Molecular Hallmarks Of Cancer
Cancer hallmarks are defined as acquired characteristics which transform phenotypically normal cells into malignant cells and promote the progression of malignant cells while damaging the host tissue.
Read And Learn More: General Pathology Notes
Given recent advances in our knowledge and understanding of molecular carcinogenesis having translational benefits to the clinic, cancer hallmarks have been revised and reclassified into seven hallmarks, by merging some, omitting a few and adding some.

A general discussion on normal cell growth and controls in the cell cycle has been given in the Following discussion correlates the role of cancer-related genes at a molecular level with those in physiological cellular growth.
1. Growth And Proliferation-Permissive Components: Altered Cell-Signalling:
Normal cell growth is regulated by growth-signalling pathways allowing the cells to proliferate in a controlled manner which gets disrupted in cancer.
- In cancer, ‘growth-promoting’ signals (oncogenes) are aberrantly produced in excess favouring tumour progression, while ‘growth inhibitory’ signals (anti-oncogenes or tumour-suppressor genes) are shut down or become insensitive allowing the tumour to proliferate due to failure of brakes.
- The net effect of both these signalling components is balanced in favour of cell proliferation in cancer.
1. Excessive and Autonomous Growth: Growth Promoting Oncogenes:
A mutated form of normal proto-oncogenes in cancer is called oncogenes. In general, the overactivity of oncogenes enhances cell proliferation and promotes the development of human cancer.
About 100 different oncogenes have been described in various cancers. Transformation of proto-oncogene (i.e. normal cell proliferation gene) to oncogenes (i.e. cancer cell proliferation gene) may occur by three mechanisms:
- Point mutations: An alteration of a single base in the DNA chain. The most important example is RAS oncogene carried in many human tumours such as bladder cancer, pancreatic adenocarcinoma, and cholangiocarcinoma.
- Chromosomal translocations: Transfer of a portion of one chromosome carrying protooncogene to another chromosome and making it independent of growth controls. This is implicated in the pathogenesis of leukaemias and lymphomas for example,
Molecular Hallmarks of Cancer:

-
- Philadelphia chromosome is seen in 95% of cases of chronic myelogenous leukaemia in which cABL proto-oncogene on chromosome 9 is translocated to BCR of chromosome 22.
- In 75% of cases of Burkitt’s lymphoma, translocation of c-MYC proto-oncogene from its site on chromosome 8 to a portion on chromosome 14.

- Gene amplification: Gene amplification increases the number of copies of DNA sequence in proto-oncogene leading to increased mRNA and thus increased or overexpressed gene product (i.e. oncoproteins). Examples of gene amplification are found in some solid human tumours for example,
- Neuroblastoma having n-MYC HSR region.
- ERB-B2 (or HER2/neu) in breast and ovarian cancer.
Most of the oncogenes encode components of the cell signalling system for promoting cell proliferation.
Accordingly, these are discussed below under the following groups about different components of cell proliferation signalling systems and are schematically shown below.
- Growth factors
- Receptors of growth factors
- Cytoplasmic signal transduction proteins
- Nuclear transcription factors
- Cell cycle regulatory proteins
Growth factors (GFs):
GFs were the first proto-oncogenes to be discovered which encode for cell proliferation cascade. They act by binding to cell surface receptors to activate the cell proliferation cascade within the cell.
- GFs are small polypeptides secreted by many cells and they normally act on another cell than the one which synthesised it to stimulate its proliferation i.e. paracrine action.
- However, a cancer cell may synthesise a GF and respond to it as well; this way cancer cells acquire growth self-sufficiency.
- Most often, growth factor genes in cancer act by overexpression which stimulates large secretion of GFs that stimulate cell proliferation. The examples of such GFs are as under:
- Platelet-derived growth factor-β-(PDGF-β) Overexpression of SIS proto-oncogene that encodes for PDGF-β and thus there is increased secretion of PDGF-β for example, Gliomas and sarcomas.
- Transforming growth factor-α (TGF-α) Overexpression of the TGF-β gene occurs by stimulation of RAS proto-oncogene and induces cell proliferation by binding to epidermal growth factor (EGF) receptors for example, Carcinoma and astrocytoma.
- Fibroblast growth factor (FGF) Overexpression of HST-1proto-oncogene in osteosarcoma and amplification of FGS3 proto-oncogene causes excess secretion of GF in cancer of the breast and stomach.
- Hepatocyte growth factor (HGF) Overexpression by binding to its receptor c-MET for example, Familial follicular carcinoma thyroid, hepatocellular carcinoma.
GF Receptors Growth:
GF Receptors Growth factors cannot penetrate the cell directly and require to be transported into the cell by GF-specific cell surface receptors.
- These receptors are transmembrane proteins, having extracellular, intramembranous and intracellular ligand-binding domains, for example, Receptor tyrosine kinase.
- Normally, binding of GF to the extracellular domain of the receptor tyrosine kinase activates the cytoplasmic component of the receptor and induces signalling.
- However, mutated forms of growth factor receptors stimulate cell proliferation independent of their binding to growth factors. Activation of oncogenes encoding for GF receptors may occur by overexpression, mutation and gene rearrangement.
- Examples of tumours by mutated receptors for growth factors are as under:
Important oncogenes, their mechanism of activation and associated human tumours:
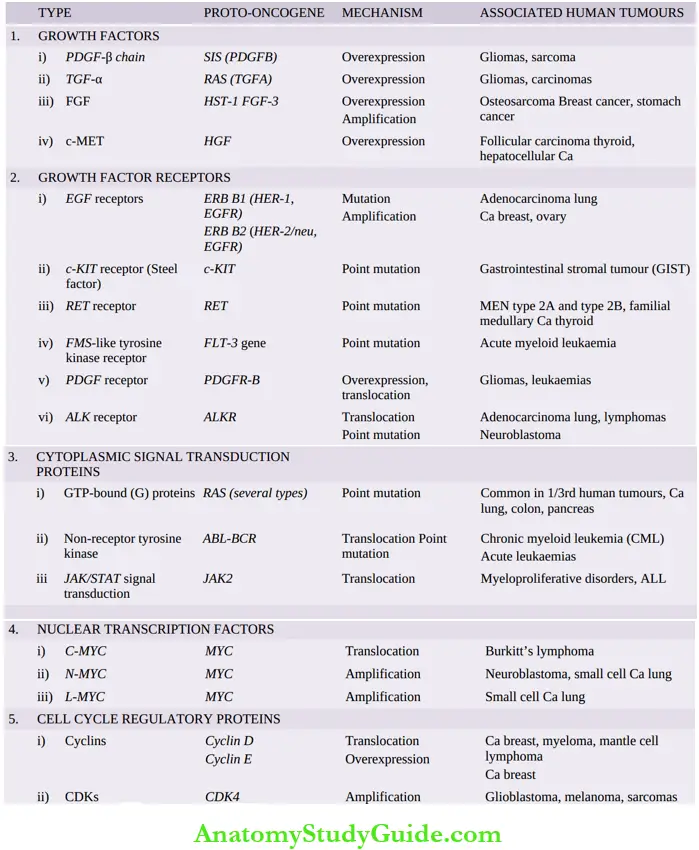

- EGF receptors The Normal EGF receptor gene is ERB B1, and hence this receptor is termed EGFR or HER1 (i.e. human epidermal growth factor receptor type 1). EGFR (or HER1) acts by mutation of normal GF receptors, for example, in 80% of adenocarcinoma of the lung.
- Another EGF receptor gene called ERB B2 (or HER2/neu or CD340) acts by gene amplification for example, in Breast cancer (25% cases), and carcinoma ovary.
- c-KIT receptor The gene coding for the receptor for stem cell factor (or steel factor) is c-KIT, which activates the tyrosine kinase pathway in cell proliferation.
- A mutated form of c-KIT by point mutation activates receptors for tyrosine kinase for example., in gastrointestinal stromal tumours (GIST).
- RET receptor RET (abbreviation of ‘rearranged during transfection’) proto-oncogene is a receptor for tyrosine kinase normally expressed in neuroendocrine cells of different tissues.
- Mutated form by point mutation is seen in MEN type 2A and 2B and in familial medullary
carcinoma thyroid. - FMS-like tyrosine kinase receptor Point mutation of FLT-3 gene (CD 135) that encodes for FMS-like tyrosine kinase receptor has been seen in acute myeloid leukaemia.
- PDGF-receptor overexpression or translocation is seen in gliomas and leukaemias.
- ALK receptor This acts as an oncogene by different mutations—by translocation in adenocarcinoma lung, and by point mutation in neuroblastoma.
Cytoplasmic signal transduction proteins:
The normal signal transduction proteins in the cytoplasm transduce signal from the GF receptors present on the cell surface, to the nucleus of the cell, to activate intracellular growth signalling pathways.
There are examples of oncogenes having mutated forms of cytoplasmic signalling pathways located in the inner surface of cell membranes in some cancers. These are as under:
- Mutated RAS gene This is the most common form of an oncogene in human tumours, the abnormality being induced by a point mutation in the RAS gene.
- About a third of all human tumours carry the mutated RAS gene (RAS for Rat Sarcoma gene where it was first described), notably in carcinoma colon, lung and pancreas.
- Normally, the inactive form of RAS protein is GDP (guanosine diphosphate)-bound while the activated form is bound to guanosine triphosphate (GTP).
- GDP/GTP are homologous to G proteins and take part in signal transduction in a similar way just as G proteins act as an ‘on-off switch’ for signal transduction.
- Normally, active RAS protein is inactivated by GTPase activity, while the mutated form of the RAS gene remains unaffected by
- GTPase, and therefore, continues to signal cell proliferation.
- ABL-BCR hybrid gene ABL (Abelson) is a non-receptor proto-oncogene having tyrosine kinase activity. The ABL gene from its normal location on chromosome 9 is translocated to chromosome 22 where it fuses with the BCR (breakpoint cluster region) gene and forms an ABLBCR hybrid gene which is more potent in the signal transduction pathway.
- ABL-BCR hybrid gene by translocation is seen in chronic myeloid leukaemia and by a point mutation in acute lymphoblastic leukaemias.
- JAK/STAT signal transduction Janus-kinase (JAK) is cytosolic non-receptor tyrosine kinase for cytokines. STAT (signal transducer and activator of transcription) is a latent transcription factor in the cytoplasm which gets activated by JAK i.e. JAK/STAT signalling pathway.
- This way, growth-promoting activity is stimulated by growth factors and cytokine receptors that lack tyrosine kinase (i.e. phosphorylation) activity. Point mutations in JAK2 causing growth-promoting JAK/STAT signal transduction are seen in various examples of myeloproliferative disorders and ALL.
Nuclear transcription factors:
The signal transduction pathway that starts with GFs ultimately reaches the nucleus where it regulates DNA transcription and induces the cell to enter into the S phase. Out of the various nuclear regulatory transcription proteins described, the most important is the MYC gene located on the long arm of chromosome 8.
- Normally MYC protein binds to the DNA and regulates the cell cycle by transcriptional activation and its levels fall immediately after the cell enters the cell cycle.
- MYC oncogene (originally isolated from myelocytomatosis virus and accordingly abbreviated) is commonly seen in human tumours.
- It is associated with the persistence of or overexpression of MYC oncoproteins which, in turn, causes autonomous cell proliferation.
The examples of tumours carrying MYC oncogene are as under:
- C-MYC oncogene Mutated MYC gene due to translocation t(8;14) seen in Burkitt’s lymphoma.
- N-MYC oncogene Mutated MYC gene due to amplification seen in neuroblastoma, small cell carcinoma lung.
- L-MYC oncogene Mutated MYC gene due to amplification seen in small cell carcinoma lung.
Cell cycle regulatory proteins:
Normally, the cell cycle is under the regulatory control of cyclins and cyclin-dependent kinases (CDKs) A, B, E and D. Cyclins activate as well as work together with CDKs, while many inhibitors of CDKs (CDKIs) are also known.
- Although all steps in the cell cycle are under regulatory controls, the G1 → S phase checkpoint is most significant for regulation by oncogenes as well as anti-oncogenes (discussed below).
- Gain-of-function mutations in cyclins (in particular cyclin D) and CDKs (in particular CDK4) are the most important growth-promoting signals in cancers.
The examples of tumours having such mutated oncogenes are as under:
- A mutated form of cyclin D proto-oncogene by translocation is seen in mantle cell lymphoma and myeloma.
- A mutated form of cyclin E by overexpression is seen in breast cancer.
- A mutated form of CDK4 by gene amplification is seen in malignant melanoma, glioblastoma and sarcomas.
2. Refractoriness to Growth-Inhibitory Signals: Tumour-suppressor Genes:
The mutation of tumour-suppressor genes (or antioncogenes having reverse function than oncogenes) results in the removal of the brakes for growth; thus the inhibitory effect on cell growth is removed and the abnormal growth continues unchecked.
- In other words, mutated tumour suppressor genes behave like growth-promoting oncogenes. As compared to the signals and signal transduction pathways for oncogenes described above, the steps in mechanisms of action by growth suppressors are not so well understood.
- In general, the point of action by tumour-suppressor genes is the G1 → S phase transition.
- Normally, tumour-suppressor genes act by either inducing the dividing cell from the cell cycle to enter into the G0 (resting) phase, or by acting in a way that the cell lies in the post-mitotic pool losing its dividing capability.
- Just as proto-oncogenes are activated by mutations to become oncogenes, the mechanisms of loss of tumour-suppressor actions of genes is by mutations which are commonly chromosomal deletions, point mutations and loss-of-function mutation inhibiting G1-S phase progression.
Major tumour-suppressor genes implicated in human cancers are as under:
RB gene:
RB gene is located on the long arm (q) of chromosome 13. This is the first-ever tumour suppressor gene identified and thus has been amply studied. RB gene codes for a nuclear transcription protein called pRB. RB gene is termed as ‘master brake in the cell cycle’ and is virtually present in every human cell.
It can exist in both an active and an inactive form:
The active form of the RB gene It blocks cell division by binding to a transcription factor, E2F, and thus inhibits the cell from the transcription of cell cycle-related genes, thereby inhibiting the cell cycle at G1 → S phase transition i.e. cell cycle is arrested at G1 phase.
- Inactive form of RB gene This takes place when the RB gene is hyperphosphorylated by CDKs and growth factors bind to their receptors. This removes the pRB function from the cell (i.e. the ‘brake’ on cell division is removed).
- Resultantly, the cell proliferation pathway is stimulated by permitting the cell to cross the G1 → S checkpoint. The activity of CDKs is inhibited by the activation of an inhibitory signal, transforming growth factor-β (TGF-β), on the cell through the activation of inhibitory protein p16
Important tumour-suppressor genes and associated human tumours:

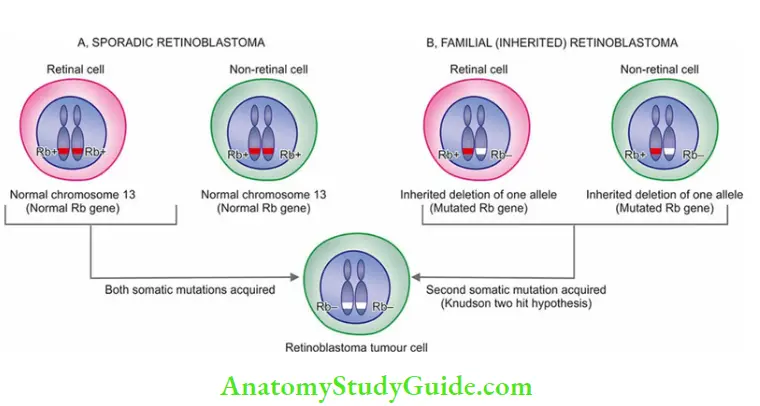
The mutant form of RB gene (i.e. inactivating mutation of RB gene) is involved in several human tumours, most commonly in retinoblastoma, the most common intraocular tumour in young children. The tumour occurs in two forms: sporadic and inherited/familial :
- Sporadic retinoblastoma constitutes about half the cases and affects one eye. These cases have acquired both somatic mutations in the two alleles in retinal cells after birth.
- Inherited/Familial retinoblastoma comprises 40% of cases and may be bilateral. In these cases, all somatic cells (retinal as well as non-retinal cells) inherit one mutant RB gene from a carrier parent (i.e. germline mutation).
- Later in life, the other mutational event of the second allele affecting the somatic cells occurs. This forms the basis of the two-hit hypothesis given by Knudson in 1971.
- Besides retinoblastoma, children inheriting the mutant RB gene have a 200 times greater risk of developing of other cancers in early adult life, notably osteosarcoma; others are cancers of the breast, colon and lungs.
p53 gene:
(TP53) Located on the short arm (p) of chromosome 17, the p53 gene (also termed TP53 because of molecular weight of 53 kd for the protein) similar to pRB is inhibitory to the cell cycle. However, p53 is normally present in very small amounts and accumulates only after DNA damage.
The two major functions of p53 in the normal cell cycle are as under:
- In blocking mitotic activity p53 inhibits the cyclins and CDKs and prevents the cell from entering the G1 phase transiently. This breathing time in the cell cycle is utilised by the cell to repair DNA damage.
- In promoting apoptosis Normally, p53 acts together with another anti-oncogene, the RB gene, and identifies the genes that have damaged DNA which cannot be repaired by an inbuilt system.
- p53 directs such cells to apoptosis by activating the apoptosis-inducing BAX gene, and thus bringing the defective cells to an end by apoptosis. This process operates in the cell cycle at the G1 and G2 phases before the cell enters the S or M phase.
Because of these significant maintenance roles in the cell cycle, p53 is called as ‘guardian of the genome’.
- In its mutated form, p53 ceases to act as a protector or as a growth suppressor but instead acts like a growth promoter or oncogene. Homozygous loss of the p53 gene allows genetically damaged and unrepaired cells to survive and proliferate resulting in malignant transformation.
- More than 70% of human cancers have a homozygous loss of p53 by acquired mutations in somatic cells; some common examples are cancers of the lung, head and neck, colon and breast.
- Besides, mutated p53 is also seen in the sequential development stages of cancer from hyperplasia to carcinoma in situ and into invasive carcinoma.
- Less commonly, both alleles of the p53 gene become defective by another way: one allele of p53 is mutated by inheritance in germ cell lines rendering the individual to another hit of somatic mutation on the second allele.
- Just as in the RB gene, this defect predisposes the individual to develop cancers of multiple organs (breast, bone, brain, sarcomas etc), termed Li-Fraumeni syndrome.
Transforming growth factor-β (TGF-β) and its receptor:
Normally, TGF-β is a significant inhibitor of cell proliferation, especially in epithelial, endothelial and haematopoietic cells. It acts by binding to the TGF-β receptor and then the complex so formed acts in the G1 phase of the cell cycle at two levels:
- It activates CDK inhibitors with a growth-inhibitory effect.
- It suppresses the growth-promoter genes such as MYC, CDKs and cyclins.
A mutant form of the TGF-β gene or its receptor impairs the growth-inhibiting effect and thus permits cell proliferation. Examples of mutated forms of TGF-β are seen in cancers of the pancreas, colon, stomach and endometrium.
Adenomatous polyposis coli (APC) gene and β-catenin protein:
The APC gene is normally inhibitory to mitosis, which takes place by a cytoplasmic protein, β-catenin. β-catenin normally has dual functions:
- Firstly, it binds to cytoplasmic E-cadherin which is involved in intercellular interactions.
- Secondly, it can activate the cell proliferation signalling pathway.
- APC gene is a component of the WNT signalling pathway that signals through frizzled cell surface receptors which activate several pathways including β-catenin. In colon cancer cells,
- APC gene is lost and thus β-catenin fails to get degraded, allowing the cancer cells to undergo mitosis without the inhibitory influence of β-catenin.
- Patients born with one mutant APC gene allele develop a large number of polyps in the colon early in life, while after the age of 20 years, these cases start developing loss of a second APC gene allele.
- It is then that almost all patients having both mutated alleles invariably develop malignant transformation of one or more polyps.
Other tumour-suppressor genes:
A few other tumour-suppressor genes having mutated germlines in various tumours are as under:
1. BRCA1 and BRCA2 genes These are two breasts (BR) cancer (CA) susceptibility genes BRCA1 located on chromosome 17q21 and BRCA2 on chromosome 13q12-13.
2. Women with inherited defects in the BRCA1 gene have a very high risk (85%) of developing breast cancer and ovarian cancer (40%). Inherited breast cancer constitutes about 5-10% of cases, it tends to occur at a relatively younger age and more often tends to be bilateral.
3. VHL gene von-Hippel-Lindau (VHL) disease is a rare autosomal dominant disease characterised by benign and malignant tumours of multiple tissues. The disease is inherited as a mutation in the VHL tumour suppressor gene located on chromosome 3p. Product of VHL gene,
4. VHL protein, is a component of ubiquitin ligase that acts as transcription factor HIF1α (hypoxia-inducible factor) i.e. undergoes gene expression in response to hypoxia. In the inactivated VHL gene, HIF1α escapes ubiquitination and degradation even in normoxic conditions and activates genes that promote angiogenesis, cell survival and proliferation.
5. The VHL gene is found inactivated in 60% of cases of renal cell carcinoma.
6. Wilms’ tumour (WT) gene WT1 and WT2 genes are both located on chromosome 11 and normally prevent neoplastic proliferation of cells in embryonic kidney. WT1 protein acts as a transcription factor. Mutant forms of WT-1 and 2 are seen in hereditary Wilms’ tumours.
7. Neurofibroma (NF) gene NF genes normally prevent the proliferation of Schwann cells. Two mutant forms are described: NF1 and NF2 seen in neurofibromatosis type 1 and type 2.
8. PTEN gene PTEN (phosphatase and tensin homologue) gene encodes for a phosphatase on the cell membrane. It is a tumour-suppressor that acts as a brake on the P13K/AKT component of receptor tyrosine kinase. Mutation of the PTEN tumour suppressor gene is by loss of function (by deletion or point mutation) in many epithelial cancers, notably breast cancer.
9. PTCH1 gene PTCH tumour-suppressor gene encodes for PATCHED-1 transmembrane protein (PTCH =protein PATCHED homolog). It acts as a brake at the level of the Hedgehog signalling pathway. Its loss-of-function in tumours permits cell proliferation by allowing the proproliferation of genes such as cyclin D for example, Gorlin syndrome (naevoid basal cell carcinoma).
10. CDKN2A gene This tumour-suppressor gene encodes for inhibitors in the cell cycle pathway at the level of p16/INK4 and CDK4/cyclin D pathways, thereby acting as RB gene checkpoint.
Deletion or point mutation in CDKN2A results in loss-of-function, hence removing the brakes in proliferation (particularly silencing of p16) for example Bladder cancer and carcinoma cervix.
The contrasting features of oncogenes and tumour-suppressor genes are summarised in
2. Favouring Overall Cell Survival: Altered Stress Response:
Cancer cells are faced with stresses of a variety of types such as excessive cell-signalling, DNA damage, hypoxia and nutrient deficiency, besides facing the onslaught of anticancer therapy.
Just as normal cells physiologically adapt to stresses for the larger good of the health of the tissue, cancer cells respond to stresses by subverting the process in favour of cancer growth and survival.
These include:
- Avoiding DNA repair
- Escaping cell death by apoptosis,
- Avoiding cell senescence and
- Recycling intracellular components by autophagy.
These are discussed below:
Avoiding DNA Repair System: Mutator Genes
Normal cells during complex mitosis suffer from minor damage to the DNA which is detected and repaired before mitosis is completed so that integrity of the genome is maintained. Similarly, small mutational damage to the dividing cell by exogenous factors (for example, By radiation, chemical carcinogens etc) is also repaired.
p53 gene is held responsible for the detection and repair of DNA damage. However, if this system of DNA repair is defective either due to germline mutations or by acquired mutations (mutator genes), the defect in unrepaired DNA is passed to the next progeny of cells and cancer results.
Oncogenes versus tumour-suppressor genes (antioncogenes)

Examples of mutator genes exist in the following inherited disorders associated with increased propensity to cancer:
- Hereditary non-polyposis colon cancer (HNPCC or Lynch syndrome) is characterised by a hereditary predisposition to develop colorectal cancer. It is due to defects in genes involved in DNA mismatch repair which results in the accumulation of errors in the form of mutations in many genes.
- Ataxia telangiectasia (AT) has an ATM (M for mutated) gene. These patients have multiple cancers besides other features such as cerebellar degeneration, immunologic derangements and oculo-cutaneous manifestations.
- Xeroderma pigmentosum is an inherited disorder in which there is a defect in the DNA repair mechanism. Upon exposure to sunlight, the damage to DNA by UV radiation cannot be repaired.
- Thus, such patients are more prone to various forms of skin cancers.
- Bloom syndrome is an example of damage by ionising radiation which cannot be repaired due to inherited defects and the patients have an increased risk of developing cancers, particularly leukaemia.
- Hereditary breast cancer patients having mutated BRCA1 and BRCA2 genes carry inherited defects in the DNA repair mechanism. These patients are not only predisposed to develop breast cancer but also cancers of various other organs.
- Paradoxically, overexpression of repair genes by upregulation of repair pathways has also been observed as a mechanism in some tumours for example in Leukaemias, Cancer of Breast and Pancreas.
Escaping Cell Death by Apoptosis: Apoptosis Regulatory:
Genes Besides the role of mutant forms of growth-promoting oncogenes and tumour-suppressor genes, another mechanism of tumour growth is by escaping cell death by apoptosis.
Apoptosis in normal cells is guided by intrinsic and extrinsic pathways resulting in DNA damage. In this process, there is the role of cell death receptor, CD95, some pro-apoptotic factors, apoptosis inhibitors and cell death-inducing signals (page 59).
Cancer cells escape cell death by subverting normal pathways of apoptosis at different levels:
- Loss of p53 leads to reduced function of proapoptotic factors such as BAX.
- By reduced release of cytochrome c from mitochondria due to upregulated antiapoptotic factors such as BCL2, BCL-XL, and MCL-1.
- By loss of APAF-1 (apoptotic peptidase activating factor-1).
- By upregulating apoptotic inhibitors.
- By reduced level of surface receptor CD95 molecule.
- By inactivation of cell death-inducing signals FADD and FasL.
- The examples of tumours by this mechanism are as under:
- BCL2 gene is seen in normal lymphocytes, but its mutant form with characteristic translocation (t14;18) (q32;q21) was first described in B-cell lymphoma and hence the name
- BCL. It is also seen in many other human cancers for example., cancers of the breast, thyroid and prostate.
- Mutation in the BCL2 gene removes the apoptosis-inhibitory control on cancer cells, thus more live cells undergoing mitosis are added to tumour growth. Besides, MYC oncogene and p53 tumour suppressor genes are also connected to apoptosis.
- While MYC allows cell growth, BCL2 inhibits cell death; thus MYC and BCL2 together allow cell proliferation. Normally, p53 activates proapoptotic gene BAX but mutated p53 (i.e. absence of p53) reduces apoptotic activity and thus allows cell proliferation.
- CD95/FasL receptors are depleted in hepatocellular carcinoma and certain lymphomas and hence the tumour cells escape apoptosis.
Evading Cell Senescence: Telomeres and Telomerase
As discussed in the pathology of ageing, after each mitosis (cell doubling) there is progressive shortening of telomeres which are the terminal tips of chromosomes.
- Telomerase is the RNA enzyme that helps in the repair of such damage to DNA and maintains normal telomere length in successive cell divisions.
- However, it has been seen that after repetitive mitosis for a maximum of 60–70 times, telomeres are lost in normal cells and the cells cease to undergo mitosis. This forms the basis of cellular ageing.
- Telomerase is active in normal stem cells but not in normal somatic cells.
- Cancer cells in most malignancies have markedly upregulated telomerase enzymes, and are thus capable of reconstituting telomere length.
- A few other stresses can also induce cellular senescence in cancers by non-telomeric DNA damage such as by reactive oxygen species, anticancer therapy, continuous cell-signalling in favour of mitosis etc.
- Overall, cancer cells avoid ageing by disrupting tumour suppressors such as p53 and INK4a/p16 in a way that mitosis does not slow down or cease, thereby immortalising the cancer cells.

Recycling Intracellular Components: Autophagy
As discussed on page 60, autophagy is a physiologic recycling process of intracellular components by cannibalism of misfolded proteins and long-lived organelles. The energy so produced by autophagy is utilised for cell metabolism.
Autophagy is upregulated in response to a variety of stresses. In cancer, autophagy may have different stress responses in different settings:
- Role as a tumour suppressor: By eliminating damaged mitochondria, it reduces the burden of reactive oxygen species, degrades overexpressed proteins and acts as a defence against oncogenic microbes.
- Role in tumour survival and progression: Upregulated autophagy in cancer stress enables cancer cells to survive harsh metabolic conditions of nutrient deficiency, and hypoxia or may secrete tumour microenvironment (TME)-reshaping proteins.
Thus, the role of autophagy in cancer remains context-dependent.
3. Sustained Perfusion Of Cancer: Vascularisation:
Cancer cells cannot thrive and metastasise without neovascularisation which supplies them with nourishment and oxygen. This process includes tumour angiogenesis as the most important mechanism but there are a few other modes of vascularisation.
Tumour Angiogenesis:
Angiogenesis is the process of formation of new vessels by sprouting from the pre-existing vessels that regenerate to form new endothelial cells by mitosis and migration, for example, Chronic inflammation, wound healing, and embryogenesis.
Angiogenesis is regulated by several pro- and anti-angiogenic factors:
- Promoters of tumour angiogenesis: Include vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF).
- Anti-angiogenesis factors: Inhibiting angiogenesis include thrombospondin-1, angiostatin, endostatin and vasculostatin. A mutated form of the p53 gene in both alleles in various cancers results in the removal of the anti-angiogenic role of thrombospondin-1, thus favouring continued angiogenesis.
In physiologic conditions, the process is controlled and self-limited and is switched off after its expected function is complete.
However, in malignancies, the angiogenic process is continuously activated due to the following features in cancers:
- The major trigger for angiogenesis is hypoxia present in many cancers that are associated with a raised level of HIF which activates the vascular endothelial growth factor (VEGF) signalling pathway.
- Another angiogenic switch in cancers is acidic TME (tumour microenvironment) induced by metabolic changes in cancer (discussed later).
- Newly formed vessels in cancer are dilated, tortuous and leaky due to loose contact of endothelial cells with each other and with basement membrane which results in accentuating hypoxia and acidosis, promoting angiogenesis.
Other Modes of Tumour Vascularisation:
- Besides angiogenesis, some non-angiogenic modes of vascularisation have been described in tumours which have clinical implications since they escape from anti-angiogenic therapy. These are as under:
- Vascular co-option During the early stage of tumour development (micro-tumours), tumour cells may obtain blood supply by diverting existing vasculature.
- Intussusceptive microvascular growth This is the process in which pre-existing vessels split to form daughter vessels and expand their capillary network i.e. splitting angiogenesis.
- Vascular mimicry This is the phenomenon in which aggressive cancer cells express stem celllike phenotype that transdifferentiates to form endothelial cells and new vascular networks.
4. Cancer Spread: Invasion And Metastasis
One of the most important characteristics of cancers is local invasion and distant metastasis. The mechanisms of spread involve a series of complex cell biological events called ‘Invasion metastasis cascade’.
It includes:
- Aggressive clonal proliferation in the primary tumour
- Loss of cell-cell and cell-ECM interaction, Degradation of ECM
- Loss of basal polarity
- Intravasation of cancer cells
- Tumour cells in circulation
Extravasation of tumour cells and organ predilection, and Micro-metastasis, dormancy and colonisation to develop into clinically detectable metastasis.
5. Pro-Growth Metabolic Changes: The Warburg Effect:
Cancer cells demonstrate changes in metabolic pathways in such a way that promotes initiation and progression of tumours.
These include the following:
Excessive Nutrient Acquisition:
For growing cells, glucose and glutamine are essential nutrients required for several metabolic needs such as ATP generation, cell signalling, gene expression and nitrogen donation in nucleotide biosynthesis.
- Cancer cells utilising glucose in the presence of adequate oxygen was discovered by Warburg in 1927, called the Warburg effect or aerobic glycolysis, for which he was conferred the Nobel Prize in 1931.
- However, in recent times, this observation has been used by injecting a metabolite of glucose, 18F-fluorodeoxyglucose, in PET (positron emission tomography) scan that shows a high level of GLUT (glucose transporter) protein in many cancers.
The following mechanisms may explain high glucose uptake and increased expression of GLUT by cancer cells:
- High level of HIF
- Aberrant P13K/AKT signalling
- Mutated K-RAS and BRAF
- Upregulated MYC transcription factor
- RB gene is a negative regulator of glutamine synthesis.
- Besides glucose and glutamine, tumour cells may also adopt alternate pathways to acquire nutrients for example,
- Entosis i.e. engulfment and digestion of living cells.
- Phagocytosis of apoptotic bodies.
- Macropinocytosis of extracellular proteins.
Altered Metabolic Pathways:
As a consequence of the Warburg effect, the increased uptake of nutrients proceeds into different metabolic pathways in cancer cells further promoting tumorigenesis.
These alterations in metabolic pathways are as under:
- Mitochondrial oxidative phosphorylation along with glycolysis serves the higher anabolic needs of cancer cells.
- A higher influx into the citric acid (Krebs) cycle produces more of reactive oxygen species in a controlled manner and without toxic accumulation.
- The availability of a higher amount of glutamine gets converted into glutamate in mitochondria which is utilised for fatty synthesis required for the lipid bilayer of proliferating tumour cells.
Oncometabolites in Tumorigenesis:
In addition to the growth-promoting effects of nutrients and oxygen, some intermediate metabolites have pro-growth effects in cancer:
- 2-HG (hydroxy glutamate), an oncometabolite, inhibits dioxygenases with hypermethylation silencing and thus promotes growth for example., in triple-negative breast cancer.
- Metabolites play a role in regulating epigenetic phenomena of acetylation and methylation by the addition or removal of acetyl or methyl groups respectively.
6. Dynamic Tumour Microenvironment: The Stromal Cells
- Although mutations are the driving component of tumour growth and progression, various stages of cancer need contribution from bone marrow-derived stromal cells that constitute the tumour microenvironment (TME).
- Components of TME have growth-promoting functions as well as play a role in enabling other cancer hallmarks.
There are three classes of mesenchyme-derived stromal cells that comprise TME:
- Angiogenic vascular cells for example., endothelial cells in tumour angiogenesis.
- Cancer-associated fibroblastic cells i.e. mesenchymal stem cells and connective tissue fibroblasts in close proximity of growth that can be recruited and induced to transdifferentiate and contribute to tumour-promoting functions.
- Infiltrating immune cells i.e. presence of cells of myeloid-lymphoid lineage which have an immunosuppressive role than acting as inflammatory cells in cancer.
The importance of each type of activated stromal cells varies in different tumours and depends upon other underlying oncogenic alterations in the primary tumour, invasiveness and its colonisation to develop metastasis. These components of TME are increasingly being used for developing therapeutic targets to eliminate primary and metastatic disease.
7. Evading Immune Destruction: Immune Modulation:
The existence of host immunity against cancer cells has been known for a long time, recognising them as ‘non-self or foreign’ and it attempts to eliminate them.
The following clinical observations support the presence of host immune surveillance in cancer:
- Certain cancers evoke significant lymphocytic infiltrate composed of immunocompetent cells and such tumours have some what better prognoses for example., medullary carcinoma breast (as compared with infiltrating ductal carcinoma), seminoma testis (as compared with other germ cell tumours of testis).
- Infrequently, a cancer may spontaneously regress partially or completely, probably under the influence of a host defence mechanism. For example, rare spontaneous disappearance of malignant melanoma temporarily from the primary site which may then reappear as metastasis.
- There is higher susceptibility of tumours in immunodeficient hosts for example, Primary immunodeficiency, HI-infected patients, and development of post-transplant lymphoproliferative disease.
- However, it is also known that despite an intact immune system in the host, cancer continues to develop and progress in the body.
- The subject is discussed below in the context of immune surveillance in the physiologic state and cancer under three headings: tumour antigens, antitumour immune responses, and escape from immune surveillance.
Tumour Antigens:
Tumour antigens are distinguished by their molecular features and their specific recognition by host immune cells.
Currently, various groups of tumour antigens are as follows:
- Oncoproteins from mutated oncogenes: Protein products derived from mutated oncogenes result in the expression of cell surface antigens on tumour cells. The examples include products of RAS, BCR/ABL and CDK4.
- Protein products of tumour-suppressor genes: In some tumours, protein products of mutated tumour-suppressor genes cause the expression of tumour antigens on the cell surface. The proteins p53 and β-catenin.
- Overexpressed cellular proteins: Some tumours are associated with a normal cellular protein but is excessively expressed in tumour cells and incite host immune response. For example, in melanoma, the tumour antigen is structurally normal melanocyte-specific protein, tyrosinase, which is overexpressed compared with normal cells. Similarly, HER2/neu protein is overexpressed in many cases of breast cancer.
- Abnormally expressed cellular proteins: Sometimes, a cellular protein is present in some normal cells but is abnormally expressed on the surface of tumour cells of some cancers. The classic example is the presence of the MAGE gene silent in normal adult tissues except in male germ line but MAGE genes are expressed on surface of many tumours such as melanoma (abbreviation Mage from ‘melanoma antigen’ in which it was first found), cancers of liver, lung, stomach and oesophagus. Other examples of similar aberrantly expressed gene products in cancers are GAGE (G antigen), BAGE (B melanoma antigen) and RAGE (renal tumour antigen).
- Tumour antigens from viral oncoproteins: Many oncogenic viruses express viral oncoproteins which result in expression of antigens on tumour cells for example,Viral oncoproteins of HPV (E6, E7) in cervical cancer and EBNA proteins of EBV in Burkitt’s lymphoma.
- Tumour antigens from randomly mutated genes: Various other carcinogens such as chemicals and radiation induce random mutations in the target cells. These mutated cells elaborate protein products targeted by the CTL of the immune system causing the expression of tumour antigens.
- Cell-specific differentiation antigens: Normally differentiated cells have cellular antigens which forms the basis of diagnostic immunohistochemistry. Cancers have varying degrees of loss of differentiation but particular lineage of the tumour cells can be identified by tumour antigens.
- For example, various CD markers for various subtypes of lymphomas, and prostate-specific antigen (PSA) in carcinoma of the prostate.
- Oncofoetal antigens: Oncofoetal antigens such as α-foetoprotein (AFP) and carcinoembryonic antigen (CEA) are normally expressed in embryonic life. But these antigens appear in certain cancers—AFP in liver cancer and CEA in colon cancer which can be detected in serum as cancer markers.
- Abnormal cell surface molecules: The normal cell expresses surface molecules of glycolipids, glycoproteins, mucins and blood group antigens. In some cancers, there is an abnormally changed expression of these molecules. For example, there may be changed blood group antigen or abnormal expression of mucin in ovarian cancer (CA-125) and in breast cancer(MUC-1).
Leave a Reply