Hematology
Table of Contents
Disorders Of Red Cells
Anemia:
Definition of Anemia:
Define anemia. Theory
- Decrease below the normal limit (below the reference level for the age and sex of the individual) of the hemoglobin concentration (Hb)/RBC count/hematocrit (packed cell volume).
- Reduction of the total circulating red cell mass below normal limits.
- Decrease in the oxygen-carrying capacity of the blood, which leads to tissue hypoxia.
Anemia may be absolute (decreased RBC mass), or relative (associated with a higher plasma volume). Anemia is conventionally used for absolute anemia.
Read And Learn More: Pathology for Dental Students Notes
Classification of Anemia:
Classify anemia.
Or
Etiological classification of anemia.
- Morphological classification: It is based on:
- Red cell size (normocytic, microcytic, or macrocytic), and
- Degree of hemoglobinization (normochromic or hypochromic).
- Etiological classification: The etiological classification of anemia is listed in Table.
Red Cell Indices:
Write a short note on red cell indices.
Red cell indices are useful in morphological characterization and diagnosis of anemias. Thy are either directly measured or automatically calculated by specialized instruments. Red cell indices include:
1. Mean Corpuscular Volume (MCV):
- MCV is indicative of the average volume of the RBC and is expressed in femtoliters (fL).
- It is used for the classification and differential diagnosis of anemias.
- Normal range: 8298 fL.

2. Mean Corpuscular Hemoglobin (MCH):
- MCH indicates the amount of Hb (weight) per RBC and is expressed as picograms (1 pg 10 -12g).
- It is of limited value in the differential diagnosis of anemias.
- Normal range: 2732 pg
- MCH = Hb (in g/L)/RBC (in millions/μL) = 15 × 10/5 = 30 pg
Morphological classification of anemia:

Etiological classification of anemia:

3. Mean Corpuscular Hemoglobin Concentration (MCHC):
- MCHC denotes the average concentration of hemoglobin in the RBC taking volume into account. It is expressed as g/dL (earlier it was expressed as %).
- It is a better indicator of hypochromasia than MCH.
- Normal range: 3135 g/dL.
- MCHC = Hb (in g/dL)/PCV = 15/0.45 = 33 g/dL
4. Red Cell Distribution Width (RDW):
- RDW is a quantitative measure of anisocytosis:
- Normal RDW is 11.5% to 14.5%.
- A normal RDW indicates that RBCs are relatively uniform in size. A raised RDW indicates that red cells are heterogeneous in size and/or shape. In early iron deficiency anemia,
- RDW increases along with low MCV while in the thalassemia trait, RDW is normal with low MCV.
RDW = (Standard deviation ÷ mean cell volume) × 100
Iron Deficiency Anemia
Discuss the etiopathogenesis of iron deficiency anemia.
Iron deficiency anemia (IDA) is the most common nutritional disorder.
Etiology of Iron Deficiency Anemia:
IDA is due to deficiency of iron causing defective heme synthesis.
Pathogenesis of Iron Deficiency Anemia:
It is due to decreased synthesis of heme and can be divided into 3 stages.
- Stage 1 (Iron depletion): Iron is adequate to maintain normal hemoglobin levels and only serum ferritin decreased.
- Stage 2 (Iron deficient erythropoiesis): Lowering of serum iron and transferrin saturation levels without anemia (Hb, MCV, and MCH within normal range). Bone marrow shows iron
deficient erythropoiesis. - Stage 3 (Iron deficiency anemia): Low serum iron, serum ferritin, and transferrin saturation. Impaired hemoglobin production. Morphologically, first reduction in the size (microcytic) and later increase in the central pallor (hypochromic) of RBCs.
Laboratory Findings of Iron Deficiency Anemia:
1. Peripheral Blood:
- Hemoglobin and hematocrit (PCV): Decrease
- Red cell indices:
- MCV: <80 fL (normal 82–98 fL)
- MCH: <25 pg (normal 27–32 pg)
- MCHC: <27 g/dL(31–36 g/dL)
- RDW: Increased and >15%. It is the earliest sign of iron deficiency (normal 11.5–14.5%).
Causes of iron deficiency anemia:

Write short note on peripheral smear findings in iron deficiency anemia.

2. Reticulocyte count: Low for the degree of anemia.
Bone Marrow:
- Cellularity: Moderately hypercellular.
- M: E (Myeloid: Erythroid) ratio: Varies from 2:1 to 1:2 (normal 2:1 to 4:1).
- Erythropoiesis: Hyperplasia and micronormoblastic maturation.
- Myelopoiesis: Normal.
- Megakaryopoiesis: Normal.
- Absence of bone marrow iron: “Gold standard” test, demonstrated by negative Prussian blue reaction.
Serum Iron Profile:
Hematological and biochemical investigations in iron deficiency anemia

Serum iron profie in IDA:

Reticulocyte Hemoglobin:
It is decreased and is an early feature of IDA.
Clinical Features of IDA:
- Nonspecific and related to both severity and the cause of the anemia (for example, Gastrointestinal disease)
- Onset: Insidious.
- Nonspecific symptoms: Fatigue, palpitations, breathlessness, weakness, and irritability.
- Pharyngeal/esophageal webs formed cause dysphagia.
- Patterson-Kelly or Plummer-Vinson syndrome:
- Microcytic hypochromic anemia
- Atrophic glossitis
- Esophageal webs
- Congestive heart failure in severe anemia.
- Central nervous system: Pica-unusual craving for substances with no nutritional value
- like clay or chalk. Craving for ice (pagophagia) specific to iron deficiency. Pica may be the cause rather than the effect of IDA.
Physical Findings of IDA:
Diminished tissue enzymes cause characteristic epithelial changes of iron deficiency anemia.
- Angular stomatitis and glossitis
- Chronic atrophic gastritis
- Koilonychia (spoon nails)
Causes of Microcytic Hypochromic Anemia:
Enumerate the causes of microcytic hypochromic anemia.
- Iron deficiency anemia
- The thalassemia major
- Anemia of chronic disorders
- Others: Alcohol, lead poisoning, and drugs
- Sideroblastic anemia (rare cause).
Megaloblastic Anemia
Discuss the causes and pathogenesis of megaloblastic anemia.
- Anemias are characterized by defective/impaired DNA synthesis and distinct megaloblasts in the bone marrow.
- Megaloblastic anemias are common among anemias due to impaired red cell production.
Etiology of Megaloblastic Anemia:
Causes of megaloblastic anemia:

Pathogenesis of Megaloblastic Change:
1. Impaired DNA synthesis: Megaloblastic anemia is commonly due to a deficiency of vitamin B12 (cyanocobalamin) or folic acid. Both vitamins are coenzymes necessary for the synthesis of thymidine (one of the four bases found in DNA).
- Delayed maturation of nucleus: The nuclear maturation lags behind the cytoplasmic maturation and results in abnormally large nucleated erythroid precursors named as megaloblasts.
- Cytoplasm matures normally. RBCs are larger than normal → macrocytes.
- Affects all rapidly dividing cells of the body (including skin, GI tract, and bone marrow).
2. Ineffective erythropoiesis: Megaloblast precursors undergo intramedullary destruction.
Laboratory Findings of Megaloblastic Anemia:
Write a short note on the laboratory findings in megaloblastic anemia.
Blood findings in vitamin B12 and/or folic acid deficiency are similar.
1. Peripheral Blood:
Peripheral smear and bone marrow findings in megaloblastic anemia
- Hemoglobin and hematocrit (PCV): Reduced
- Red cell indices:
- MCV: Above 100 fL (normal 8298 fL)
- MCH (normal 2732 pg)
- Normal MCHC (3136 g/dL)
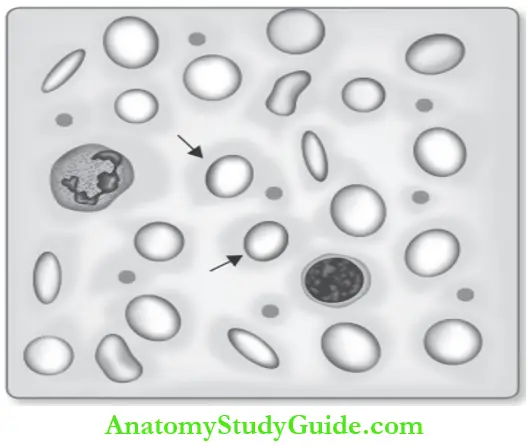
Peripheral smear: Pancytopenia (decreased RBC, WBCs, and platelets).
- RBCs:
- Macrocytic and oval (egg-shaped macro-ovalocytes)-diagnostic.
- Most macrocytes lack the central pallor.
- Marked variation in the size and shape of red cells (anisopoikilocytosis).
- Evidence of dyserythropoiesis: Basophilic stippling, (precipitated ribosomal RNA). Cabot ring nuclear remnants and Howell Jolly bodies nuclear remnants.
- WBCs:
- Decreased WBC count (leukopenia).
- Hypersegmented neutrophils (more than five nuclear lobes): First and specific morphological sign of megaloblastic anemia. These neutrophils are also larger than normal (macropods).
- Platelets: Decreased.
2. Reticulocyte count: Normal or low.
Dimorphic Anemia:
- Combined vitamin B
- 12/folic acid and iron deficiency.
- Peripheral smear shows two populations of RBCs namely: Macro-ovalocytes and microcytic hypochromic.





Bone Marrow:
- Cellularity: Moderately to markedly hypercellular.
- M: E ratio: Due to marked erythroid hyperplasia, the M: E ratio is reversed ranging from 1:1 to 1:6 (normal
2:1 to 4:1). - Erythropoiesis: Megaloblastic type.
- Megaloblasts: Large, abnormal counterparts of normal normoblasts. Megaloblast shows asynchrony of nuclear and cytoplasmic maturation. The cytoplasm shows normal hemoglobinization.
- Ineffective erythropoiesis: Developing megaloblasts die in the marrow (intramedullary hemolysis).
- Myelopoiesis:
- Myeloid cells are adequate in number.
- Granulocytic precursors display nuclear-cytoplasmic asynchrony in the form of giant metamyelocytes and band forms.
- Megakaryopoiesis: Normal or increased in number.
- Bone marrow iron: Moderately
Biochemical Tests for Megaloblastic Anemia:
Common for both vitamin B and folic acid deficiency
1. Deoxyuridine suppression test:
It is a sensitive measure of deficiency of 5, 10-methylene THF, which occurs in both folic acid and vitamin B12 deficiency.

Diagnostic tests for vitamin B12 deficiency
- Serum vitamin B12 levels: Decreased
- Schilling tests for vitamin B12 absorption.

3. Specific tests for folic acid deficiency:
- Serum folic acid levels: Decreased
- FIGLU in urine: Excessively excreted.
Pernicious Anemia
Discuss the etiopathogenesis and morphology of pernicious anemia.
Pernicious anemia (PA) is an autoimmune disease due to a deficiency of the intrinsic factor causing impaired absorption of vitamin B12 and megaloblastic anemia.
Rare in India. A genetic predisposition is suspected.
- Age: Older age — fifth to eighth decades of life
- Sex: Females are more involved than males (F: M is 1.5: 1).
Etiopathogenesis of Pernicious Anemia:
- An autoimmune disease due to the destruction of the gastric mucosa.
- Stomach shows damage to parietal cells, dense infiltration by lymphocytes and plasma cells → chronic atrophic gastritis → failure of production of intrinsic factor.
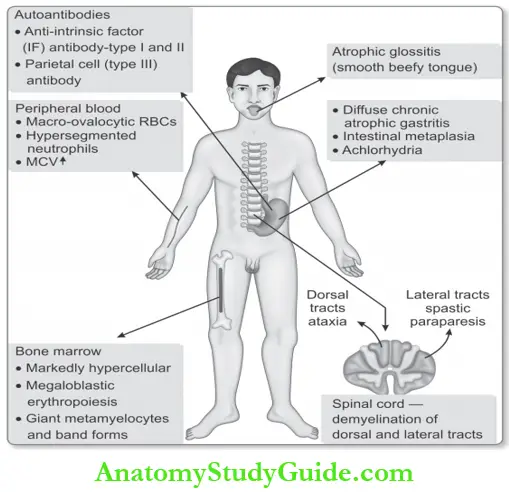
- Presence of autoantibodies: Two major types of autoantibodies
- Anti-intrinsic factor (IF) antibody:
- Type 1 (blocking) antibody: Blocks the binding of vitamin B12 to IF. Present in 5075% of the cases.
- Type 2 (binding) antibody: Attaches to the IFvitamin B12 complex and prevents its binding to receptors in the ileum. Present in about 40% of patients.
- Parietal cell (Type III) antibody: Neither specific for PA nor other autoimmune disorders. It is found in 90% of patients.
- Anti-intrinsic factor (IF) antibody:
Morphology of Pernicious Anemia:
1. Alimentary System:
- Atrophic glossitis: Tongue shiny, glazed, and beefy.
- Stomach:
- Diffuse chronic atrophic gastritis and impaired secretion of hydrochloric acid, pepsin and intrinsic factor.
- Intestinal metaplasia.
2. Central Nervous System:
- Found in 75% of cases.
- Demyelination in the dorsal and lateral tracts: Subacute combined degeneration
- Peripheral neuropathy.
Laboratory Findings of Pernicious Anemia:
Write a short note on laboratory findings in pernicious anemia.
- Blood, bone marrow, and biochemical test findings are similar to those described earlier for megaloblastic anemias.
Specific Diagnostic Tests for Pernicious Anemia:
- Schilling test for vitamin B12 absorption: Abnormal
- Radioactive vitamin B12 is used to assess the status of intrinsic factor (IF) and vitamin B12.
- Helps in distinguishing megaloblastic anemia due to IF deficiency (pernicious anemia) from other causes of vitamin B12 deficiency.
- Serum antibodies to intrinsic factors are highly specific for pernicious anemia.
- Achlorhydria with histamine/pentagastrin stimulation.
- Severe deficiency of the intrinsic factor.
Clinical Features of Pernicious Anemia:
Mention the various clinical features of megaloblastic anemia.
The clinical features of vitamin B12 deficiency anemia and pernicious anemia are:
- Onset: Insidious and progresses slowly.
- The classic triad of presentation: Weakness, sore throat, and paresthesias.
- Tongue: Painful red “beefy” tongue.
- Neurological manifestations:
- Bilateral peripheral neuropathy: Glove and sock distribution of numbness or paresthesia.
- Demyelination of spinal cord: Subacute combined demyelination/degeneration of dorsal and lateral tracts—ataxia, uncoordinated gait, impairment of vibration and position sense.
- Atherosclerosis: Serum homocysteine level is raised and is a risk factor for atherosclerosis
and thrombosis.
Aplastic Anemia
Write a short note on aplastic anemia.
Hematopoietic stem cell (HSC) disorder characterized by:
- Pancytopenia (anemia, neutropenia, and thrombocytopenia).
- With markedly hypocellular bone marrow (less than 30% cellularity).
Etiology of Aplastic Anemia:
The most common causes associated with aplastic anemia are shown in Table.
Common causes of Aplastic Anemia:

Clinical Features of Aplastic Anemia:
- Any age of both sexes
- Insidious
- Progressive weakness, pallor, and dyspnea due to anemia.
- Frequent (mucocutaneous bacterial infections) or fatal infections due to neutropenia.
- Bleeding manifestations in the form of petechiae, bruises, and ecchymoses due to thrombocytopenia.
Laboratory Findings of Aplastic Anemia:

Peripheral smear: Pancytopenia, i.e. decreased red cells, neutrophils, and platelets.
- RBCs: Normocytic normochromic anemia
- WBCs: Total leukocyte count decreased. Neutrophils are markedly diminished and neutropenia is a reflection of the severity of aplasia. Initial stages, lymphocytes are normal in number as the disease progresses their count decreases.
- Platelets: Count is decreased.
Bone Marrow of Aplastic Anemia :
- Marrow aplasia: Best appreciated in a bone marrow (trephine) biopsy
- Cellularity: Marked hypocellularity.
- Hematopoiesis: Paucity of all erythroid, myeloid, and megakaryocytic precursors.
- Other cells: Lymphocytes and plasma cells are prominent.
No Splenomegaly of Aplastic Anemia :
- Diagnosis: Diagnosis is made with peripheral blood and bone marrow biopsy findings.
- Prognosis: Unpredictable.
Hereditary Defects In Hemoglobin
Classify hereditary disorders of hemoglobin
Hemoglobin defects may be quantitative (reduced production of normal hemoglobin) or qualitative (production of abnormal hemoglobin).
- Quantitative defect: Characterized by reduced a-globin or b-globin chain (for example, Thalassemia). It leads to a net reduction of hemoglobin.
- Qualitative defect: Characterized by the production of abnormal hemoglobin (for example, Sickle cell anemia).
Thalassemia Syndrome of Hereditary Hemoglobin:
- These are a group of inherited disorders due to the abnormality of globin production.
- It is characterized by decreased or absence of synthesis of either a or b-globin chain of adult hemoglobin, HbA (α2β2).
Classification of Hereditary Hemoglobin:
They are mainly classified as:
- Thalassemia syndromes: Impaired synthesis of b-chains of globin.
- α – Thalassemia syndromes: Impaired synthesis of a-chains of globin.
- Miscellaneous thalassemia syndromes.
β – Thalassemia:
- Autosomal recessive hereditary disorder.
- Diminished synthesis of b-globin chains and the normal synthesis of a-chains. It may be classified as b thalassemia major, intermedia, and minor.
- b-globin chains are encoded by a single gene.
- The molecular errors of over 200 genetic defects leading to b-thalassemia have been identified.
- Diffrent types of mutations in b-globin gene.
β-Thalassemia Major
Write short note on β -thalassemia major.
- It is a hereditary hemolytic anemia due to the absence of synthesis of the β-globin chain of hemoglobin. The synthesis of the α-globin chain is not affected.
- Homozygous form of β0/β0 or β+/β+ or double heterozygous β0/β+
- Most common in Mediterranean countries, parts of Africa, and South East Asia.
- Hemolytic anemia is of severe degree.
Pathophysiology of β-thalassemia Major:
1. Consequence of Defective or Absent β-chains:
- Severe hemolytic anemia due to:
Absence of b-globin chain.
- Ineffective erythropoiesis: Unpaired and excess a-chains aggregate into insoluble precipitates, which bind to and damage the membrane of erythroid precursors. These erythroid precursors fail to mature and undergo apoptosis in the marrow.
- Extravascular hemolysis: RBCs with a-chain inclusions are removed by macrophages of the spleen (extravascular hemolysis).
- Synthesis of fetal hemoglobin (HbF): The ϒ-globin chain synthesis continues even 6 months after birth and combines with a-globin leading to increased levels of HbF (α2ϒ2). The level of HbF varies from 30% to 90%.
2. Consequences of Ineffective Erythropoiesis:
- Changes in bone marrow: Marked erythroid hyperplasia.
- Changes in bone:
- Skull X-ray: Hair on end (“crew-cut”) appearance
- Typical facies: Thlassemic facies—prominent forehead, cheekbones and upper jaw.
- Extramedullary hematopoiesis: In liver and spleen → consequent hepatosplenomegaly.
- Cachexia: Develops in untreated patients.

3. Iron Overload and its Consequences:
Causes of iron overload:
- Increased absorption of dietary iron from the duodenum
- Hemolysis
- Repeated transfusions (usual mode of treatment).
Consequences: Iron overload produces hemosiderosis and secondary hemochromatosis and damages to parenchyma of organs (for example, Heart, Liver, and pancreas).
Clinical Features of β-thalassemia Major:
- Age: Infants develop moderate to severe anemia 69 months after birth.
- Growth and development: Untreated/untransfused children fail to thrive and die within 45 years of age.
- Bone changes: Those who survive longer develop distortion of skull and facial bones.
- X-ray skull shows hair on end appearance and face shows a characteristic thalassemic facies.
- Marked splenomegaly: Up to 1500 grams due to hyperplasia and extramedullary hematopoiesis.
- Extramedullary hemopoiesis: Liver and lymph nodes may show extramedullary hematopoiesis.
- Iron overload: Multiple blood transfusions may lead to iron overload and result in hemosiderosis and secondary hemochromatosis (heart, liver, and pancreas).
Laboratory Findings of β-thalassemia Major:
Peripheral Blood:
Mention the laboratory findings in the β-thalassemia major.
- Hemoglobin (ranges from 3 to 8 g/dL) and hematocrit (ranges from 8 to 23%): Markedly
reduced. - RBC count increased/normal (in contrast to iron deficiency anemia).
- Reticulocyte count increased in the range of 5 to 15%.
- Red cell indices:
- MCV decreased and in the range of 45 – 70 fL (normal range 8298 fL).
- MCHC decreased and in the range of 22 – 30 g/dL (normal range 3135 g/dL)
- MCH decreased and in the range of 20 – 28 pg
- RDW-within normal limits (in contrast to iron deficiency anemia where it is increased).
Write short note on peripheral smear findings in β-thalassemia major.
Peripheral smear:
- RBCs:
- Microcytic hypochromic anemia
- Moderate to marked anisocytosis and poikilocytosis
- Many target cells
- Basophilic stippling
- Nucleated red cell precursors (normoblasts) in variable numbers (540%).
- WBCs: Leukocytosis with mild left shift.
- Platelets: Normal.
Bone Marrow of β-thalassemia Major:
- Cellularity: Markedly hypercellular.
- M: E ratio: Reversed from 1:1 to 1:5 depending upon the degree of erythroid hyperplasia.
- Erythropoiesis: Normoblastic with marked erythroid hyperplasia.
- Myelopoiesis: Normal.
- Megakaryopoiesis: Normal.
- Bone marrow iron: Markedly increased due to increased dietary absorption and hemolysis.


Sickle Cell Anemia
Sickle cell anemia is a hereditary disorder of hemoglobin characterized by the production of defective hemoglobin called sickle hemoglobin (HbS). On low oxygen tension or deoxygenation, HbS imparts sickle shape to RBCs. HbS is produced due to a qualitative defect in hemoglobin production caused by a mutation in the β-globin gene.
Characteristic Features of Sickle Cell Anemia:
- The autosomal recessive disorder manifests early in life.
- Homozygous state (SS) is caused by a mutation in the b-globin gene.
- HbS constitutes more than 70% of hemoglobin in their RBCs with no HbA.
Etiopathogenesis of Sickle Cell Anemia:
Discuss the etiopathogenesis of sickle cell anemia.
- Production of abnormal hemoglobin called sickle hemoglobin (HbS).
- Missense point mutation: In HbS, there is the substitution of glutamic acid by valine in the 6th position of the β – globin chain of hemoglobin. It alters the solubility or stability of the hemoglobin and produces hemolytic anemia.
- HbS is responsible for the characteristics of the disease.
Molecular Basis of Sickle Cell Anemia:
- During low O2 tension or deoxygenation, HbS molecules undergo aggregation and polymerization.
- If deoxygenation continues, the aggregated HbS molecules form long needle-like fibers (or pseudocrystalline structures known as tactoids) within RBCs.
- The tactoids grow in length beyond the diameter of RBCs and distort RBC shape.
- RBC becomes elongated and assumes a shape like sickle (or crescent moon or holly-leaf or boat) and predisposes to stasis and vascular occlusion.
- When the oxygen tension returns to normal, the sickled red cell returns to normal shape.
- Recurrent sickling causes red cell membrane damage and these RBCs become irreversibly sickled cells (ISC).
Clinical Features of Sickle Cell Anemia:
- The presence of HbF in the first 6 months of life has a protective role.
- Symptoms appear after 6 months of age as the HbF disappears.
- Infants and children present with acute problems like severe infection, acute chest syndrome, splenic sequestration, and stroke.
- Chronic hypoxia in children is responsible for generalized impairment of growth and development. Adults manifest with chronic organ damage.
Chronic Hemolytic Anemia:
Lifelong hemolysis (mainly extravascular) causes chronic hemolytic anemia, which is of moderate degree. Thus produces raised unconjugated (indirect) bilirubin, and predisposes to pigment bilirubin gallstones (cholelithiasis) and cholecystitis.
Crises of Sickle Cell Anemia:
Four types of crises are encountered. These are:
1. Sickling crisis (vaso-occlusive/pain/painful/infarction crisis):
- Most common
- Blockage of microcirculation by sickled red cells produces hypoxic injury and infarction.
- Bone: Manifest as the hand-foot syndrome, dactylitis of the bones of the hands or feet or both.
- Lung: Acute chest syndrome (dangerous).
- Spleen: Acute abdominal pain due to infarcts of abdominal viscera caused by occlusion of vessels. Recurrent splenic infarction results in auto splenectomy.
2. Hemolytic crisis: Rare type and presents with a marked increase in hemolysis.
3. Aplastic crisis:
- Associated with parvovirus B19.
- Reticulocytopenia.
4. Sequestration crisis:
- Usually occurs in children.
- Sudden trapping of blood in the spleen or liver causes rapid enlargement of the organ and a drop in hematocrit leading to hypovolemic shock.
Increased Susceptibility to Infections:
- Common infections are pneumonia due to Pneumococcus, meningitis due to S. pneumonia, and osteomyelitis due to Salmonella. The increased frequency of osteomyelitis is due to bone infarcts, which act as a nidus for infection.
- Septicemia and meningitis are the most common causes of death in children.
Chronic Organ Damage:
Particularly seen in the spleen, bones, kidneys, heart, lungs, brain, and skin.
- Spleen:
- Children after 6 months of life present with splenomegaly (up to 500 g).
- After 5–6 years of age, the spleen gets fired and gradually reduces in size due to multiple infarcts.
- Gradual loss of splenic function secondary to infarcts results in auto splenectomy.
- Bone: Osteomyelitis, particularly with Salmonella typhimurium.
- Extremities: Skin ulcers over the lower extremities.
Laboratory Findings of Sickle Cell Anemia:
Laboratory findings in sickle cell anemia.
Peripheral Blood:
- Hemoglobin: Decreased.
- Hematocrit (PCV): Decreased.
- ESR: Reduced.
- Reticulocyte count: Increased and ranged from 3 to 10%.


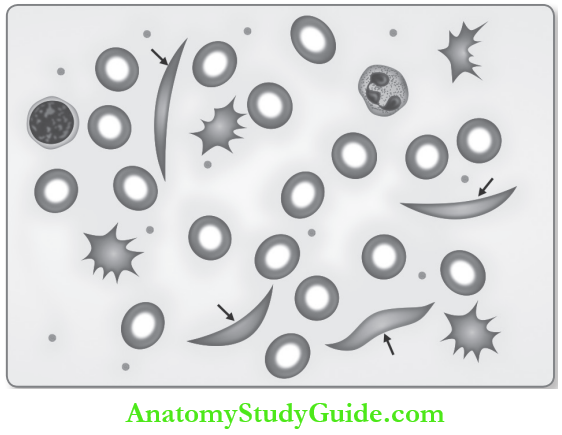
Bone Marrow of Sickle Cell Anemia:
- Cellularity: Hypercellular.
- Erythropoiesis: Compensatory normoblastic erythroid hyperplasia, which expands the marrow and causes resorption of bone and secondary new bone formation.
- Myelopoiesis: Normal.
- Megakaryopoiesis: Normal.
- Iron stores: Usually increased.
Serum Findings of Sickle Cell Anemia:
- Serum bilirubin: Raised and predisposes to pigment gallstones.
- Iron status: Raised serum iron, serum ferritin, and transferrin saturation.
- Serum haptoglobin: Reduced.
- Urine Urobilinogen: Increased.
Diagnostic/Confirmatory Tests
- Sickling test:
- Sickling is induced by adding a reducing (oxygen-consuming) agent like 2% sodium metabisulfite or sodium dithionite to the blood sample.
- Red cells with HbS show sickled and holly leaf appearance.
- It is diagnostic of sickle cell anemia.
- Hemoglobin electrophoresis: HbS is slow-moving compared to HbA and HbF.
- Estimation of HbF: In homozygous state constitutes about 1030% of hemoglobin.
- HPLC: Useful for confirmation of diagnosis.
- Prenatal diagnosis: By analysis of fetal DNA obtained by amniocentesis or chorionic villous biopsy, to detect the point mutations.

Disorders Of White Cells
Normal Differential Leukocyte Count (DLC):
The normal range of DLC in an adult is presented in Table
Normal range of different leukocytes in an adult:

Quantitative Disorders Of Leukocytes
1. Leukocytosis:
Define leukocytosis and list its causes.
An increase in the total number of leukocytes in the blood is more than 11,000/cu mm (11 × 109/L).
Causes of Leukocytosis: Common causes of leukocytosis are shown in Table.
Common causes of leukocytosis:

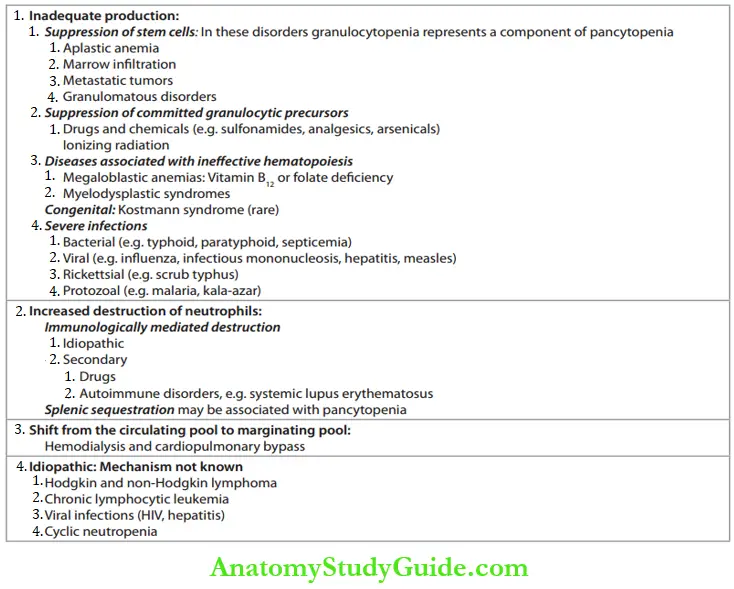
2. Leukopenia:
Total leukocyte count is less than 4,000/cu mm (4 × 109/L).
Causes of Leukopenia: Common causes of leukopenia are shown in Table.
Common causes of leukopenia:

Disorders of Neutrophils:
Define neutrophilia and mention its causes.
1. Neutrophilia:
An absolute neutrophil count of more than 8,000/cu mm (8 × 109/L). Differential count shows more than 70% neutrophils and is usually accompanied by leukocytosis (15–30 × 10/L).

Causes of neutrophilia:
Major causes of neutrophilia are shown in Table
2. Leukemoid Reaction:
- Benign leukocytic proliferation is characterized by a total leukocyte count of more than 25 ×109/L with immature white cells (like band forms, metamyelocytes, and myelocytes).
- It is different from chronic myelocytic/myeloid leukemia.
3. Neutropenia (Agranulocytosis):
- Reduction in the absolute neutrophil count (total WBC × % segmented neutrophils and band forms) below 1.5 × 109/L (1,500/cu mm).
- Etiology: The causes of neutropenia are presented in Table.
Eosinophilia:
Eosinophil count of more than 450/cu mm (0.45 × 109/L).
The causes of eosinophilia are presented in Table.
Major causes of neutrophilia:

Diffrences between leukemoid reaction and chronic myeloid leukemia
Tabulate The Differences between leukemia and leukemoid reaction.

Causes of neutropenia:
Causes of eosinophilia:

Causes of Lymphocytosis:

Lymphocytosis:
Lymphocyte count is more than 4,000/cu mm (4 × 109/L) in adults and more than 8,000/cu mm (8 × 109/L) in children. Common causes of lymphocytosis are given.

Acute Leukemia
Define and classify leukemia.
Acute leukemia is a malignant disease of the bone marrow stem cell and its characteristic features are:
- Bone marrow: Diffse replacement with proliferating neoplastic blast cells that fail to mature. Blast cells more than 20% (WHO criteria) of the nucleated cells in the marrow.
- Peripheral blood: Abnormal numbers and forms of immature white blood cells.
- Aleukemic/sub-leukemic leukemia is characterized by very few/no blasts in the peripheral blood.
- Acute leukemia is mainly divided into two groups namely acute lymphoblastic leukemia (ALL) and acute myeloblastic leukemia (AML).
Etiology and Pathogenesis:
Risk factors: Risk factors may cause mutations in the proto-oncogenes and tumor suppressor genes.
Risk factors for acute leukemia:

Classification of Acute Leukemia:
Classify acute leukemia.
Traditional classification depending on the microscopic appearance of the involved cell and the course of leukemias is presented in Table.
The traditional classification of leukemia:

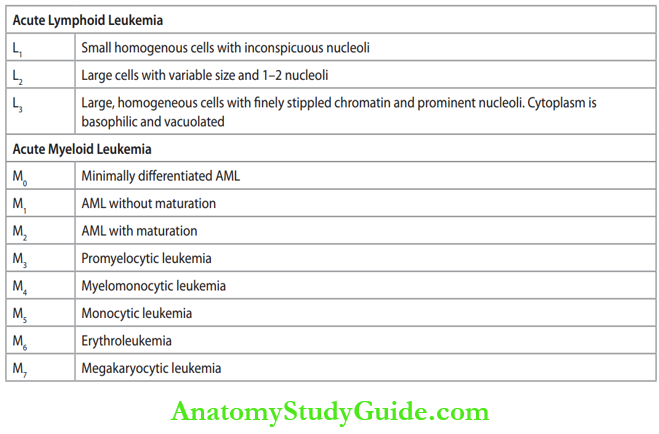
FAB Classification of Acute Leukemias:
- First French, American, and British (FAB) classification (1976) was based on the 1. morphological and 2. cytochemical characteristics of blast cells.
- Revised FAB classification: It includes
- M: Morphology and cytochemistry of blast cells
- I: Immunophenotyping
- C: Cytogenetics
- M: Molecular genetics.
Revised French, American, and British (FAB) classification of acute leukemias:
WHO Classification (2008) of Acute Leukemia:
WHO classification (2008) of acute lymphoblastic and myeloid leukemia:

WHO classification:
- Precursor B cells ALL (about 85%) are seen in childhood and present as acute leukemias.
- Precursor T cells ALL (15%) present in adolescent males as lymphomas, often with the involvement of mediastinum (thymus).
Molecular Pathogenesis:
- Chromosomal abnormalities are found in about 90% of ALLs.
- Numerical abnormality: Hyperploidy (>50 chromosomes) and hypoploidy.
- Structural abnormality: Balanced chromosomal translocations (for example, Philadelphia chromosome).
Acute Lymphoblastic Leukemia/Lymphoma
Acute Lymphoblastic Leukemia/Lymphoma (ALL) is a group of neoplasms consisting of
lymphoblasts.
Lymphoblast is an immature, precursor B (pre – B) or T (pre-T) Lymphocyte.


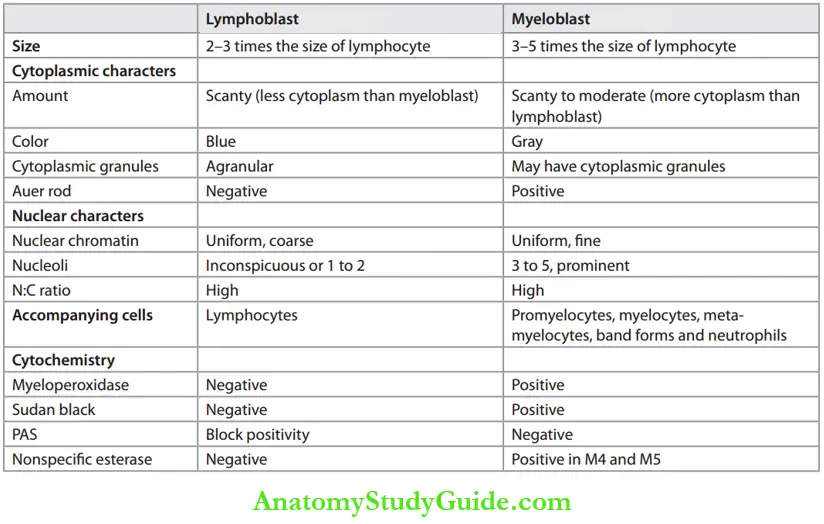
List the differences between myeloblast and lymphoblast.
Or
Differences between myeloblast and lymphoblast based on morphology and cytochemistry.
Differences between Myeloblast and Lymphoblast:
Classification of Acute Lymphoblastic: Leukemia
Clinical Features of Acute Lymphoblastic:
- Age: Most common hematological malignancy of children. Most common between 1 and 5 years of age and between 30 and 40 years.
- Sex: Slight male preponderance.
- Onset: Abrupt.
Symptoms of Acute Lymphoblastic:
- Bone marrow failure:
- Anemia: Causes fatigue, weakness.
- Neutropenia: Infections by bacteria or opportunistic fungi. Develop sore throat and
respiratory infections. - Thrombocytopenia: Bleeding into the skin and mucosa in the form of purpura or
ecchymoses. - Bone pain and sternal tenderness.
- Extramedullary infiltration:
- Lymphadenopathy: 75% of patients, usually involve cervical lymph nodes.
- Bone pain and tenderness.
- Hepatosplenomegaly: Splenomegaly is more common than hepatomegaly.
- Mediastinal thymic mass: More common in T-ALL.
- CNS involvement: Spread into the meninges causes leukemic meningitis ALL (pre-B).
- Testicular involvement (ALL).
Laboratory Findings of Acute Lymphoblastic:
Peripheral Blood:
Write a short note on laboratory/peripheral smear findings in acute lymphoblastic leukemia.
- Total WBC count: Markedly raised ranging from 20 × 109/L to 200 × 109/L
- Platelet count: Reduced (thrombocytopenia).
- Hemoglobin: Decreased and may be as low as 3 g/dL.

Cytochemistry of lymphoblasts:
- PAS: Cytoplasmic aggregates of PAS-positive material (block positivity).
- Myeloperoxidase (MPO) negative.
- Sudan black B negative.
Write short note on acute myeloid leukemia
Bone Marrow on acute myeloid leukemia:
- Cellularity: Markedly hypercellular due to the proliferation of blasts.
- Erythropoiesis and myelopoiesis: Reduced.
- Megakaryopoiesis: Megakaryocytes gradually decrease.
- Blasts: Constitute 20–100% of the marrow cells.


Immunophenotyping on acute myeloid leukemia:
- Terminal-deoxynucleotidyl-transferase (TdT) + in pre-B and pre-T lymphoblasts.
- Immature B cells + positive for pan B cell marker CD19 and CD10 (CALLA—common ALL antigen).
- Precursor T ALL cells are positive for CD2, CD5, and CD8.
Biochemical Findings on acute myeloid leukemia:
- Serum uric acid: Raised due to the destruction of leukemic cells during chemotherapy leading to hyperuricemia.
- LDH: Raised, because of increased turnover of leukemic cells.
CSF Examination on acute myeloid leukemia:
To know/rule out CNS involvement.
Acute Myelogenous Leukemia
Write a short note on the classification of acute myeloid leukemia
Or
Write a short note on acute myeloid leukemia
Definition of acute myeloid leukemia: Neoplasm of hematopoietic progenitors characterized by proliferation resulting in accumulation of immature myeloblasts in the marrow.
Classification of acute myelogenous leukemia (AML)
Molecular Pathogenesis:
- Many recurrent genetic abnormalities can disrupt genes encoding transcription factors involved in normal myeloid differentiation.
- Mutated tyrosine kinase activation is common.
Clinical Features of acute myeloid leukemia:
- Age: AML may develop at any age, but is more common in adults.
- Onset: Acute leukemias are abrupt in onset.
- Symptoms: Related to depressed marrow function.
Bone marrow failure:
- Anemia: Fatigue and weakness.
- Neutropenia: Life-threatening infections by bacteria or opportunistic fungi.
- Thrombocytopenia: Bleeding, the patient may also develop disseminated intravascular
coagulation (DIC) in AML M3 and primary fibrinolysis. - Bone pain and tenderness.
Extramedullary infiltration:
- Gingival hypertrophy (M4 and M5) and infiltration of skin (leukemia cutis).
- Hepatosplenomegaly: Usually more than in ALL.
Laboratory Findings of acute myeloid leukemia:
Peripheral Blood:
Write short note on laboratory/peripheral smear findings in AML.
- Total WBC Count: Markedly raised ranging from 20 × 109/L to 100 × 109/L.
- Hemoglobin: Decreased and ranges from 5 to 9 g/dL.
Peripheral smear :

Cytochemistry of Myeloblasts:
- Stain positively with myeloperoxidase (MPO) and Sudan black B.
- Monoblasts stain with nonspecific esterases.
Bone Marrow of Myeloblasts:
- Cellularity: Markedly hypercellular.
- Erythropoiesis: Markedly suppressed.
- Myelopoiesis: Suppression of myeloid maturation and myeloblasts constitute more than 20% of marrow cells.
- Megakaryopoiesis: Gradually decreased.
Immunophenotyping of Myeloblasts:
Diagnosis of AML is confirmed by using stains for myeloid specific antigens.
Cytogenetics:
Very important in the WHO classification of AML.


Chronic Myelogenous Leukemia
Definition of Chronic myelogenous leukemia:
Chronic myelogenous leukemia (CML) is one of the myeloproliferative neoplasm (MPN) of pluripotent hematopoietic stem cells characterized by overproduction of cells of the myeloid series which results in marked splenomegaly and leukocytosis.
Distinguished from other myeloproliferative neoplasms by the presence of:
- Chimeric fusion BCR-ABL gene.
- Philadelphia (Ph) chromosome in more than 90% of cases.
Etiology and Pathogenesis:
Write a short note on the etiology of chronic myeloid leukemia
- Risk factor: Exposure to ionizing radiation and benzene.
Molecular Pathogenesis of Chronic myelogenous leukemia:
Write a short note on the Philadelphia chromosome and its clinical significance.
Philadelphia (Ph) Chromosome:
- Acquired chromosomal abnormality in all proliferating hematopoietic stem cells (erythroid, myeloid, monocytic, and megakaryocytic precursors).
- Balanced reciprocal translocation between the long arm of chromosome 9 and 22, i.e. t (9; 22)
- It increases the length of chromosome 9 and the shortening of 22. This shortened chromosome 22 is known as the Philadelphia chromosome.
BCR-ABL Fusion Gene:
ABL proto-oncogene from chromosome 9 joins the BCR on chromosome 22.
- It produces a new chimeric (fusion) gene called BCR-ABL, thus converting ABL proto-oncogene into an oncogene. The product of the fusion gene plays a central role in the development of CML.
- The product of this oncogene, i.e. oncoprotein (for example, p210) causes cell division and inhibition of apoptosis.

Clinical Features of Chronic myelogenous leukemia:
Write a short note on the clinical features of chronic myeloid leukemia
- Age: Usually occurs between 40 to 60 years of age.
- Sex: Males are slightly more affected than females.
- Onset: Insidious.
Symptoms:
- Nonspecific symptoms: Fatigue, weakness, weight loss, anorexia.
- The fullness of the abdomen due to splenomegaly (caused by leukemic infitration and extramedullary hematopoiesis). Splenomegaly is moderate to severe and is a characteristic feature in the majority (8090%) of patients.
- Hepatomegaly: Mild or moderate seen in 6070% of cases.
Natural History of Chronic Myeloid Leukemia:
Three diffrent phases
- Chronic phase
- Accelerated phase and
- Blastic phase.
Chronic/Stable/Indolent Phase (CP):
- Most are diagnosed in this phase.
- Lasts for 2 to 6 years.
- If not treated, progresses gradually to the accelerated phase or abruptly to the blastic phase.
Laboratory Findings of Chronic Myeloid Leukemia:
Write a short note on laboratory findings /peripheral smear in CML.
Peripheral blood:
Hemoglobin: Usually less than 11 g/dL.



Bone Marrow of chronic myeloid leukemia:
Write a short note on bone marrow findings in chronic myeloid leukemia
- Cellularity: Markedly hypercellular due to myeloid hyperplasia.
- M: E ratio: Often exceeds 20:1.
- Erythropoiesis: Diminished erythropoiesis as disease progresses.
- Myelopoiesis: Marked hyperplasia. Blast cells are usually less than 10%. Basophils, eosinophils, and their precursors are usually found.
- Megakaryopoiesis: Megakaryocytes are either normal or increased. Dwarf megakaryocytes.
- Sea-blue histiocytes (Gaucher-like cells/pseudo-Gaucher cells) are seen.
Biochemical fidings of chronic myeloid leukemia:
- Serum uric acid raised
- Serum LDH raised.
Philadelphia chromosome and BCR-ABL fusion gene: Demonstrated either by chromosomal analysis or fluorescent in situ hybridization (FISH) or PCR-based tests.
- Accelerated Phase (AP)
- More aggressive and lasts for a few months.
- Blast Phase/Crisis (BP):
- Blood picture resembles acute leukemia and has a poor prognosis.
Chronic Lymphocytic Leukemia
Definition of Chronic lymphocytic leukemia:
Write a short note on chronic lymphocytic leukemia.
Chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) is a tumor composed of monomorphic small B lymphocytes in the peripheral blood, bone marrow and lymphoid organs (spleen and lymph nodes).
- Both CLL and SLL is a single entities with different presentations.
- Small lymphocytic lymphoma (SLL) is the tissue equivalent of chronic lymphocytic leukemia (CLL).
- CLL/SLL tumor cells coexpress CD5 and CD23.
Etiology and Pathogenesis:
- Environmental factors: Suggested but none proved.
- Hereditary factors: Families with a higher risk of CLL or other lymphoid neoplasms.
Clinical Features of Chronic lymphocytic leukemia:
- Age: Between 5060 years of age.
- Sex: More in males than in females (2:1).
Symptoms of Chronic lymphocytic leukemia:
- Asymptomatic in about 2530%
- Nonspecific symptoms: Fatigue, loss of weight, and anorexia
- Generalized lymphadenopathy
- Immunological defects either as immune deficiency or autoimmunity.
Laboratory Findings of Chronic lymphocytic leukemia:
Peripheral Blood:
- Hemoglobin: Decreased and usually below 13 g/dL.
- Total leukocyte count is increased (2050 × 109/L).


Peripheral smear:

Bone Marrow of Chronic lymphocytic leukemia:
- Cellularity: Hypercellular marrow due to infiltration by mature lymphocytes.
- Erythropoiesis: Normal.
- Myelopoiesis: Normal.
- Megakaryopoiesis: Normal.
- Lymphocytic infiltrate: As the disease advances neoplastic lymphocytes replace the normal erythroid, myeloid and megakaryocytic series in the bone marrow resulting in anemia, neutropenia and thrombocytopenia.
Lymph Node of Chronic lymphocytic leukemia:
- Show loss of normal architecture
- Diffse infiltration by monomorphic, small, round lymphocytes
- Lymphocytes have nuclei with coarse chromatin and scanty cytoplasm
- Small, nodular aggregates of medium to large-sized lymphocytes known as proliferation centers or pseudo-follicles, or growth centers and when found are pathognomonic for CLL/SLL.
Course and prognosis: Median survival rate is 4 to 6 years. They may progress to B cell prolymphocytic transformation or into diffuse large B cell lymphoma (Richter syndrome).
Disorders Of Hemostasis
Normal Hemostasis:
- Hemostasis is the body’s response to vascular damage/injury.
- Includes several sequences of events at the site of vascular injury. They are as follows:
- Primary Hemostatic Plug: Platelet adhere to subendothelial structures at the site of injury. The platelets change their shape and release granule contents. The released contents cause platelet aggregation and form a primary hemostatic plug.
- Secondary Hemostatic Plug: Exposure of tissue factor at the site of vascular injury activates the extrinsic coagulation system. The firin formed develops into a secondary hemostatic plug.
Classification Of Hemostatic Disorders
Classify bleeding disorders.
- Bleeding disorders (hemorrhagic disorders/hemorrhagic diathesis): Bleeding disorders have an abnormal tendency to bleed (hemorrhage) due to failure of hemostasis.
- Thrombotic disorders: They cause thrombus formation.
Bleeding Disorders Caused by Vessel Wall Abnormalities:
Vascular purpura (nonthrombocytopenic purpura) is a group of disorders of blood vessels that results in bleeding. They should be distinguished from bleeding disorders due to abnormalities of platelets.
Classification of disorders of hemostasis:

Classification of bleeding disorders caused by vessel wall abnormalities are presented in
Classification of bleeding disorders caused by vessel wall abnormalities:

Bleeding Disorders Due to Abnormalities of Platelet
Classification of Platelet Disorders.
Classification of platelet disorders:

Thrombocytopenia:
Decrease in the platelet count below the lower limit of 150,000/cu mm (150 × 109/L).
Clinical Features of Thrombocytopenia:
- Cutaneous bleeding appears as pinpoint hemorrhages (petechiae) and ecchymoses.
- Mucosal bleeding.
- Intracranial bleeding (subarachnoid and intracerebral hemorrhage) is rare but serious.
Severity of Bleeding:
- Post-traumatic bleeding — When the platelet count is 20,000 to 50,000/cu mm.
- Spontaneous bleeding — When the platelet count falls below 20,000/cu mm.
- Intracranial bleeding — When platelet count is <10,000/cu mm.
Causes of Thrombocytopenia

Immune Thrombocytopenic Purpura
- Most common form of thrombocytopenia.
- Due to increased destruction of platelets by immune mechanisms—mainly autoimmune
mechanism.
Types of Immune Thrombocytopenic Purpura (ITP):
Acute Immune Thrombocytopenic Purpura
Write a short note on immune thrombocytopenic purpura.
- Self-limited disease.
- Children: 2 to 4 years and seen equally in both sexes.
- Presents 1 to 3 weeks after viral (measles, rubella, EBV) infection.
- Platelet destruction by antiplatelet autoantibodies.
- Platelet count is decreased, sometimes even below 10,000/cu mm (10 × 109/L).
Clinical Features of immune thrombocytopenic purpura:
- Sudden onset.
- Petechiae, gum bleeding, epistaxis and mild fever.
- Usually resolve spontaneously within 6 months.
- Excellent prognosis.
Chronic Immune Thrombocytopenic Purpura (ITP):
- Persistent thrombocytopenia for more than 6 to 12 months.
- Indolently, females are more affected than males (F: M = 3:1).
- More common and usually seen in adults (20 to 40 years).
Pathogenesis of ITP :
- An autoimmune disorder is characterized by the formation of antiplatelet antibodies, directed against membrane glycoproteins (most often IIb-IIIa or Ib-IX of platelets).
- Antiplatelet antibodies in about 80% of patients and are of the IgG type.
- Antiplatelet antibodies act as opsonins and are recognized by IgG Fc receptors present on mononuclear phagocytes of RE system (mainly the spleen) and are destroyed there resulting in thrombocytopenia.
- Splenectomy causes marked improvement in 75% to 80% of patients.
Clinical Features of ITP :
- More common in females (F: M ratio is 3:1).
- Age between 20 and 40 years.
- Clinical features are due to thrombocytopenia: Skin bleeding, mucosal bleeding, menorrhagia in females, etc.

Laboratory Findings of ITP :
Write short notes on laboratory findings in ITP.
Peripheral Blood:
- Platelet count: Markedly reduced—below 80,000/cu mm (80 × 109/L).
- Hemoglobin: Ranges from 7 to 12 g/dL.

Bone Marrow of ITP :
- Cellularity: Hypercellular.
- Megakaryopoiesis:
- A moderate increase in the number of both immature and mature forms of megakaryocytes.
- Immature megakaryocytes predominate large non lobulated single nuclei and basophilic cytoplasm.
Erythropoiesis of ITP:
-
- Prolonged bleeding may cause anemia leading to normoblastic erythroid hyperplasia.
- Constant bleeding leads to iron deficiency and micronormoblastic erythroid hyperplasia.
- Myelopoiesis: Normal.
- Storage iron: Severe and chronic bleeding causes iron deficiency with reduced iron stores.
- Bleeding time (BT): Prolonged, but PT and PTT are normal.
- Tourniquet test: Positive.
- Clotting time (CT): Normal.
- Tests for platelet autoantibodies: May be positive.
- Spleen: Normal size.
Coagulation Disorders
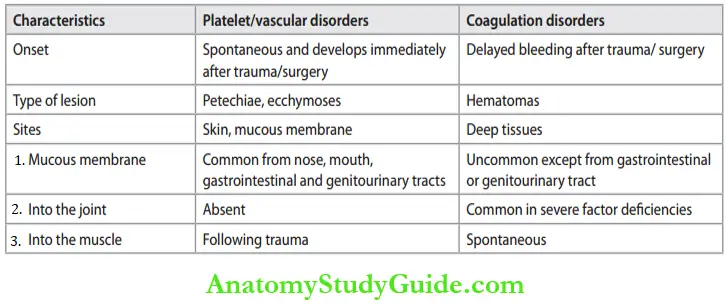
Bleeding due to coagulation disorders must be distinguished from those due to platelet/ vascular disorders.
Distinguishing patterns of bleeding in platelet/vascular and coagulation disorders:
Classifiation of Coagulation Disorders:

Hereditary Coagulation Disorders
Usually, due to a deficiency of the single coagulation factor.
Factor 8-vWF Complex:
- Factor 8-vWF complex has two components:
- Plasma factor 8
- von Willebrand factor.
- vWF protects factor VIII and is important for its stability.
- Subendothelial vWF promotes platelet adhesion.
- Whenever there is vascular endothelial injury, plasma vWF gets adsorbed to the exposed subendothelial matrix and augments the adhesion of platelets.
Hemophilia
Write a short note on hemophilia
- Hemophilia A and B are similar in both clinical and pathological features, the difference being in the deficient factor.
- Both are sex-linked recessive disorders resulting in an inherited deficiency of the clotting factor or synthesis of a defective clotting factor.
- Males are affected and females are carriers.
Hemophilia A (Factor 8 Deficiency):
- Common hereditary X-linked recessive disease.
- About 30% of hemophiliacs may be due to acquired mutations.
- Reduced amount or activity of factor VIII is associated with life-threatening bleeding.
- Bleeding is due to both inadequate coagulation and inappropriate clot removal (fibrinolysis).
Mode of Inheritance:
- X-linked recessive disease. Genes for factor VIII are located on the long arm of the
X-chromosome. - Does not manifest when there is a normal copy of the X-chromosome.
- Males with a defective/mutant factor VIII gene (hemophiliac gene) on their single X chromosome (XH) suffer from hemophilia.
- Heterozygous females are carriers and do not express the full clinical disease because of the paired normal X-chromosome.
- However, females with two copies of the defective XH chromosome may rarely suffr from hemophilia.
Clinical Features of Inheritance:
Clinical severity depends on the level of factor 8 activity with a normal range expressed as a percentage. Severe cases have less than 1% residual factor VIII activity.
Common clinical presentations include:
- Frequent and spontaneous hemorrhage into the joints-hemarthrosis.
- Hemorrhage into soft tissues.
- Prolonged bleeding following trauma.
Laboratory Findings of Inheritance:
- Bleeding time: Normal
- Clotting time: Prolonged, but is not a sensitive test
- Platelet count: Normal
- Prothrombin time: Normal
- Activated partial thromboplastin time (APTT): Increased (normal 3040 seconds)
- Factor 8 assay: Essential for the diagnosis and to assess the levels and severity of disease.
- Fibrinogen assay: Normal
- FDP: Negative
- Detection of carriers: By DNA markers
- To detect female carriers
- Prenatal diagnosis of affected fetuses.

Complications of Inheritance:
- Due to Hemophilia:
- Deforming arthritis and contractures: This is due to repeated bleeding into the joints.
- Organization and fibrosis of intramuscular hematomas → contractures of involved muscles.
- Anemia: Excessive, spontaneous, or repeated bleeding leads to anemia.
- Due to Therapy:
- Viral hepatitis: Hepatitis B, C, and D in patients who received multiple transfusions of FFP/ cryoprecipitate.
- AIDS: In individuals who received fresh frozen plasma (FFP) or cryoprecipitate, when screening tests for HIV were not available.
- Factor 8 inhibitors: Makes further management difficult.
Hemophilia B (Christmas Disease, Factor 9 Deficiency):
- Clinically indistinguishable from hemophilia A
- X-linked recessive disorder
- Variable clinical severity
- Asay of factor IX should be done to diagnose Christmas disease (named after the fist
patient).
Laboratory Findings of Inheritance:
- Similar to hemophilia A.
- Bleeding time: Normal
- Clotting time: Prolonged
- Platelet count: Normal
- Prothrombin time: Normal
- Activated partial thromboplastin time (APTT): Increased (normal 3040 seconds)
- Factor 9 assay: Factor IX is decreased.
Von Willebrand Disease (vWD)
Write a short note on von Willebrand disease
- Most common inherited bleeding disorders.
- Most cases are autosomal dominant disorders.
- Variable clinical picture with more than 20 variants.
Categories of von Willebrand disease:
Grouped into two major categories:
- Quantitative deficiency of vWF: Decreased circulating vWF
- Type 1- autosomal dominant, mild disorder and form about 75% of all cases.
- Type 3-autosomal recessive, severe disorder, and least common type.
- Qualitative defects in vWF:
- Type 2-autosomal dominant, accounts for 25% with several subtypes.
Clinical Features of von Willebrand disease:
- Most cases are of mild bleeding
- Common symptoms
- Spontaneous bleeding from mucous membranes (for example, Epistaxis)
- Excessive bleeding from wounds or menorrhagia
- In severe cases, similar to hemophilia A.
Laboratory Findings of von Willebrand disease:
- Platelet count: Normal
- Bleeding time: Prolonged
- Clotting time: Prolonged
- Tourniquet test (Hess test): Positive due to defect in platelet adhesion
- APTT: Prolonged APTT
- PT: Normal
- vWF assay: Plasma level of active vWF is decreased
- Platelet function test: Defective ristocetin-induced platelet aggregation test is diagnostic of vWF.
Laboratory tests in hereditary disorders are summarized in Table.
Summary of laboratory tests in hereditary coagulation disorders:

Acquired Coagulation Disorders
Coagulation Factor Abnormalities:
Usually characterized by multiple clotting abnormalities
- Vitamin K deficiency: In neonates, low levels of vitamin K levels may produce life-threatening hemorrhage during the first week of life known as hemorrhagic disease of the newborn.
- Liver disease: Liver synthesizes all the clotting factors and severe liver disease is associated with a hemorrhagic diathesis.
- Other causes: Disseminated intravascular coagulation that involves a deficiency of several coagulation factors.
Disseminated Intravascular Coagulation:
Write a short note on DIC
Widespread disorder with a combination of thrombosis and hemorrhage.
Etiology of DIC:
Develops as a secondary complication of a wide variety of disorders.
Major disorders associated with disseminated intravascular coagulation:

Clinical Features of DIC:
- Serious, often fatal, clinical condition
- Signs and symptoms are related to:
- Hemorrhagic diathesis/bleeding: Most common, manifest as ecchymoses, petechiae, or bleeding from mucous membranes or at the sites of venipuncture.
- Microvascular thrombi: Tissue hypoxia and infarction of the organ leading to multiorgan failure.
Laboratory Findings in DIC:
- Screening Assays
- Coagulation abnormalities
- APTT: Increased as a result of consumption and inhibition of the function of clotting
factors. - Prothrombin time: Increased.
- Thrombin time (TT): Increased because of decreased fibrinogen.
- Fibrinogen: Decreased.
- APTT: Increased as a result of consumption and inhibition of the function of clotting
- Bleeding time: Increased due to decreased platelet count.
- Platelet count: Decreased due to utilization of platelets in microthrombi.
- Peripheral smear: Microangiopathic hemolytic anemia with schistocytes.
Confirmatory Tests on DIC:
- Fibrinolysis abnormalities
- FDP (fibrin degradation/split products): Secondary fibrinolysis results in the generation of FDPs, which can be measured by latex agglutination
- D-dimer test: It is specifi for diagnosing DIC.
Leave a Reply