers competitively block the β-mediated actions of catecholamines and other adrenergic agonists.

Pharmacological properties of β-blockers
- Cardiovascular system:
- Heart: β-Blockers depress all the cardiac properties.
- Decrease heart rate (negative chronotropic effect).
- Decrease the force of myocardial contractility (negative inotropic effect).
- Decrease cardiac output.
- Depress SA node and AV nodal activity.
- Increase the refractory period of the AV node.
- Decrease conduction in atria and A–V node (negative dromotropic effect).
- Decrease automaticity of ectopic foci.
- Decrease cardiac work, thus reducing the O2 requirement of the myocardium.
- Only in high doses, do some of them have a membrane-stabilizing effect.
- Blood vessels: Blockade of β2-receptors of the blood vessels initially may cause a rise in peripheral vascular resistance (PVR) due to the unopposed α1– action. However, continued administration of these drugs leads to a fall in peripheral vascular resistance in patients with hypertension due to chronic reduction in cardiac output. Both systolic and diastolic BP are reduced.
- They also reduce the release of renin from the juxtaglomerular apparatus due to the blockade of β1-receptors and decrease central sympathetic outflow.
- Heart: β-Blockers depress all the cardiac properties.
- Respiratory system: Blockade of β2-receptors in bronchial smooth muscle can produce severe bronchospasm in patients with COPD and asthma. Therefore, β- blockers should be avoided in patients with asthma and COPD. Selective β1– blockers such as atenolol and metoprolol are less likely to cause bronchospasm.
- Skeletal muscle: On chronic use, β-blockers may cause skeletal muscle weakness and tiredness due to the blockade of β2-receptors of the skeletal muscle and blood vessels supplying it. They also reduce stress-induced tremors.
- Metabolic effects: β-Blockers inhibit glycogenolysis and delay recovery from hypoglycemia. They also mask the warning signs and symptoms of hypoglycemia. Therefore, β-blockers should be used cautiously in diabetics on hypoglycaemic agents. Chronic use of nonselective β-blockers decreases HDL (high-density lipoprotein) cholesterol and LDL (low-density lipoprotein) cholesterol ratio which may increase the risk of coronary artery disease.
- Eye: β-Blockers on topical administration decrease IOP by reducing the secretion of aqueous humor.
- Pharmacokinetics
Propranolol is highly lipid soluble and is well absorbed from the GI tract. However, the bioavailability of propranolol is low because of its extensive first-pass metabolism. It is highly bound to plasma proteins, has a large volume of distribution, freely crosses BBB, and metabolites are excreted in urine.
- Adverse effects of β-blockers
They are mainly an extension of pharmacological actions.
- CVS:
- Bradycardia, heart block, and may precipitate CHF in patients with low cardiac reserve.
- Blockade of vascular β2-receptors causes unopposed α1 action, reduces further blood supply, and may worsen peripheral vascular disease.
- β-Blockers can exacerbate Prinzmetal angina (variant angina) due to unopposed α1 action, hence are contraindicated in this condition.
- Respiratory system: Blockade of β2-receptors in the bronchial smooth muscle can cause severe bronchospasm in patients with asthma and COPD. Hence, β- blockers are contraindicated in the above conditions.
- CNS: Sleep disturbances, hallucinations, fatigue, and mental depression.
- Metabolic: Recovery from hypoglycemia (induced by antidiabetic drugs) is delayed by β-blockers. β-Blockers may also mask the warning signs and symptoms of hypoglycemia.
- Muscular weakness and tiredness: These are due to reduced blood flow to skeletal muscle.
- Withdrawal symptoms: Abrupt withdrawal of β-blockers after chronic use is dangerous because angina or frank myocardial infarction and even sudden death can occur. This is due to the upregulation (supersensitivity) of β-receptors in response to prolonged blockade.
- Drug interactions
- Propranolol × verapamil: They produce additive cardiac depressant effects and may cause CCF, bradyarrhythmias, heart block, or even cardiac arrest.
- Propranolol × lignocaine: Propranolol reduces the clearance of lignocaine by decreasing hepatic blood flow.
- Insulin/sulfonylureas × β-blockers: Nonselective β-blockers inhibit glycogenolysis and delay recovery from hypoglycemia.

- Propranolol × nonsteroidal anti-inflammatory drugs (NSAIDs): NSAIDs, by inhibiting prostaglandin synthesis, promote Na+ and water retention on chronic use. Thus, they decrease the antihypertensive effect of β-blockers.
Therapeutic uses of β-blockers
- Hypertension: β-Blockers are useful for all grades of hypertension. These drugs are preferred especially in patients with angina, myocardial infarction, or cardiac arrhythmias. The advantages of β-blockers are:
- Sodium and water retention is rare.
- Cheaper.
- Have a long duration of action.
- Well tolerated.
- Angina pectoris and Myocardial infarction (MI): β-Blockers reduce myocardial O2 demand by decreasing heart rate, myocardial contractility, and arterial pressure. They improve exercise tolerance and reduce the frequency of anginal episodes. The use of β-blockers early in the acute phase of MI may limit infarct size. Long-term use of β-blockers may reduce mortality and reinfarction.
- Cardiac arrhythmias: β-Blockers are mainly used in atrial arrhythmias such as atrial fibrillation and atrial flutter, but rarely for ventricular arrhythmias.
- Congestive cardiac failure: Chronic use of β-blockers such as carvedilol, metoprolol, and bisoprolol has been shown to reduce the mortality rate in chronic heart failure
- Pheochromocytoma: β-Blockers are used to control the cardiac manifestations of pheochromocytoma.
- Glaucoma: β-Blockers decrease IOP by reducing the production of aqueous humor. They are useful in the treatment of glaucoma. Timolol, carteolol, levobunolol, betaxolol, etc. are used topically in glaucoma. Timolol is the most frequently used β-blocker in glaucoma.
- Prophylaxis of migraines: Propranolol and metoprolol are effective in reducing the frequency of migraine headaches. The mechanism is not known.
- Hyperthyroidism: The signs and symptoms of hyperthyroidism such as tachycardia, palpitation, tremors, and anxiety are reduced due to the blockade of β- receptors. Propranolol inhibits the peripheral conversion of T4–T3. It is used in thyroid storm.
- Essential tremors: Oral propranolol may give some benefit in patients with essential tremors.
- Acute anxiety states: β-Blockers are useful in controlling the symptoms of acute anxiety such as palpitation, tachycardia, tremor, and sweating.
Important features of β-blockers are given in Table
Β-Blockers With Important Features
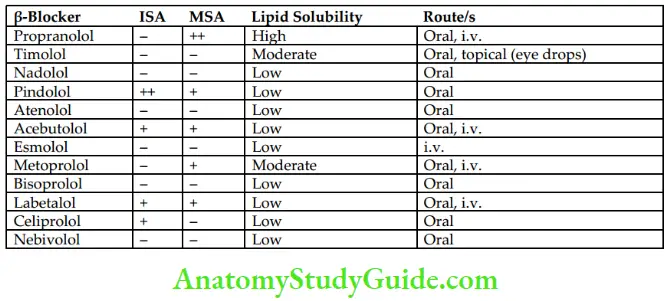
Selective β1-Adrenergic Blockers
Selective β1-blockers have a lower risk of bronchoconstriction, and less effect on carbohydrate metabolism, lipid profile, and exercise capacity.
- Esmolol
- It is administered intravenously.
- It is rapidly metabolized by esterases in RBCs; t1/2 is about 10 min.
- It has no membrane-stabilizing effect; and no intrinsic sympathomimetic activity.
- It is a selective β1-blocker and has a short duration of action.
- Esmolol is used in hypertensive emergencies and for rapid control of ventricular rate in supraventricular arrhythmias.
- Atenolol
Differences between Propranolol and Atenolol
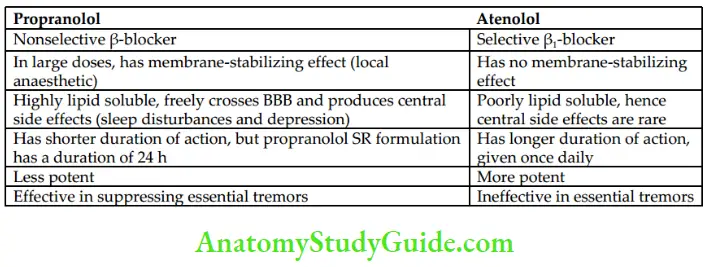
β-Blockers With Additional Vasodilatory Action
- Labetalol It is a competitive blocker at β1-, β2-, and α1-adrenergic receptors. It is administered orally or intravenously. It undergoes extensive first-pass metabolism after oral administration; hence, its bioavailability is poor. Oral labetalol is useful in the treatment of essential hypertension and i.v. labetalol for hypertensive emergencies. It is safe for use during pregnancy. The important side effects are postural hypotension and hepatotoxicity.
- Carvedilol Like labetalol, it also blocks β1-, β2-, and α1-adrenergic receptors. In addition, carvedilol has antioxidant, antiproliferative, membrane-stabilizing, and vasodilatory properties; it has no intrinsic sympathomimetic activity. It has a cardioprotective effect; hence, long-term use reduces mortality in patients with CHF.
- Celiprolol is a third-generation selective β1-blocker and has weak vasodilating and broncho-dilating effects (β2-agonism). It is effective in the treatment of hypertension and angina.
- Nebivolol
- Has (NO-mediated) vasodilating activity
- No unfavorable effect on lipid p
Leave a Reply