Genetics
Question 1. Write a note on autosomal dominant disorders with examples.
Answer:
Autosomal dominant disorders
- Manifests in a heterozygous state and one parent of an index case are usually affected
- Males and females, both can be affected and can transmit the condition
- If an affected person marries an unaffected one, every child has a 50% chance of having the disease
- A proportion of patients do not have affected parents (these patients have new mutations involving either the egg or the sperm, from which they were derived)
- Incomplete penetrance: Individuals inherit the mutant gene but are phenotypically normal (BRCA-2 associated breast cancers)
Read and Learn More Preparatory Manual of Pathology Question and Answers

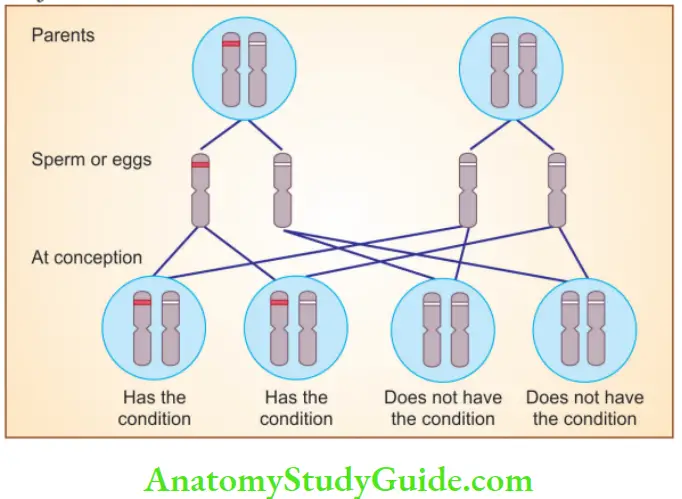
Question 2. Write salient features of autosomal recessive disorders with examples.
Answer:
Autosomal Recessive disorders
- Occur when both alleles at a given gene locus are mutated
- Parents of an affected individual can be normal, but siblings may show the disease
- Siblings have one chance in four of having the trait (i.e. recurrence risk is 25% for each birth)
- Increased risk of transmission to siblings produced as a result of consanguineous marriages
- Complete penetrance is seen and expression of disease is more uniform

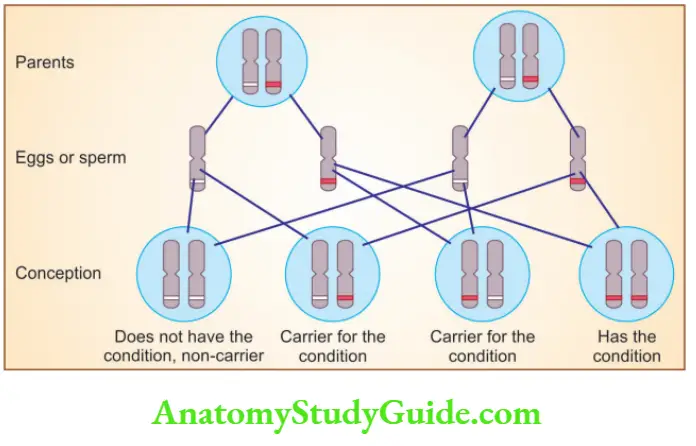
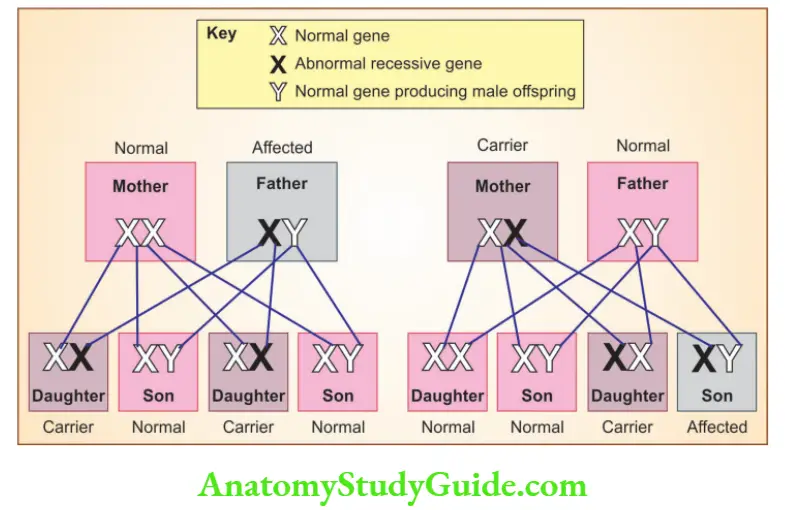
Question 3. Discuss X-linked recessive disorders with examples.
Answer:
X-linked recessive disorders
- Disorders are expressed in males
- An affected male does not transmit the disorder to his sons, but all daughters are carriers
- Sons of heterozygous women have, one chance in two of receiving the mutant gene
- Because of the paired normal allele, heterozygous females are carriers (do not express full phenotypic change)
- Heterozygous females can show the disease if there is random inactivation of the normal X-chromosome

Question 4. Write a note on Marfan’s syndrome.
Answer:
Marfan’s syndrome
- Shows autosomal dominant inheritance
Pathogenesis:
- An inherited defect in the fibrillin-1 gene
- Aorta, ligaments, and ciliary zonules that support the lens are affected (as microfibrils are made of fibrillin)
- Loss of microfibrils stimulates transforming growth factor- (TGF-), which increases the activity of metalloproteases, resulting in loss of extracellular matrix
Morphology:
1. Skeletal abnormalities
- The patient is tall with long extremities and long tapering fingers and toes
- Lax joint ligaments of hands and feet, the thumb can be hyperextended back to the wrist
- Bossing of the frontal eminences and prominent supraorbital ridges
- Kyphosis, scoliosis, rotation or slipping of the dorsal or lumbar vertebrae
- Pectus excavatum (deeply depressed sternum) or a pigeon-breast deformity
2. Ocular changes
- Ectopia lentis: Bilateral subluxation or dislocation (usually outward and upward) of the lens
3. Cardiovascular lesions:
- Mitral valve prolapse
- Aortic dissection
Question 5. Write a note on familial hypercholesterolemia.
Answer:
Familial hypercholesterolemia
- Occurs due to mutation in the gene encoding LDL receptor, involved in the transport and metabolism of cholesterol
- Elevated plasma cholesterol levels result in tendinous xanthomas and premature atherosclerosis
LDL metabolism
- The liver secretes very-low-density lipoproteins (VLDLs) into the bloodstream
- Lipolysis of VLDL molecule in capillaries occurs by lipoprotein lipase, resulting in the formation of intermediate-density lipoprotein (IDL)
- VLDL molecule comprises Apo C, E, B-100, whereas IDL molecule comprises Apo E, B-100
- IDL can be taken up by the liver by LDL receptors, resulting in the formation of VLDL, or is converted to LDL, which is taken up by the liver
- IDL is the immediate and major source of plasma LDL
- LDL molecule comprises Apo B-100
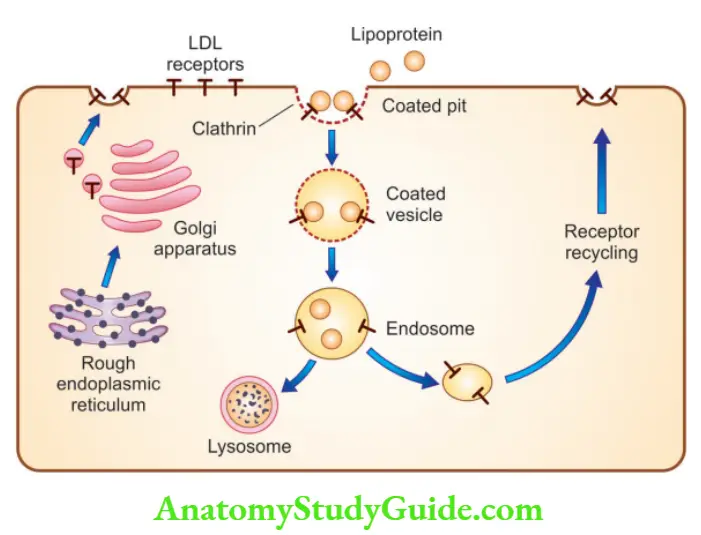
LDL receptor pathway and regulation of cholesterol metabolism
- 70% of plasma LDL is cleared by the liver
- Binding of LDL to cell surface receptors, present in coated pits
- Receptor-bound LDL is internalized by invagination to form coated vesicles
- These coated vesicles inside the cytoplasm of the cell fuse with the lysosomes
- In the lysosome, the LDL molecule is degraded into cholesterol and ApoB-100 is degraded into amino acids
- Free cholesterol exits through the lysosome, with the help of NPC1 and NPC2 proteins
- LDL receptor mutations result in increased LDL levels in the blood, resulting in increased deposition of cholesterol in tissues (hypercholesterolemia) and atherosclerosis
Question 6. Write a short note on Tay-Sachs disease.
Answer:
Tay-Sachs disease (GM2 gangliosidosis)
- Lysosomal storage disorder, which occurs due to deficiency of hexosaminidase, subunit.
- Characterized by the accumulation of GM2 ganglioside
- Accumulation can be seen in the heart, liver, and spleen, however, involvement of neurons and retina dominates the clinical picture
Morphology
- The cytoplasm of neurons shows inclusions
- Inclusions are made up of lysosomes filled with gangliosides
- These gangliosides are stained with oil red O or Sudan black
- Electron microscopy: cytoplasmic inclusions are visualized as whorled configurations within the lysosomes
- A cherry-red spot appears in the macula
Question 7. Write a short note on Niemann-Pick disease.
Answer:
Niemann-Pick disease type A and B
- Lysosomal accumulation of sphingomyelin due to an inherited deficiency of sphingomyelinase
Types
- Type A (severe infantile form): The patient presents with CNS manifestations, resulting in death within the first 3 years of life.
- Type B patients have organomegaly, without CNS involvement
Morphology
- Affected cells appear enlarged due to the distention of lysosomes with sphingomyelin in
- Electron microscopy (EM)—“zebra” bodies: Cytoplasmic bodies resembling concentric lamellated myelin figures in lysosomes
- Numerous foamy vacuoles in the cytoplasm, which can be demonstrated by a frozen section
- Spleen: Massive splenomegaly
- Lymphadenopathy
Niemann-Pick disease type C
- Due to mutations in the genes NPC1 and NPC2
- NPC gene: It is responsible for the transport of free cholesterol from the lysosomes to the cytoplasm
- A child presents with ataxia, dystonia, dysarthria
Question 8. Write a short note on Gaucher’s disease.
Answer:
Gaucher’s disease
- Autosomal recessive disorder
- Most common lysosomal storage disorder
- Occurs due to mutations in the gene encoding the enzyme glucocerebrosidase
- Resulting in the accumulation of glucocerebroside in macrophages
- Thus resulting in activation of macrophages, and secretion of IL-1, IL-6, and tumor necrosis factor (TNF)
Three subtypes
- Type I, or chronic non-neuronopathic form, in which glucocerebroside accumulates in the spleen and skeletal system
- Type II, or acute neuronopathic Gaucher disease, death occurs at an early age
- Type III, intermediate between types I and II
Morphology
- Massive accumulation of glucocerebrosides within the phagocytic cells throughout the body (Gaucher cells)
- Gaucher cells are found in the spleen, liver, bone marrow, lymph nodes, tonsils, thymus, and Peyer patches
- Gaucher cells have fibrillary cytoplasm, resembling crumpled tissue paper, with dark eccentrically placed nuclei
- Gaucher cells show periodic acid–Schiff stain positivity
- Electron microscopy: Fibrillary cytoplasm shows lysosomes, containing the stored lipid in the stacks of bilayers.
Diagnosis
- By measuring glucocerebrosidase activity in the peripheral blood leukocytes or in extracts of cultured skin fibroblasts
Treatment
- Replacement therapy with recombinant enzymes
- Allogeneic hematopoietic stem cell transplantation can be curative
Question 9. What is the principle of Karyotyping? Enumerate the stains used in the technique.
Answer:
Karyotyping
- Definition: Defined as the study of chromosomes
- Chromosomes are examined by arresting the dividing cells in metaphase with mitotic spindle inhibitors
- Metaphase spread: Individual chromosomes take the form of two chromatids connected at the centromere
- Karyotype is obtained by arranging each pair of autosomes according to their length (in decreasing order), followed by sex chromosomes
- G banding: Giemsa stain is used for the identification of individual chromosomes based on their alternative light and dark areas
Other staining patterns
- Q-Quinacrine banding demonstrates bands along a chromosome
- C-banding (constitutive) demonstrates heterochromatin (chromosome material with increased density than normal)
Normal karyotyping
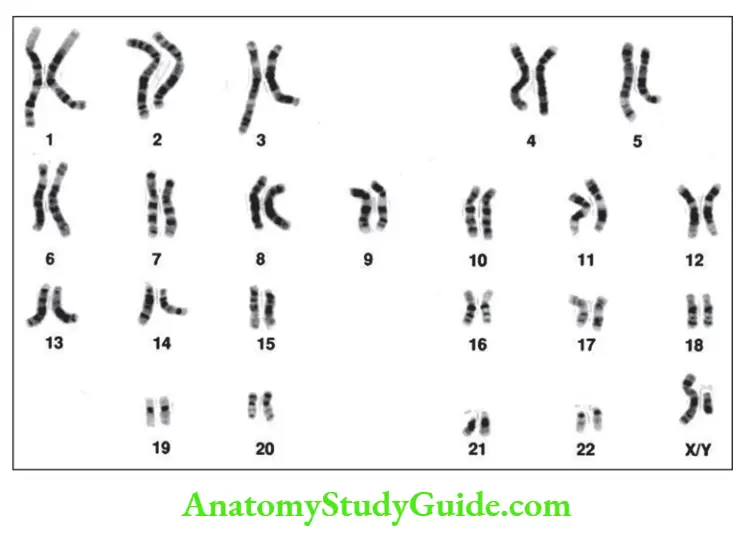
Question 10. Enumerate different types of chromosomal rearrangements.
Answer:
Chromosomal rearrangements
- Deletion: Refers to the loss of a portion of a chromosome, which can be interstitial or terminal
- Ring chromosome: A special form of deletion, which is produced when a break occurs at both ends of the chromosome with a fusion of the damaged ends
- Inversion: Rearrangement that involves two breaks within a single chromosome with reincorporation of the inverted, intervening segment
- Iso-chromosome: When one arm of a chromosome is lost and the remaining arm is duplicated, resulting in a chromosome consisting of two short arms only or of two long arms. Most commonly seen in the long arm of the X and is designated i(X)(q10)
- Balanced translocation: A segment of one chromosome is transferred to another
- Robertsonian translocation: Breaks occur close to the centromeres of each chromosome. Transfer of the segments then leads to one very large chromosome and one extremely small one (which is lost)
Question 11. Write a short note on trisomy 21.
Answer:
Trisomy 21 (Down syndrome)
- Most common chromosomal disorder and the major cause of mental retardation
- 95% of affected individuals have 47 chromosomes
- Maternal age has a strong influence on the incidence of trisomy 21
Causes
- The most common cause is meiotic non-disjunction, of chromosome 21, seen in the ovum
- Robertsonian translocation of the long arm of chromosome 21 to another acrocentric chromosome (example 22 or 14)
- Mosaicism: Mixture of cells with 46 or 47 chromosomes
Clinical features
- The most common congenital heart defect seen is endocardial cushion defects
- Children have an increased risk of developing acute leukemia (acute megakaryoblastic leukemia being the most common)
- Individuals more than 40 years old develop Alzheimer’s disease
- Patients are prone to serious infections
Question 12. Write a short note on Klinefelter syndrome.
Answer:
Klinefelter syndrome
- The common cause of hypogonadism in males
- Manifests after puberty, patients have 47, XXY karyotype which results from nondisjunction during meiotic division
Clinical features
- Eunuchoid body habitus with abnormally long legs
- Small atrophic testes, lack of secondary male characteristics, gynecomastia
- Mean IQ is lower than normal, but mental retardation is not seen
- Increased incidence of type 2 diabetes and metabolic syndrome
- Mitral valve prolapse is commonly seen in adults
- Elevated FSH levels, with reduced testosterone levels
- Increased risk for breast cancer, extra-gonadal germ cell tumors, and autoimmune diseases like SLE
- An important genetic cause of reduced spermatogenesis and male infertility
Question 13. Write a short note on Turner’s syndrome.
Answer:
Turner’s syndrome
- Results from complete or partial monosomy of the X chromosome
- Characterized by hypogonadism in phenotypic females
- The most common sex chromosome abnormality in females
- The most common structural abnormality of the X chromosome includes 45, X
Clinical features
In infants
- Presents with edema of the dorsum of the hand and foot due to lymph stasis
- Swelling of the nape of the neck (cystic hygroma)
- Congenital heart disease—pre-ductal coarctation of the aorta and bicuspid aortic In adults
- Short stature, webbing of neck, low posterior hairline
- Streak gonads, primary infertility (most common cause)
- Broad chest, widely separated nipples, and pigmented nevi
Note:
Short stature homeobox (SHOX) gene at Xp22.33 is responsible for short-stature
Question 14. Discuss Fragile-X syndrome.
Answer:
Fragile-X syndrome
- Second most common genetic cause of mental retardation, after Down syndrome
- Caused by mutation in familial mental retardation-1 (FMR-1) gene, located on chromosome Xq27.3
- There appears a constriction or discontinuity of staining in the long arm of the X chromosome, and it appears that the chromosome is “broken”, hence it was named a fragile site
Salient features
- Anticipation: Clinical features of Fragile X syndrome worsen with each successive generation
- FMR1 gene located on chromosome Xq27.3, in normal population encodes CGG trinucleotide repeats, number varying from 6-55
- Permutations: Normal transmitting males and carrier females carry 55 to 200 CGG repeats
- Full mutations: Affected individuals show 200 to 4000 CGG repeats
Pedigree analysis
- In the first generation, all sons are normal and all females are carriers
- During oogenesis in the carrier female, premutation expands to full mutation; hence, in the next generation all males who inherit the X with full mutation are affected
- Carrier male, transmits the repeats, with a small number of changes
- When a carrier female, transmits the repeats, dramatic amplification of the CGG repeats are seen, leading to mental retardation in male offspring and 50% of female offspring
- Hence, pre-mutations are converted to mutations by triple nucleotide repeat amplification, which occurs during the process of oogenesis
Clinical features
- Affected males have long faces with large mandibles, large everted ears, and large testicles (macro-orchid-ism, most distinctive feature)
- Hyperextensible joints, high-arched palate, and mitral valve prolapse
Question 15. Write a note on mitochondrial inheritance.
Answer:
Mitochondrial genes mutations
- Mitochondrial DNA is maternally inherited, as ova contains numerous mitochondria within its cytoplasm
- Mothers transmit the disease to their sons and daughters, and sons do not transmit the disease to their progeny
- As mtDNA encodes enzymes involved in oxidative phosphorylation, mutations affecting these genes exert their deleterious effects primarily on the organs most dependent on oxidative phosphorylation such as the central nervous system, skeletal muscle, cardiac muscle, liver, and kidneys
- For example, CPEO (Chronic progressive external ophthalmoplegia), KSS (Kearns-Sayre syndrome), Pearson syndrome, Leigh syndrome, NARP (neurogenic weakness with ataxia and retinitis pigmentosa), MELAS (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes), MERRF (myoclonic epilepsy with ragged red fibers), LHON (Leber hereditary optic neuropathy)
Question 16. Write a note on genomic imprinting with examples.
Answer:
Genomic imprinting
- Every individual inherits two copies of each autosomal gene, from the mother (maternal chromosome) and from the father (paternal chromosome)
- Imprinting selectively inactivates either the maternal or paternal allele
- Maternal imprinting refers to the silencing of the maternal allele
- Paternal imprinting refers to the silencing of the paternal allele
- Imprinting occurs in the ovum or the sperm, before fertilization, and is transmitted to all somatic cells through mitosis
1. Prader-Willi syndrome
- Occurs due to deletion of paternally derived chromosome 15
- Characterized by mental retardation, short stature, hypotonia, profound hyperphagia, obesity, small hands and feet, and hypogonadism
2. Angelman syndrome
- Occurs due to deletion of maternally derived chromosome 15
- Characterized by mental retardation, ataxic gait, seizures, and inappropriate laughter
- Because of their laughter and ataxia, they have been referred to as “happy puppets”
Question 17. Write a short note on Fluorescent in situ hybridization.
Answer:
Fluorescence in situ hybridization (FISH)
Uses DNA probes that recognize sequences specific to particular chromosomal regions Samples on which FISH can be performed
Samples on which FISH can be performed
Prenatal samples, peripheral blood cells, touch preparations from cancer biopsies
Uses
- To detect aneuploidy, microdeletions, translocations, and gene amplification (for example HER2 in breast cancer or NMYC amplification in neuroblastomas)
- Determination of treatment efficacy, for example in BCR-ABL positive CML
- Definitive diagnosis of HPV
Leave a Reply