The Heart Normal Structure:
Heart Anatomy And Physiology:
Table of Contents
The average weight of the heart in an adult male is 300-350 gm while that of an adult female is 250-300 gm.
The heart is divided into four chambers: a right and a left atrium both lying superiorly and a right and a left ventricle both lying inferiorly and are larger.
The atria are separated by a thin interatrial partition called the interatrial septum, while the ventricles are separated by a thick muscular partition called the interventricular septum.
The thickness of the right ventricular wall is 0.3 to 0.5cm while that of the left ventricular wall is 1.3 to 1.5 cm. The blood in the heart chambers moves
Read And Learn More: Systemic Pathology Notes
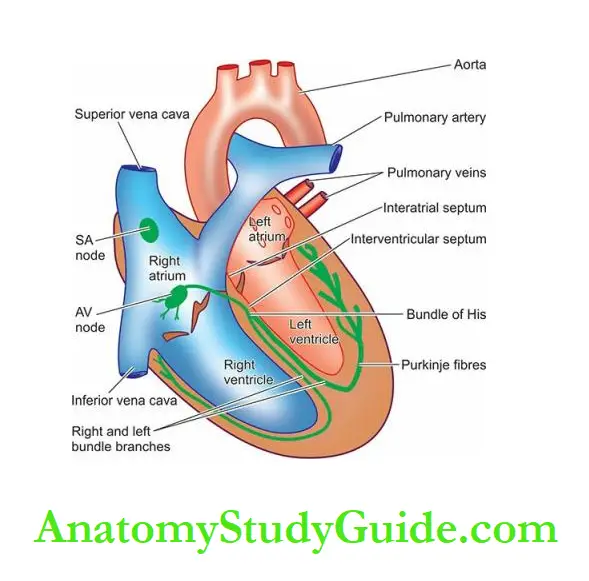
1. The sinoatrial (SA) node is located in the posterior wall of the right atrium adjacent to the point at which the superior vena cava enters the heart.
It is also called a cardiac pacemaker since it is responsible for determining the rate of contraction for all cardiac muscles.
2. The atrioventricular (AV) bundle conducts the impulse from the SA node to the AV node.
3. The atrioventricular (AV) node is located on the top of the interventricular septum and receives impulses from the SA node via the AV bundle and transmits them to the bundle of His.
4. The bundle of His extends through the interventricular septum and divides into right and left bundle branches which arborise in the respective ventricular walls.
These fibres transmit impulses from the AV node to the ventricular walls.
The pericardium consists of a closely apposed layer, the visceral pericardium or epicardium, and an outer fibrous sac, the parietal pericardium.
The two layers enclose a narrow pericardial, cavity which is lined by mesothelial cells and normally contains 10-30 ml of clear, watery serous fluid.
This fluid functions as a lubricant and shock absorbent to the heart.
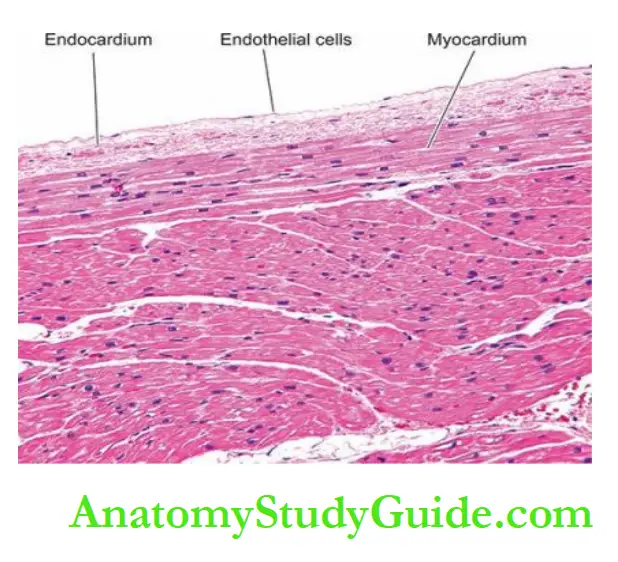
The endocardium is the smooth shiny inner lining of the myocardium that covers all the cardiac chambers, the cardiac valves, the chordae tendineae and the papillary muscles.
It is lined by endothelium with connective tissue and elastic fibres in its deeper part.
The valve cusps and semilunar leaflets are delicate and translucent structures.
The valves are strengthened by collagen and elastic tissue and covered by a layer of endothelium (valvular endocardium).
Myocardial Blood Supply:
The cardiac muscle, in order to function properly, must receive an adequate supply of oxygen and nutrients.
Blood is transported to myocardial cells by the coronary arteries which originate immediately above the aortic semilunar valve.
Most of the blood flow to the myocardium occurs during diastole.
There are three major coronary trunks, each supplying blood to specific segments of the heart:
1. The anterior descending branch of the left coronary artery, commonly called LAD (left anterior descending coronary)
supplies most of the apex of the heart, the anterior surface of the left ventricle, the adjacent third of the anterior wall of the right ventricle, and the anterior two third of the interventricular septum.
2. The circumflex branch of the left coronary artery, commonly called LCX (left circumflex coronary) supplies the left atrium and a small portion of the lateral aspect of the left ventricle.
3. The right coronary artery, abbreviated as RCA supplies the right atrium, the remainder of the anterior surface of the right ventricle, the adjacent half of the posterior wall of the left ventricle and the posterior third of the interventricular septum.
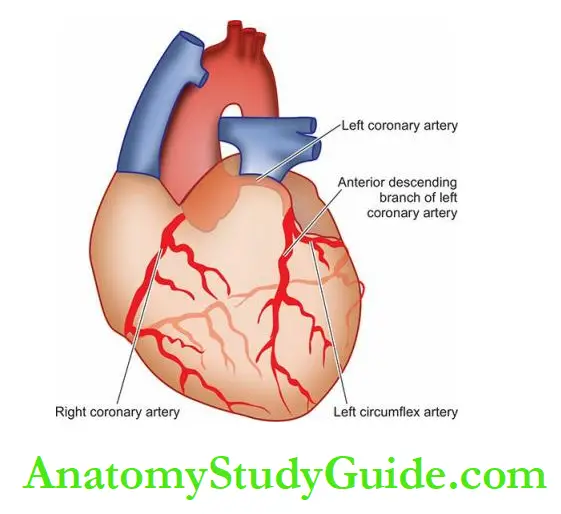
There are 3 anatomic patterns of distribution of the coronary blood supply, depending upon which coronary artery crosses the crux.
Crux is the region on the posterior surface of the heart
where all the four cardiac chambers and the interatrial and interventricular septa meet.
These patterns are as under:
Right coronary artery preponderance: is the most common pattern. In this, the right coronary artery supplies blood to the whole of the right ventricle, the posterior half of the interventricular septum and a part of the posterior wall of the left ventricle by crossing the crux.
Balanced cardiac circulation: is the next most frequent pattern. In this, the right and left ventricles receive blood supply entirely from the right and left coronary arteries respectively.
The posterior part of the interventricular septum is supplied by a branch of the right coronary while
the anterior part is supplied by a branch of the left coronary artery.
Left coronary preponderance: is the least frequent pattern. In this, the left coronary artery supplies blood to the entire left ventricle, the whole of the interventricular septum and also supplies blood to a part of the posterior wall of the right ventricle by crossing the crux.
Coronary veins run parallel to the major coronary arteries to collect blood after the cellular needs of the heart are met. Subsequently, these veins drain into the coronary sinus.
Before describing diseases of the heart, it may be mentioned here that patterns of heart diseases in developing and developed countries are distinct due to differences in living standards:
In children, valvular diseases are common all over the world.
But in developing countries including India, infectious origin, particularly rheumatic valvular disease, is the dominant cause compared to congenital valvular disease in developed countries.
In adults, cardiovascular diseases due to ischaemic heart disease and hypertensive cardiomyopathy are the major heart diseases in adults in high-income group countries compared
to low-income group countries.
Overall, cardiovascular disease accounts for about 30% of deaths worldwide which is expected to increase.
Normal Structure:
- The average weight of the heart is 300-350 gm in adult males and 250-300 gm in adult females.
- The thickness of the right ventricular wall is 0.3-0.5 cm while that of the left ventricular wall is 1.3-1.5 cm.
- The average normal circumference of the valvular openings measures about 12 cm in the tricuspid, 8.5 cm in the pulmonary, 10 cm in the mitral and 7.5 cm in the aortic valve.
- The wall of the heart consists mainly of the myocardium which is covered externally by the epicardium, and lined internally by the endocardium.
- There are three major coronary trunks, each supplying blood to specific segments of the heart: left anterior descending coronary (LAD), left circumflex coronary (LCX) and right
coronary artery (RCA). - There are 3 anatomic patterns of distribution of the coronary blood supply, depending upon which coronary artery crosses the crux: right coronary artery preponderance (the most common), balanced cardiac circulation, and left coronary preponderance.
Heart Failure
Heart failure is defined as the pathophysiologic state in which impaired cardiac function is unable to maintain adequate circulation for the metabolic needs of the tissues of the body.
It may be acute or chronic. The term congestive heart failure (CHF) is used for the chronic form of heart failure in which the patient has evidence of congestion of peripheral circulation and of the lungs.
CHF is the end result of various forms of serious heart disease.
Heart failure Eiology:
Heart failure may be caused by one of the following factors, either singly or in combination:
1. Intrinsic Pump Failure: The most common and most important cause of heart failure is the weakening of the ventricular muscle due to disease so that the heart fails to act as an efficient pump.
The various diseases which may culminate in pump failure by this mechanism are as under:
- Ischaemic heart disease
- Myocarditis
- Cardiomyopathies
- Metabolic disorders e.g. beriberi
- Disorders of the rhythm e.g. atrial fibrillation and flutter.
2. Increased Workload On The Heart:
Increased mechanical load on the heart results in increased myocardial demand resulting in myocardial failure.
Increased load on the heart may be in the form of pressure load or volume load.
Increased pressure load may occur in the following states:
- Systemic and pulmonary arterial hypertension.
- Valvular disease e.g. mitral stenosis, aortic stenosis, pulmonary stenosis.
- Chronic lung diseases.
Increased volume load occurs when a ventricle is required to eject more than the normal volume of the blood resulting in cardiac failure.
This is seen in the following conditions:
- Valvular insufficiency
- Severe anaemia
- Thyrotoxicosis
- Arteriovenous shunts
- Hypoxia due to lung diseases.
3. Impaired Filling Of Cardiac Chambers Decreased cardiac output and cardiac failure may result from extracardiac causes or defects in the filling of the heart:
Cardiac tamponade e.g. haemopericardium, hydropericardium
Constrictive pericarditis.
Types Of Heart Failure:
Heart failure may be acute or chronic, right-sided or left-sided, and forward or backward failure.
Acute And Chronic Heart Failure:
Depending upon whether the heart failure develops rapidly or slowly, it may be acute or chronic.
Acute heart failure Sudden and rapid development of heart failure occurs in the following conditions:
- Larger myocardial infarction
- Valve rupture
- Cardiac tamponade
- Massive pulmonary embolism
- Acute viral myocarditis
- Acute bacterial toxaemia.
In acute heart failure, there is a sudden reduction in cardiac output resulting in systemic hypotension but oedema does not occur.
Instead, a state of cardiogenic shock and cerebral hypoxia develops.
Chronic heart failure More often, heart failure develops slowly as observed in the following states:
- Myocardial ischaemia from atherosclerotic coronary artery disease.
- Multivalvular heart disease.
- Systemic arterial hypertension
- Chronic lung diseases resulting in hypoxia and pulmonary arterial hypertension
- Progression of acute into chronic failure.
In chronic heart failure, compensatory mechanisms like tachycardia, cardiac dilatation and cardiac hypertrophy try to make adjustments so as to maintain adequate cardiac output.
This often results in well-maintained arterial pressure and there is an accumulation of oedema.
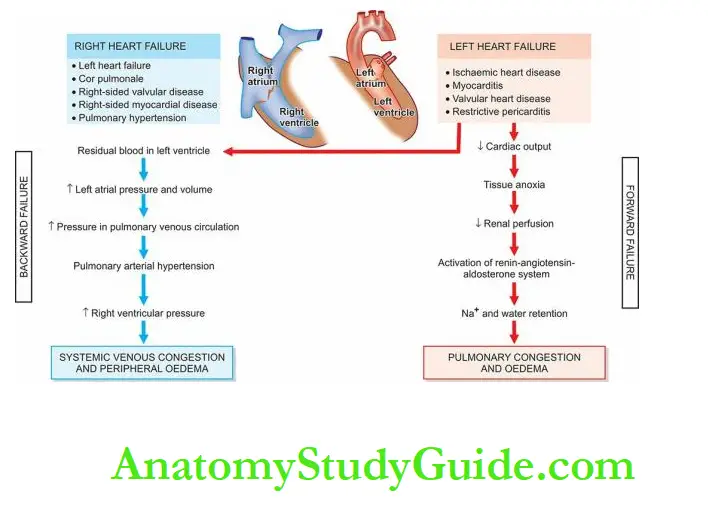
Left-Sided And Right-Sided Heart Failure:
Though the heart as an organ eventually fails as a whole, functionally, the left and right heart act as independent units.
From a clinical point of view, therefore, it is helpful to consider the failure of the left and right heart separately.
The clinical manifestations of heart failure result from the accumulation of excess fluid upstream to the left or right cardiac chamber whichever is initially affected
Left Heart Failure:
It is initiated by stress to the left heart. The major causes are as follows:
- Systemic hypertension is, the most common.
- Mitral or aortic valve disease (stenosis)
- Ischaemic heart disease
- Myocardial diseases e.g. cardiomyopathies, myocarditis.
- Restrictive pericarditis.
The clinical manifestations of left-sided heart failure result from decreased left ventricular output and hence there is an accumulation of fluid upstream in the lungs.
Accordingly, the major pathologic changes are as under:
Pulmonary congestion and oedema cause dyspnoea and orthopnoea.
Decreased left ventricular output causing hypoperfusion and diminished oxygenation of tissues e.g. In kidneys causing ischaemic acute tubular necrosis, in the brain causing hypoxic encephalopathy, and in skeletal muscles causing muscle weakness and fatigue.
Hypertensive Heart Disease:
Hypertensive heart disease or hypertensive cardiomyopathy results from systemic hypertension of prolonged duration and manifests by left ventricular hypertrophy.
Even mild hypertension (blood pressure higher than 140/90 mmHg) of sufficient duration may induce hypertensive heart disease.
It is the second most common form of heart disease after IHD.
As already pointed out, hypertension predisposes to atherosclerosis.
Therefore, most patients with hypertensive heart disease have also advanced coronary atherosclerosis and may develop progressive IHD.
Amongst the causes of death in hypertensive patients, cardiac decompensation leading to CHF accounts for about one-third of the patients; other causes of death are IHD, cerebrovascular stroke, renal failure following arteriolar nephrosclerosis, dissecting aneurysm of the aorta and
sudden cardiac death.
Pathogenesis:
The pathogenesis of systemic hypertension is discussed later.
Pathogenesis of left ventricular hypertrophy (LVH) which is most commonly caused by systemic hypertension is described here.
Stimulus to LVH is pressure overload in systemic hypertension.
Both genetic and haemodynamic factors contribute to LVH. The stress of pressure on the ventricular wall causes increased production of myofilaments, myofibrils, and other cell organelles and nuclear enlargement.
Since the adult myocardial fibres do not divide, the fibres are hypertrophied.
However, the sarcomeres may divide to increase the cell width.
LVH can be diagnosed by ECG. Aggressive control of hypertension can regress the left ventricular mass.
Abnormalities of diastolic function in hypertension are more common in hypertension and is present in about one-third of patients with normal systolic function.
Right-Sided Heart Failure:
Right-sided heart failure occurs more often as a consequence of left-sided heart failure.
However, some conditions affect the right ventricle primarily, producing right-sided heart failure.
These are as follows:
- As a consequence of left ventricular failure.
- Cor pulmonale or pulmonary heart disease in which right heart failure occurs due to intrinsic lung diseases, most common.
- Pulmonary or tricuspid valvular disease.
- Pulmonary hypertension is secondary to pulmonary thromboembolism.
- Myocardial disease affects the right heart.
- Congenital heart disease with left-to-right shunt.
Whatever the underlying cause, the clinical manifestations of right-sided heart failure are
upstream of the right heart such as systemic (due to caval blood) and portal venous congestion, and reduced cardiac output.
Accordingly, the pathologic changes are as under:
- Systemic venous congestion in different tissues and organs e.g. subcutaneous oedema on dependent parts, passive congestion of the liver, spleen, and kidneys, ascites, hydrothorax, congestion of leg veins and neck veins.
- Reduced cardiac output results in circulatory stagnation causing anoxia, cyanosis and coldness of extremities.
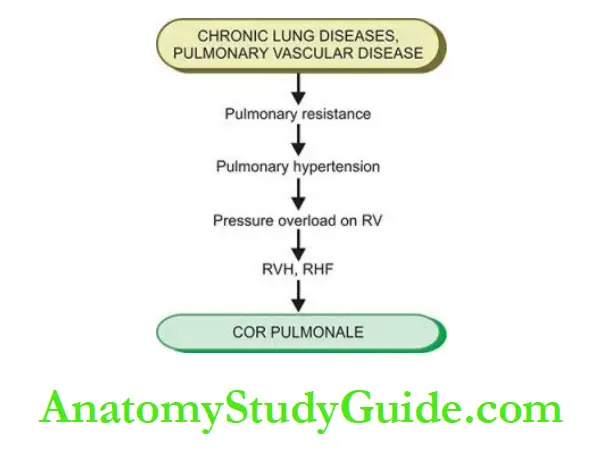
Cor pulmonale: Cor pulmonale (cor = heart; pulmonale= lung) or pulmonary heart disease is the right heart disease resulting from disorders of the lungs.
It is characterised by right ventricular dilatation or hypertrophy, or both.
Thus, cor pulmonale is the right-sided counterpart of hypertensive heart disease that affects the left heart predominantly.
Depending upon the rapidity of development, cor pulmonale may be acute or chronic:
- Acute cor pulmonale occurs following a massive pulmonary embolism resulting in sudden dilatation of the pulmonary trunk, conus and right ventricle.
- Chronic cor pulmonale is more common and is often preceded by chronic pulmonary hypertension.
Following chronic lung diseases can cause chronic pulmonary hypertension and subsequent cor pulmonale:
- Chronic emphysema
- Chronic bronchitis
- Pulmonary tuberculosis
- Pneumoconiosis
- Cystic fibrosis
- Hyperventilation in marked obesity (Pickwickian syndrome)
- Multiple organised pulmonary emboli.
Pathogenesis Chronic lung diseases as well as diseases of the pulmonary vessels cause increased pulmonary vascular resistance.
The most common underlying mechanism causing increased pulmonary blood pressure (pulmonary hypertension) is pulmonary vasoconstriction, activation of the coagulation pathway and obliteration of pulmonary arterial vessels.
Pulmonary hypertension causes pressure overload on the right ventricle and hence right ventricular enlargement.
Initially, there is right ventricular hypertrophy, but as cardiac decompensation sets in and right heart failure ensues, dilatation of the right ventricle occurs.
The sequence of events involved in the pathogenesis of cor pulmonale is summarised.
In summary, in the early stage, left heart failure manifests with features of pulmonary congestion and decreased left ventricular output, while right heart failure presents with systemic venous congestion and involvement of the liver and spleen.
CHF, however, combines the features of both left and right heart failure.
Backwards And Forward Heart Failure:
The mechanism of clinical manifestations resulting from heart failure can be explained on the
basis of a mutually interdependent backward and forward failure.
Backward heart failure: According to this concept, either of the ventricles fails to eject blood normally,
resulting in the rise of end-diastolic volume in the ventricle and an increase in volume and pressure in the atrium which is transmitted backwards producing elevated pressure in the veins.
Forward heart failure According to this hypothesis, clinical manifestations result directly from the failure of the heart to pump blood causing diminished flow of blood to the tissues, especially diminished renal perfusion and activation of the renin-angiotensin-aldosterone system.
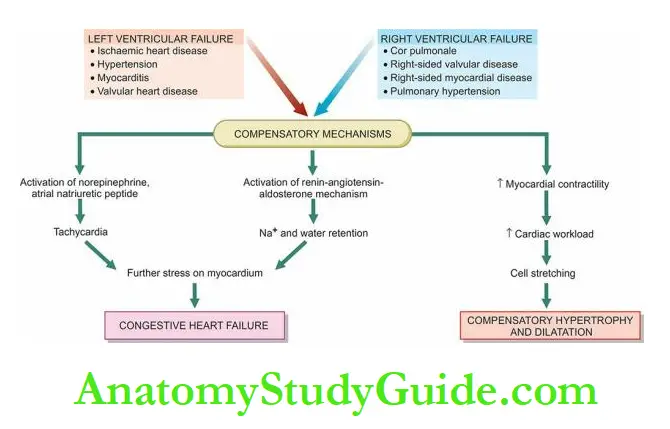
Compensatory Mechanisms: Cardiac Hypertrophy And Dilatation:
In order to maintain normal cardiac output, several compensatory mechanisms play a role:
- Compensatory enlargement in the form of cardiac hypertrophy, cardiac dilatation, or both.
- Tachycardia (i.e. increased heart rate) due to activation of the neurohumoral system e.g. release of norepinephrine and atrial natriuretic peptide, activation of renin-angiotensin-aldosterone mechanism.
According to Starling’s law on the pathophysiology of the heart, the failing dilated heart, in order to maintain cardiac performance, increases the myocardial contractility and thereby attempts to maintain stroke volume.
This is achieved by increasing the length of sarcomeres in the dilated heart.
Ultimately, however, dilatation decreases the force of contraction and leads to residual volume in the cardiac chambers causing volume overload resulting in cardiac failure that ends in death.
Cardiac Hypertrophy:
Hypertrophy of the heart is defined as an increase in the size and weight of the myocardium.
It generally results from increased pressure load while increased volume load (e.g. valvular incompetence) results in hypertrophy with dilatation of the affected chamber due to regurgitation of the blood through an incompetent valve.
The atria may also undergo compensatory changes due to increased workload.
The basic factors that stimulate the hypertrophy of the myocardial fibres are not known.
It appears that stretching of myocardial fibres in response to stress induces the cells to increase in length.
The elongated fibres receive better nutrition and thus increase in size.
Other factors which may stimulate an increase in the size of myocardial fibres are anoxia (e.g. in coronary atherosclerosis) and the influence of certain hormones (e.g. catecholamines, pituitary growth hormone).
Cardiac Hypertrophy Causes:
Hypertrophy with or without dilatation may involve predominantly the left or the right heart, or both sides.
Left ventricular hypertrophy Common causes are as under:
- Systemic hypertension, the most common
- Aortic stenosis and insufficiency
- Mitral insufficiency
- Coarctation of the aorta
- Occlusive coronary artery disease
- Congenital anomalies like septal defects and patent ductus arteriosus
- Conditions with increased cardiac output e.g. thyrotoxicosis, anaemia, arteriovenous fistulae.
- Right, ventricular hypertrophy Most of the causes of right ventricular hypertrophy are due to pulmonary arterial hypertension.
These are as follows:
Cor pulmonale from chronic lung diseases (e.g. chronic emphysema, bronchiectasis, pneumoconiosis, pulmonary vascular disease etc), the most common
- Pulmonary stenosis and insufficiency
- Tricuspid insufficiency
- Mitral stenosis and/or insufficiency
- Left ventricular hypertrophy and failure of the left ventricle.
- Cardiac Dilatation
- Quite often, hypertrophy of the heart is accompanied by cardiac dilatation.
Stress leading to the accumulation of excessive volume of blood in a chamber of the heart causes an increase in the length of myocardial fibres and hence cardiac dilatation as a compensatory mechanism.
Cardiac Hypertrophy Causes:
Accumulation of excessive volume of blood within the cardiac chambers from the following causes may result in dilatation of the respective ventricles or both:
Valvular insufficiency (mitral and/or aortic insufficiency in left ventricular dilatation, tricuspid and/or pulmonary insufficiency in right ventricular dilatation)
- Left-to-right shunts e.g. in VSD
- Conditions with high cardiac output e.g. thyrotoxicosis, arteriovenous shunt
- Myocardial diseases e.g. cardiomyopathies, myocarditis
- Systemic hypertension.
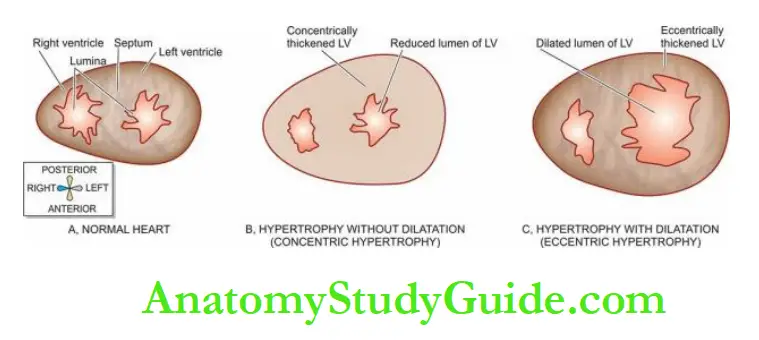
Morphologic Features:
Hypertrophy of the myocardium without dilatation is referred to as concentric, and when associated with dilatation is called eccentric.
Grossly, the most significant finding is marked hypertrophy of the heart, chiefly of the left ventricle.
The weight of the heart increases to 500 gm or more (normal weight is about 300 gm).
In LVH, the thickness of the left ventricular wall increases from its normal 13 to 15 mm up to 20 mm or more.
The papillary muscles and trabeculae carnage are rounded and prominent.
Initially, there is concentric hypertrophy of the left ventricle (without dilatation).
But when decompensation and cardiac failure supervene, there is eccentric hypertrophy (with dilatation) with thinning of the ventricular wall and there may be dilatation and hypertrophy of the right heart as well i.e. biventricular hypertrophy.
In RVH due to acute cor pulmonale, there is characteristic ovoid dilatation of the right ventricle, and sometimes of the right atrium.
In chronic cor pulmonale, there is an increase in thickness of the right ventricular wall from its normal 3 to 5 mm up to 10 mm or more i.e. right ventricular hypertrophy (RVH).
Often, there is dilatation of the right ventricle too i.e. biventricular hypertrophy.
Microscopically, there is an increase in the size of individual muscle fibres.
There may be multiple minute foci of degenerative changes and necrosis in the hypertrophied myocardium.
These changes appear to arise as a result of relative hypoxia of the hypertrophied muscle as the blood supply is inadequate to meet the demands of the increased fibre size.
Ventricular hypertrophy renders the inner part of the myocardium more liable to ischaemia.
Electron microscopy reveals an increase in the number of myofilaments comprising myofibrils, mitochondrial changes and multiple intercalated discs which are active sites for the formation of new sarcomeres.
Besides, the nucleic acid content determinations have shown an increase in total RNA and an increased ratio of RNA to DNA content of the hypertrophied myocardial fibres.
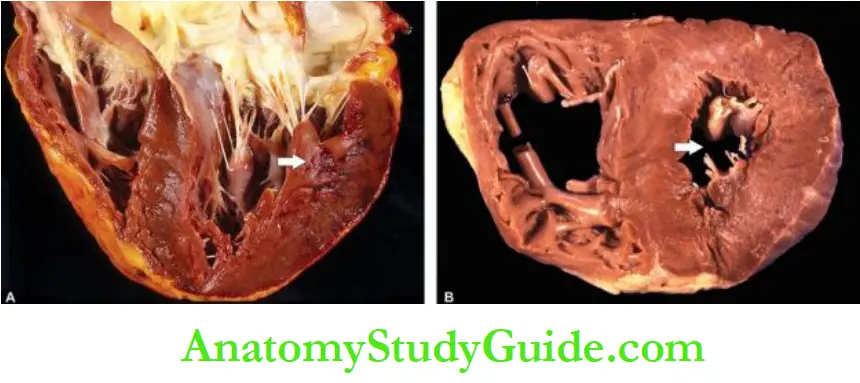
Heart Failure:
Heart failure is a pathophysiologic state of impaired cardiac function when it is unable to maintain the metabolic needs of the tissues of the body.
Heart failure may be caused by intrinsic pump failure, increased pressure or volume overload, or impaired filling.
Heart failure may be an acute or chronic, left-sided or right-sided, or backward or forward failure.
Hypertensive heart disease resulting from systemic hypertension of prolonged duration is the most common cause of left heart failure and left ventricular hypertrophy (LVH).
In LVH, initially, there is concentric hypertrophy of the left ventricle (without dilatation).
But when decompensation and cardiac failure supervene, there is eccentric hypertrophy (with dilatation).
Cor pulmonale or pulmonary heart disease is the most common cause of right heart failure and right ventricular hypertrophy (RVH) resulting from disorders of the lungs.
Right heart failure may be acute or chronic; the latter is more common.
There is thickened right ventricular wall, often with dilatation.
In both LHF and RHF, compensatory mechanisms are its enlargement in the form of cardiac hypertrophy (concentric or eccentric), cardiac dilatation, or both; eventually, there is biventricular enlargement.
Congenital Heart Disease
Congenital heart disease is the abnormality of the heart present from birth.
It is the most common and important form of heart disease in the early years of life and is present in about 0.5% of newborn children.
The incidence is higher in premature infants. The cause of congenital heart disease is unknown in the majority of cases.
It is attributed to multifactorial inheritance involving genetic and environmental influences.
Other factors like rubella infection in the mother during pregnancy, drugs taken by the mother and heavy alcohol drinking by the mother, have all been implicated in causing in-utero foetal injury resulting in congenital malformations of the heart.
Congenital heart disease Classification:
Congenital anomalies of the heart may be either shunts (left-to-right or right-to-left), or defects causing obstructions to flow.
However, complex anomalies involving combinations of shunts and obstructions are also often present.
A simple classification of important and common examples of these groups.
1. Malpositions Of The Heart:
Dextrocardia is the condition when the apex of the heart points to the right side of the chest.
It may be accompanied by situs inversus so that all other organs of the body are also transposed in a similar way and thus the heart is in a normal position in relation to them.
However, isolated dextrocardia is associated with major anomalies of the heart such as transposition of the atria in relation to ventricles or transposition of the great arteries.
2. Shunts (Cyanotic Congenital Heart Disease):
A shunt may be left-to-right side or right-to-left side of the circulation.
1. Left-To-Right Shunts (Acyanotic Or Late Cyanotic Group):
In conditions where there is shunting of blood from the left-to-right side of the heart, there is volume overload on the right heart producing pulmonary hypertension and right ventricular hypertrophy.
At a later stage, the pressure on the right side is higher than on the left side creating late cyanotic heart disease.
The important conditions included in this category are described here:
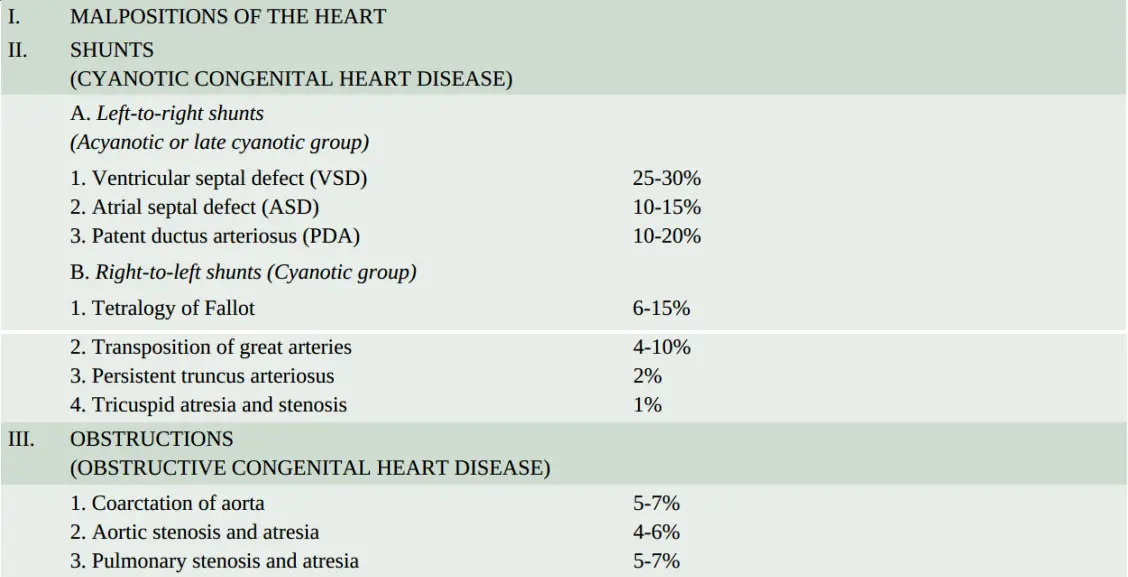
Ventricular Septal Defect:
Ventricular septal defect (VSD) is the most common congenital anomaly of the heart and comprises about 30% of all congenital heart diseases.
The condition is recognised early in life. The smaller defects often close spontaneously, while larger
defects remain patent and produce significant effects.
Depending upon the location of the defect, VSD may be of the following types:
1. In 90% of cases, the defect involves the membranous septum and is very close to the bundle of His.
2. The remaining 10% of cases have VSD immediately below the pulmonary valve (subpulmonic), below the aortic valve (subaortic), or exist in the form of multiple defects in the muscular septum.
Morphologic Features
The effects of VSD are produced due to left-to-right shunt at the ventricular level, increased pulmonary flow and increased volume in the left side of the heart.
These effects are as under:
- Volume hypertrophy of the right ventricle.
- Enlargement and haemodynamic changes in the tricuspid and pulmonary valves.
- Endocardial hypertrophy of the right ventricle.
- Pressure hypertrophy of the right atrium.
- Volume hypertrophy of the left atrium and left ventricle.
- Enlargement and haemodynamic changes in the mitral and aortic valves.
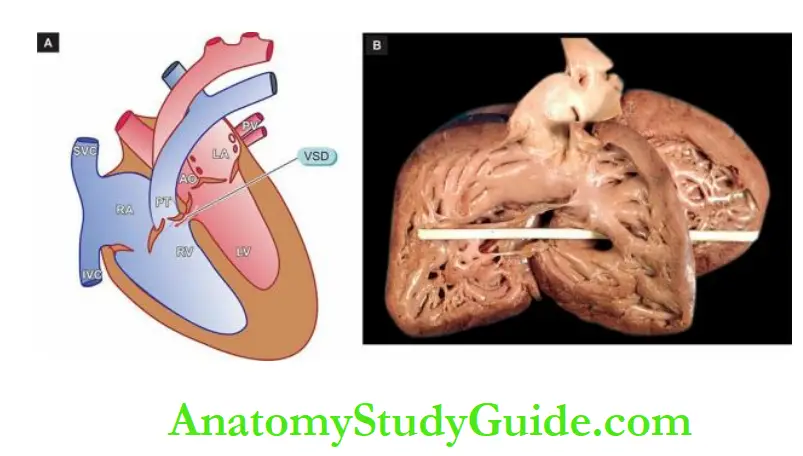
Atrial Septal Defect:
Isolated atrial septal defect (ASD) comprises about 10% of congenital heart diseases.
The condition remains unnoticed in infancy and childhood till pulmonary hypertension is induced causing late cyanotic heart disease and right-sided heart failure.
Depending upon the location of the defect, there are 3 types of ASD:
Fossa ovalis type or ostium secundum type: is the most common form comprising about 90% of cases of ASD.
The defect is situated in the region of the fossa ovalis.
Ostium primum type: comprises about 5% of cases of ASD. The defect lies low in the interatrial septum adjacent to atrioventricular valves.
There may be a cleft in the aortic leaflet of the mitral valve producing mitral insufficiency.
Sinus venous type: accounts for about 5% of cases of ASD. The defect is located high in the interatrial septum near the entry of the superior vena cava.
Morphologic Features:
The effects of ASD are produced due to left-to-right shunt at the atrial level with the increased pulmonary flow.
These effects are as follows:
- Volume hypertrophy of the right atrium and right ventricle.
- Enlargement and haemodynamic changes of tricuspid and pulmonary valves.
- Focal or diffuse endocardial hypertrophy of the right atrium and right ventricle.
- Volume atrophy of the left atrium and left ventricle.
- Small-sized mitral and aortic orifices.
Patent Ductus Arteriosus (Pda):
The ductus arteriosus is a normal vascular connection between the aorta and the bifurcation of the pulmonary artery.
Normally, the ductus closes functionally within the first or second day of life.
Its persistence after 3 months of age is considered abnormal.
The cause for the patency of ductus arteriosus is not known but possibly it is due to continued synthesis of PGE2 after birth which keeps it patent as evidenced by the association of PDA with respiratory distress syndrome in infants and pharmacologic closure of PDA with the administration of indomethacin to suppress PGE2 synthesis.
PDA constitutes about 10% of congenital malformations of the heart and great vessels.
In about 90% of cases, it occurs as an isolated defect, while in the remaining cases, it may be associated with other anomalies like VSD, coarctation of the aorta and pulmonary or aortic stenosis.
A patent ductus may be up to 2 cm in length and up to 1 cm in diameter.
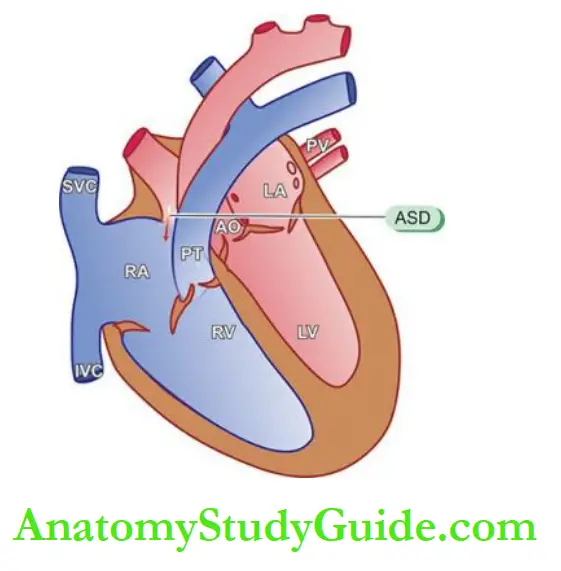
Morphologic Features:
The effects of PDA on the heart occur due to left-to-right shunt at the level of ductus resulting in increased pulmonary flow and increased volume in the left heart.
These effects are as follows:
- Volume hypertrophy of the left atrium and left ventricle.
- Enlargement and haemodynamic changes of the mitral and pulmonary valves.
- Enlargement of the ascending aorta.
2. Right-To-Left Shunts (Cyanotic Group):
In conditions where there is shunting of blood from the right side to the left side of the heart, there is entry of poorly-oxygenated blood into systemic circulation resulting in early cyanosis.
The examples described below are not pure shunts but are combinations of shunts with obstructions but are described here since there is functional shunting of blood from one to the other side of the circulation.
Tetralogy Of Fallot:
Tetralogy of Fallot is the most common cyanotic congenital heart disease, found in about 10% of children with anomalies of the heart.
Morphologic Features:
The four features of tetralogy are as under:
- Ventricular septal defect (VSD) (‘shunt’).
- Displacement of the aorta to the right so that it overrides the VSD.
- Pulmonary stenosis (‘obstruction’).
- Right ventricular hypertrophy. The severity of the clinical manifestations is related to two factors: the extent of pulmonary stenosis and the size of VSD.
Accordingly, there are two forms of tetralogy: cyanotic and cyanotic:
Easy way to remember from acronym PROVe = Pulmonary stenosis, Right ventricular hypertrophy, Overriding of the aorta, Ventricular septal defect.
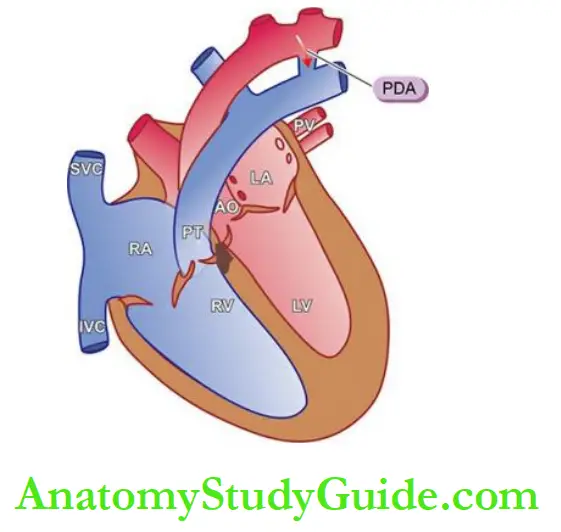
Cyanotic tetralogy Pulmonary stenosis is greater and the VSD is mild so that there is more resistance to the outflow of blood from the right ventricle resulting in right-to-left shunt at the ventricular level and cyanosis.
The effects on the heart are as follows:
- pressure hypertrophy of the right atrium and right ventricle.
- Smaller and more abnormal tricuspid valve.
- The smaller left atrium and left ventricle.
- Enlarged aortic orifice.
Acyanotic tetralogy The VSD is larger and pulmonary stenosis is mild so there is mainly a left-to-right shunt with increased pulmonary flow and increased volume in the left heart but no cyanosis.
The effects on the heart are as under:
- Pressure hypertrophy of the right ventricle and right atrium.
- Volume hypertrophy of the left atrium and left ventricle.
- Enlargement of mitral and aortic orifices.
Transposition Of Great Arteries:
The term transposition is used for complex malformations as regards the position of the aorta, pulmonary trunk, atrioventricular orifices and the position of atria in relation to ventricles.
Morphologic Features:
There are several forms of transpositions. The common ones are described below:
Regular transposition: is the most common type. In this, the aorta which is normally situated to the right and posterior with respect to the pulmonary trunk, is instead displaced anteriorly and to the right.
In regular complete transposition, the aorta emerges from the right ventricle and the pulmonary trunk from the left ventricle so that there is cyanosis from birth.
Corrected transposition: this is an uncommon anomaly. There is the complete transposition of the great arteries with the aorta arising from the right ventricle and the pulmonary trunk from the left ventricle, as well as transposition of the great veins so that the pulmonary veins enter the right atrium and the systemic veins drain into the left atrium.
This results in a physiologically corrected circulation.
Persistent Truncus Arteriosus
Persistent truncus arteriosus (PTA) is a rare anomaly.
Morphologic Features
In PTA, the arch that normally separates the aorta from the pulmonary artery fails to develop.
This results in a single large common vessel receiving blood from the right as well as the left ventricle. The orifice may have 3 to 6 cusps. There is often an associated VSD.
There is left-to-right shunt and frequently early systemic cyanosis. The prognosis is generally poor.
Tricuspid Atresia And Stenosis:
Tricuspid atresia and stenosis are rare anomalies. There is often associated pulmonary stenosis or pulmonary atresia.
Morphologic Features:
In tricuspid atresia, there is the absence of a tricuspid orifice and instead, there is a dimple in the floor of the right atrium.
In tricuspid stenosis, the tricuspid ring is small and the valve cusps are malformed.
In both conditions, there is often an interatrial defect through which the right-to-left shunt of blood takes place. Children are cyanotic since birth and live for a few weeks or months.
3. Obstructions (Obstructive Congenital Heart Disease):
Congenital obstruction to blood flow may result from an obstruction in the aorta due to narrowing (coarctation of the aorta), obstruction to outflow from the left ventricle (aortic stenosis and atresia), and obstruction to outflow from the right ventricle (pulmonary stenosis and atresia).
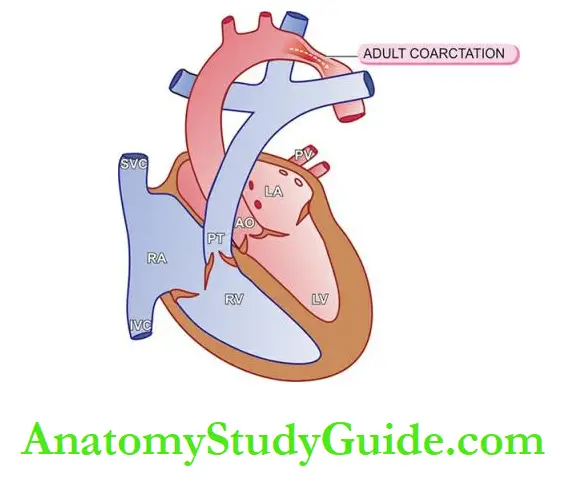
Coarctation Of Aorta:
The word ‘coarctation’ means contracted or compressed.
Coarctation of the aorta is localised narrowing in any part of the aorta, but the constriction is more often just distal to ductus arteriosus (postductal or adult), or occasionally proximal to the ductus arteriosus (preductal or infantile type) in the region of the transverse aorta
Morphologic Features:
The two common forms of coarctation of the aorta are as under:
Postductal or adult type: The obstruction is just distal to the point of entry of ductus arteriosus which is often closed.
In the stenotic segment, the aorta is drawn in as if a suture has been tied around it. The aorta is dilated on either side of the constriction.
The condition is recognised in adulthood, characterised by hypertension in the upper extremities, weak pulses and low blood pressure in the lower extremities and effects of arterial insufficiency such as claudication and coldness.
In time, there is the development of collateral circulation between pre-stenotic and post-stenotic arterial branches so that intercostal arteries are enlarged and palpable and may produce erosions on the inner surface of the ribs.
Preductal or infantile type: The manifestations are produced early in life.
The narrowing is proximal to the ductus arteriosus which usually remains patent.
The narrowing is generally gradual and involves a larger segment of the proximal aorta. There is often associated interatrial septal defect.
Preductal coarctation results in right ventricular hypertrophy while the left ventricle is small.
Cyanosis develops in the lower half of the body while the upper half remains unaffected since it is supplied by vessels originating proximal to the coarctation.
Children with this defect have a poor prognosis.
Aortic Stenosis And Atresia:
The most common congenital anomaly of the aorta is a bicuspid aortic valve which does not have much functional significance but predisposes it to calcification.
Congenital aortic atresia is rare and incompatible with survival.
Aortic stenosis may be acquired (e.g. in rheumatic heart disease, calcific aortic stenosis) or congenital.
Morphologic Features:
Congenital aortic stenosis may be of three types: valvular, subvalvular and supravalvular.
Valvular stenosis The aortic valve cusps are malformed and irregularly thickened.
The aortic valve may have one, two or three such maldeveloped cusps.
Subvalvular stenosis There is a thick fibrous ring under the aortic valve causing subaortic stenosis.
Supravalvular stenosis The most uncommon type, there is fibrous constriction above the sinuses of Valsalva.
In all these cases, there is pressure hypertrophy of the left ventricle and left atrium and dilatation of the aortic root.
Pulmonary Stenosis And Atresia:
Isolated pulmonary stenosis and atresia do not cause cyanosis and hence are included under cyanotic heart diseases.
Morphologic Features:
The changes in these conditions are as under:
Pulmonary stenosis is the commonest form of obstructive congenital heart disease comprising about 7% of all congenital heart diseases. It may occur as a component of the tetralogy of Fallot or as an isolated defect.
Pulmonary stenosis is caused by the fusion of cusps of the pulmonary valve forming a diaphragm-like obstruction to the outflow of blood from the right ventricle and dilatation of the pulmonary trunk.
Pulmonary atresia There is no communication between the right ventricle and the lungs so the blood bypasses the right ventricle through an interatrial septal defect.
It then enters the lungs via patent ductus arteriosus.
Congenital Heart Disease
- Congenital heart diseases are anomalies of the heart present since birth and are seen in ~0.5% of newborn babies.
- These anomalies may be either shunts (left-to-right or right-to-left) or defects causing obstructions to flow.
- Left-to-right shunts are cyanotic groups of heart diseases; e.g. ventricular and atrial septal defects, and patent ductus arteriosus.
- Right-to-left shunts are a cyanotic group of heart disease. Examples are tetralogy of Fallot, transposition of great arteries, persistent truncus arteriosus and tricuspid atresia and stenosis.
- Obstructive congenital heart diseases are coarctation of the aorta and stenosis and atresia of the aorta or pulmonary artery
Ischaemic Heart Disease
Ischaemic heart disease (IHD) is defined as an acute or chronic form of cardiac disability arising from an imbalance between the myocardial supply and demand for oxygenated blood.
Since narrowing or obstruction of the coronary arterial system is the most common cause of myocardial anoxia, the alternate term ‘coronary heart disease (CHD)’ is used synonymously with IHD.
IHD or CHD is the leading cause of death in high-income countries (~40%) and somewhat lower incidence is observed in low- and middle-income countries (~28%).
Overall, IHD is already the leading cause of death worldwide accounting for 30% of mortality.
Higher incidence in the industrialised world is attributed to declining physical activity while total caloric intake has
been increasing, particularly from animal fat; these account for major risk factors– lipid abnormalities, obesity, type 2 diabetes mellitus, and hypertension.
Men develop IHD earlier than women and death rates are also slightly higher for men than for women until menopause.
Etiopathogenesis:
Ischaemia heart disease (IHD) is invariably caused by disease affecting the coronary arteries, the most prevalent being atherosclerosis accounting for more than 90% of cases, while other causes are responsible for less than 10% of cases of IHD.
Therefore, it is convenient to consider the aetiology of IHD under three broad headings:
- coronary atherosclerosis;
- superadded changes in coronary atherosclerosis; and
- non-atherosclerotic causes.
1. Coronary Atherosclerosis:
Coronary atherosclerosis resulting in ‘fixed’ obstruction is the major cause of IHD in more than 90% of cases.
The general aspects of atherosclerosis as regards its etiology, pathogenesis and the morphologic features of atherosclerotic lesions have already been dealt with at length in the preceding.
Here, a brief account of the specific features in the pathology of lesions in atherosclerotic coronary artery disease, in particular, is presented.
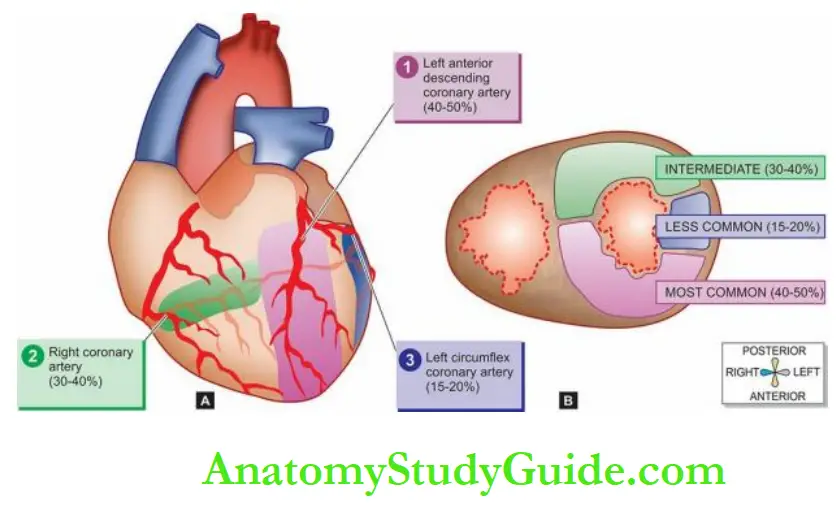
1. Distribution: Atherosclerotic lesions in coronary arteries are distributed in one or more of the three major coronary arterial trunks, the highest incidence being in the anterior descending branch of the left coronary (LAD), followed in decreasing frequency, by the right coronary artery (RCA) and still less in a circumflex branch of the left coronary (CXA).
About one-third of cases have the single-vessel disease, most often left anterior descending arterial involvement; another one-third have two-vessel disease, and the remainder has three major vessel diseases.
2. Location: Almost all adults show atherosclerotic plaques scattered throughout the coronary arterial system.
However, significant stenotic lesions that may produce chronic myocardial ischaemia show more than 75% (three-fourths) reduction in the cross-sectional area of a coronary artery or its branch.
The area of severest involvement is about 3 to 4 cm from the coronary ostia, more often at or near the bifurcation of the arteries, suggesting the role of haemodynamic forces in atherogenesis.
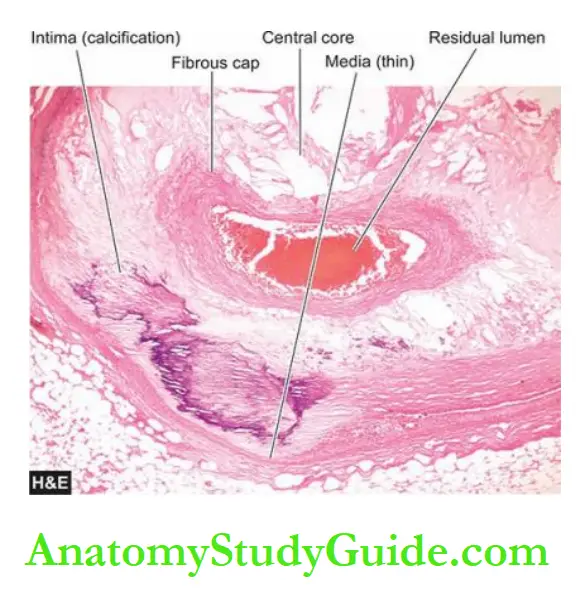
3. Fixed atherosclerotic plaques: atherosclerotic plaques in the coronaries are more often eccentrically located bulging into the lumen from one side.
Occasionally, there may be concentric thickening of the wall of the artery. Atherosclerosis produces gradual luminal narrowing that may eventually lead to ‘fixed’ coronary obstruction.
The general features of atheromas of coronary arteries are similar to those affecting elsewhere in the body and may develop similar complications like calcification, coronary thrombosis, ulceration, haemorrhage, rupture and aneurysm formation.
2. Superadded Changes In Coronary Atherosclerosis
The attacks of acute coronary syndromes, which include acute myocardial infarction, unstable angina and sudden ischaemic death, are precipitated by certain changes superimposed on a preexisting fixed coronary atheromatous plaque.
These changes are as under:
1. Acute changes in chronic atheromatous plaque: Though chronic fixed obstructions are the most frequent cause of IHD, acute coronary episodes are often precipitated by sudden changes in chronic plaques such as plaque haemorrhage, fissuring, or ulceration that result in thrombosis and embolisation of atheromatous debris.
Acute plaque changes are brought about by factors such as sudden coronary artery spasms, tachycardia, intraplaque haemorrhage and hypercholesterolaemia.
2. Coronary artery thrombosis: Transmural acute myocardial infarction is often precipitated by partial or complete coronary thrombosis.
The initiation of a thrombus occurs due to surface ulceration of fixed chronic atheromatous plaque, ultimately causing complete luminal occlusion. The lipid core of plaque, in particular, is highly thrombogenic.
Small fragments of thrombotic material are then dislodged which are embolised to terminal coronary branches and cause microinfarcts of the myocardium.
3. Local platelet aggregation and coronary artery spasm: Some cases of acute coronary episodes are caused by local aggregates of platelets on the atheromatous plaque, short of forming a thrombus.
The aggregated platelets release vasospastic mediators such as thromboxane A2 which may probably be responsible for coronary vasospasm in the already atherosclerotic vessel.
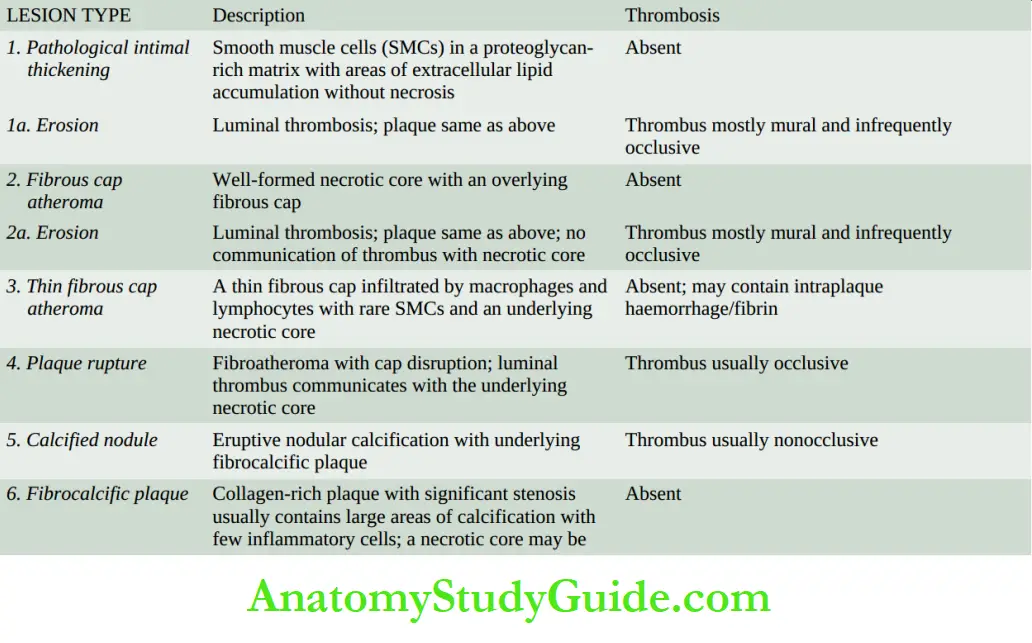
A simple modified American Heart Association (AHA) classification scheme (2000) for atherosclerosis based on morphologic features has been proposed that takes into consideration progressive stages of the disease by events that include erosion, rupture, thinning of a fibrous cap
and development of thrombosis.
3. Non-atherosclerotic Causes:
Several other coronary lesions may cause IHD in less than 10% of cases. These are as under:
1. Vasospasm: It has been possible to document vasospasm of one of the major coronary arterial trunks in patients with no significant atherosclerotic coronary narrowing which may cause angina or myocardial infarction.
2. Stenosis of coronary ostia: Coronary ostial narrowing may result from the extension of syphilitic aortitis or from aortic atherosclerotic plaques encroaching on the opening.
3. Arteritis: Various types of inflammatory involvements of coronary arteries or small branches like in rheumatic arteritis, polyarteritis nodosa, thromboangiitis obliterans (Buerger’s disease), Takayasu disease, Kawasaki disease, tuberculosis and other bacterial infections may contribute to myocardial damage.
4. Embolism: Rarely, emboli originating from elsewhere in the body may occlude the left coronary artery and its branches and produce IHD.
The emboli may originate from bland thrombi, or from vegetations of bacterial endocarditis; rarely fat embolism and air embolism of coronary circulation may occur.
5. Thrombotic diseases: Another infrequent cause of coronary occlusion is from hypercoagulability of the blood such as in shock, polycythaemia vera, sickle cell anaemia and
thrombotic thrombocytopenic purpura.
6. Trauma: Contusion of a coronary artery from penetrating injuries may produce thrombotic occlusion.
7. Aneurysms: Extension of dissecting aneurysms of the aorta into the coronary artery may produce thrombotic coronary occlusion.
Rarely, congenital, mycotic and syphilitic aneurysms may occur in coronary arteries and produce similar occlusive effects.
8. Compression: Compression of a coronary from outside by a primary or secondary tumour of the heart may result in coronary occlusion.
Effects Of Myocardial Ischaemia:
The development of lesions in the coronaries is not always accompanied by cardiac disease.
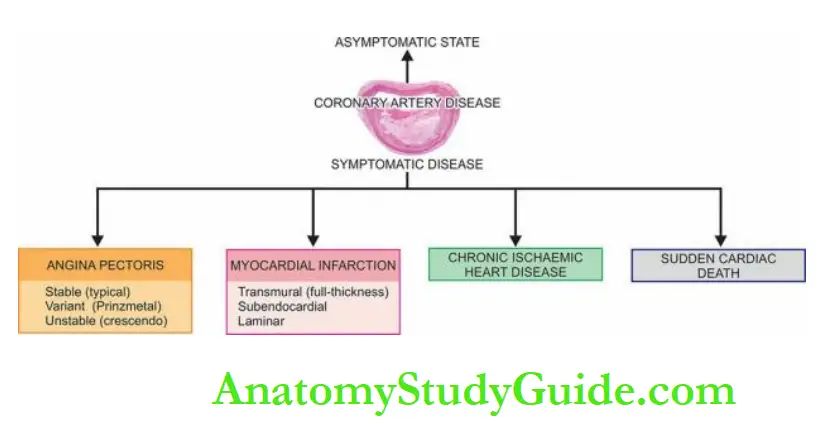
Depending upon the suddenness of onset, duration, degree, location and extent of the area affected by myocardial ischaemia, the range of changes and clinical features may range from an asymptomatic state at one extreme to immediate mortality at another:
- Asymptomatic state
- Angina pectoris (AP)
- Acute myocardial infarction (MI)
- Chronic ischaemic heart disease (CIHD)/Ischaemic cardiomyopathy/Myocardial fibrosis
- Sudden cardiac death
The term acute coronary syndromes include a triad of acute myocardial infarction, unstable angina and sudden cardiac death.
Angina Pectoris:
Angina pectoris is a clinical syndrome of IHD resulting from transient myocardial ischaemia.
It is characterised by paroxysmal pain in the substernal or precordial region of the chest which is aggravated by an increase in the demand of the heart and relieved by a decrease in the work of the heart.
Often, the pain radiates to the left arm, neck, jaw or right arm. It is more common in men past 5th decade of life.
There are 3 overlapping clinical patterns of angina pectoris with some differences in their pathogenesis:
- Stable or typical angina
- Prinzmetal’s variant angina
- Unstable or crescendo angina
Stable Or Typical Angina:
This is the most common pattern. Stable or typical angina is characterised by attacks of pain following physical exertion or emotional excitement and is relieved by rest.
The pathogenesis of the condition lies in chronic stenotic coronary atherosclerosis that cannot perfuse the myocardium adequately when the workload on the heart increases.
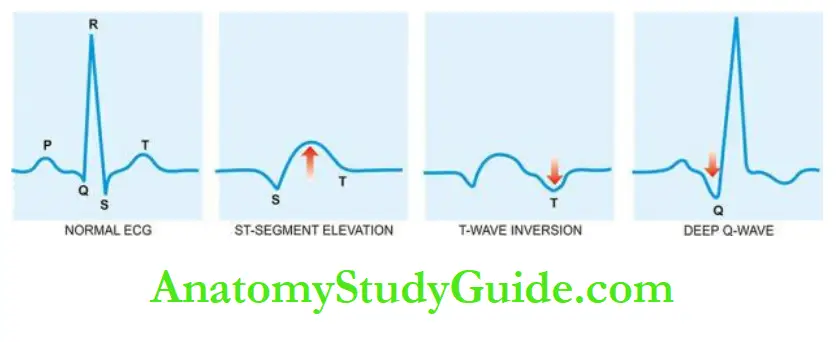
During the attacks, there is depression of the ST segment in the ECG due to poor perfusion of the subendocardial region of the left ventricle but there is no elevation of enzymes in the blood as there is no irreversible myocardial injury.
Prinzmetal’s Variant Angina: This pattern of angina is characterised by pain at rest and has no relationship with physical activity. The exact pathogenesis of Prinzmetal’s angina is not known.
It may occur due to sudden vasospasm of a coronary trunk induced by coronary atherosclerosis or may be due to the release of humoral vasoconstrictors by mast cells in the coronary adventitia.
ECG shows ST segment elevation due to transmural ischaemia. These patients respond well to vasodilators like nitroglycerin.
Unstable Or Crescendo Angina:
Also referred to as ‘pre-infarction angina’ or ‘acute coronary insufficiency’, this is the most serious pattern of angina.
It is characterised by a more frequent onset of pain of prolonged duration and occurs often at rest.
It is thus indicative of an impending acute myocardial infarction.
The distinction between unstable angina and acute MI is made by ST segment changes on ECG—acute MI characterised by ST-segment elevation while unstable angina may have non-ST segment elevation MI.
Multiple factors are involved in the pathogenesis of unstable angina which include:
Stenotic coronary atherosclerosis, complicated coronary plaques (e.g. superimposed thrombosis, haemorrhage, rupture, ulceration etc), platelet thrombi over atherosclerotic plaques and vasospasm of coronary arteries.
More often, the lesions lie in a branch of the major coronary trunk so that collaterals prevent infarction.
Acute Myocardial Infarction
Acute myocardial infarction (MI) is the most important and feared consequence of coronary artery disease.
Many patients may die within the first few hours of the onset, while the remainder suffers from the effects of impaired cardiac function.
A significant factor that may prevent or diminish myocardial damage is the development of collateral circulation through anastomotic channels over a period of time.
A regular and well-planned exercise programme encourages good collateral circulation and improved cardiac performance.
Incidence:
Acute MI accounts for 10-25% of all deaths. Due to the dominant etiologic role of coronary atherosclerosis in acute MI, the incidence of acute MI correlates well with the incidence of atherosclerosis in a geographic area.
Age Acute MI may virtually occur at all ages, though the incidence is higher in the elderly.
About 5% of heart attacks occur in young people under the age of 40 years, particularly in those with major risk factors to develop atherosclerosis like hypertension, diabetes mellitus, cigarette smoking and dyslipidaemia including familial hypercholesterolaemia.
Sex Males throughout their life are at a significantly higher risk of developing acute MI as compared to females.
Women during the reproductive period have a remarkably low incidence of acute MI, probably due to the protective influence of oestrogen.
The use of oral contraceptives is associated with a high risk of developing acute MI.
After menopause, this gender difference gradually declines but the incidence of disease among women never reaches that among men of the same age.
Etiopathogenesis:
The etiologic role of severe coronary atherosclerosis (more than 75% compromise of the lumen) of one or more of the three major coronary arterial trunks in the pathogenesis of about 90% of cases of acute MI is well documented by autopsy studies as well as by coronary angiographic studies.
A few notable features in the development of acute MI are as under:
1. Myocardial ischaemia: is brought about by one or more of the following mechanisms:
- Diminished coronary blood flow e.g. in coronary artery disease, shock.
- Increased myocardial demand e.g. in exercise, and emotions.
- Hypertrophy of the heart without simultaneous increase of coronary blood flow e.g. in hypertension, valvular heart disease.
2. Role of platelets: Rupture of an atherosclerotic plaque exposes the subendothelial collagen to platelets which undergo aggregation, activation and release reaction.
These events contribute to the build-up of the platelet mass that may give rise to emboli or initiate thrombosis.
3. Acute plaque rupture: In general, slowly developing coronary ischaemia from stenotic coronary atherosclerosis of high grade may not cause acute MI but continue to produce episodes of angina pectoris.
But acute complications in coronary atherosclerotic plaques in the form of superimposed coronary thrombosis due to plaque rupture and plaque haemorrhage are frequently encountered in cases of acute MI
- Superimposed coronary thrombosis due to disruption of plaque is seen in about half the cases of acute MI. Infusion of intracoronary fibrinolysis in the first half an hour of development of acute MI in such cases restores blood flow in the blocked vessel in the majority of cases.
- Intramural haemorrhage is found in about one-third of cases of acute MI. Plaque haemorrhage and thrombosis may occur together in some cases.
4. Non-atherosclerotic causes: About 10% of cases of acute MI are caused by non-atherosclerotic factors such as coronary vasospasm, arteritis, coronary ostial stenosis, embolism, thrombotic diseases, trauma and outside compression as already described.
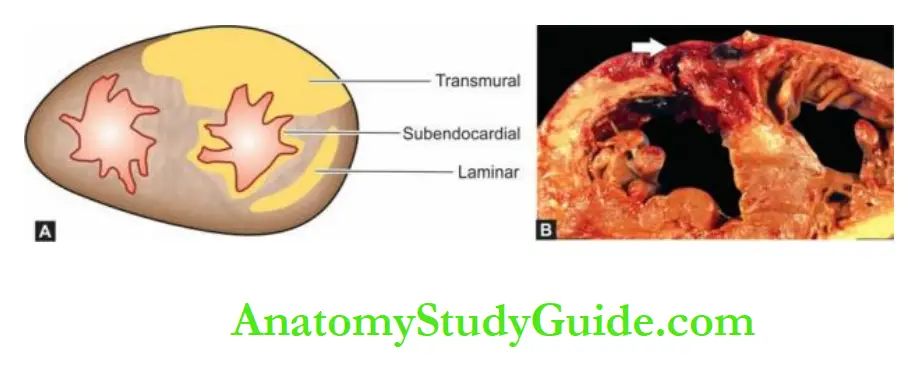
5. Transmural versus subendocardial infarcts: There are some differences in the pathogenesis of the transmural infarcts involving the full thickness of the ventricular wall and the subendocardial (laminar) infarcts affecting the inner subendocardial one-third to half.
These are as under:
Transmural (full-thickness) infarcts are the most common type seen in 95% of cases.
Critical coronary narrowing (more than 75% compromised lumen) is of great significance in the causation of such infarcts.
Atherosclerotic plaques with superimposed thrombosis and intramural haemorrhage are significant in about 90% of cases and non-atherosclerotic causes in the remaining 10% of cases.
Subendocardial (laminar) infarcts have their genesis in reduced coronary perfusion due to coronary atherosclerosis but without critical stenosis (not necessarily 75% compromised lumen), aortic stenosis or haemorrhagic shock.
This is because the subendocardial myocardium is normally least well perfused by coronaries and thus is more vulnerable to any reduction in the coronary flow.
Superimposed coronary thrombosis is frequently encountered in these cases too, hence the beneficial role of fibrinolytic treatment in such patients.
Types Of Infarcts:
Infarcts have been classified in a number of ways by physicians and pathologists:
1. According to the anatomic region of the left ventricle involved, they are called anterior, posterior (inferior), lateral, septal and circumferential, and their combinations like anterolateral, posterolateral (or inferolateral) and anteroseptal.
2. According to the degree of thickness of the ventricular wall involved, infarcts are of two types:
- Full-thickness or transmural, when they involve the entire thickness of the ventricular wall.
- Subendocardial or laminar, when they occupy the inner subendocardial half of the myocardium.

3. According to the age of infarcts, they are of two types:
- Newly-formed infarcts are called acute, recent or fresh.
- Advanced infarcts are called as old, healed or organised.
Location Of Infarcts:
Infarcts are most frequently located in the left ventricle.
The right ventricle is less susceptible to infarction due to its thin wall, having less metabolic requirements and is thus adequately nourished by the besan vessels.
Atrial infarcts, whenever present, are more often in the right atrium, usually accompanying the infarct of the left ventricle.
The left atrium is relatively protected from infarction because it is supplied by the oxygenated blood in the left atrial chamber.
The region of infarction depends upon the area of obstructed blood supply by one or more of
the three coronary arterial trunks.
Accordingly, there are three regions of myocardial infarction:
1. Stenosis of the left anterior descending coronary artery is the most common (40-50%).
The region of infarction is the anterior part of the left ventricle including the apex and the anterior two-thirds of the interventricular septum.
2. Stenosis of the right coronary artery is the next most frequent (30-40%).
It involves the posterior part of the left ventricle and the posterior one-third of the interventricular septum.
3. Stenosis of the left circumflex coronary artery is seen least frequently (15-20%).
Its area of involvement is the lateral wall of the left ventricle.
Morphologic Features:
The gross and microscopic changes in the myocardial infarction vary according to the age of the infarct and are therefore described sequentially.
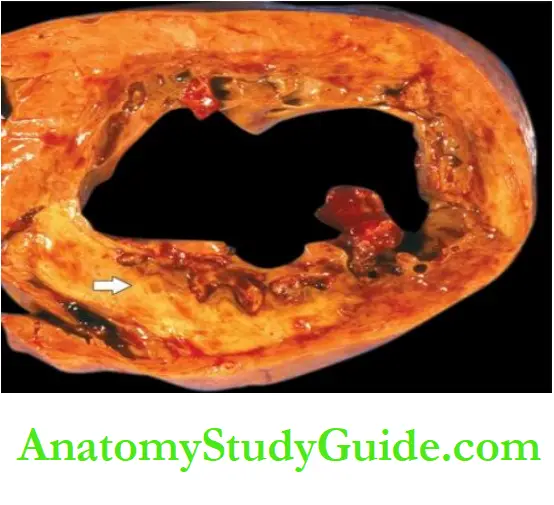
Grossly, most infarcts occur singly and vary in size from 4 to 10 cm.
As explained above, they are found most often in the left ventricle. Less often, there are multifocal lesions.
The transmural infarcts, which by definition involve the entire thickness of the ventricular wall, usually have a thin rim of preserved subendocardial myocardium which is perfused directly by the blood in the ventricular chamber.
The subendocardial infarcts which affect the inner subendocardial half of the myocardium produce less well-defined gross changes than the transmural infarcts.
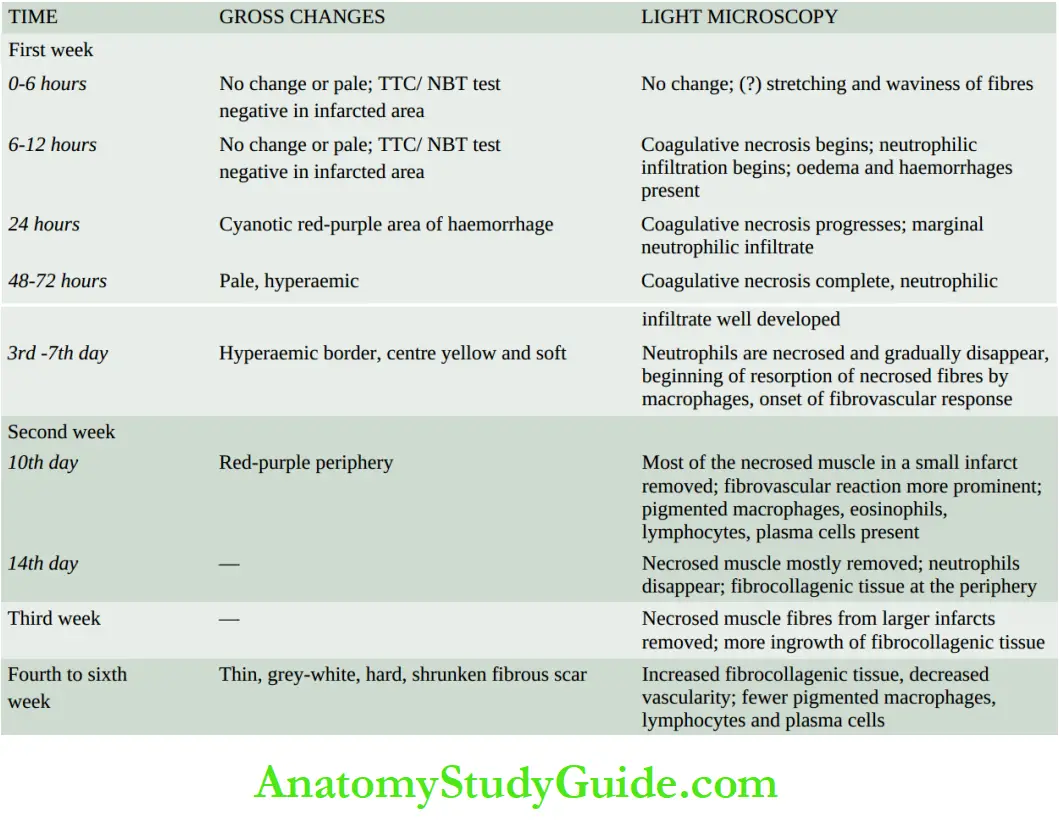
The sequence of macroscopic changes in all myocardial infarcts is as under:
1. In 6 to 12 hours old infarcts, no striking gross changes are discernible except that the affected myocardium is slightly paler and drier than normal.
However, the early infarcts (3 to 6 hours old) can be detected by histochemical staining for dehydrogenases on unfixed slices of the heart.
This consists of immersing a slice of unfixed heart in the solution of triphenyl tetrazolium chloride (TTC) which imparts red-brown colour to the normal heart muscle, while
the area of infarcted muscle fails to stain due to a lack of dehydrogenases.
The viability of cardiac muscle is nitroblue tetrazolium (NBT) dye which imparts a blue colour to unaffected cardiac muscle while infarcted myocardium remains unstained.
2. By about 24 hours, the infarct develops cyanotic, red-purple, blotchy areas of haemorrhage due to stagnation of blood.
3. During the next 48 to 72 hours, the infarct develops a yellow border due to neutrophilic infiltration and thus becomes more well-defined.
4. In 3-7 days, the infarct has a hyperaemic border while the centre is yellow and soft.
5. By 10 days, the periphery of the infarct appears reddish-purple due to the growth of granulation tissue. With the passage of time, further healing takes place; the necrotic muscle is resorbed and the infarct shrinks and becomes pale grey.
6. By the end of 6 weeks, the infarcted area is replaced by a thin, grey-white, hard, shrunken fibrous scar which is well developed in about 2 to 3 months.
However, the time taken by an infarct to heal by fibrous scar may vary depending upon the size of the infarct and adequacy of collateral circulation.
Microscopically, the changes are similar in both transmural and subendocardial infarcts.
As elsewhere in the body, myocardial ischaemia induces ischaemic coagulative necrosis of the myocardium which eventually heals by fibrosis.
However, in myocardial infarction, the following time-related sequential light microscopic changes are observed:
1. First week The progression of changes takes place in the following way:
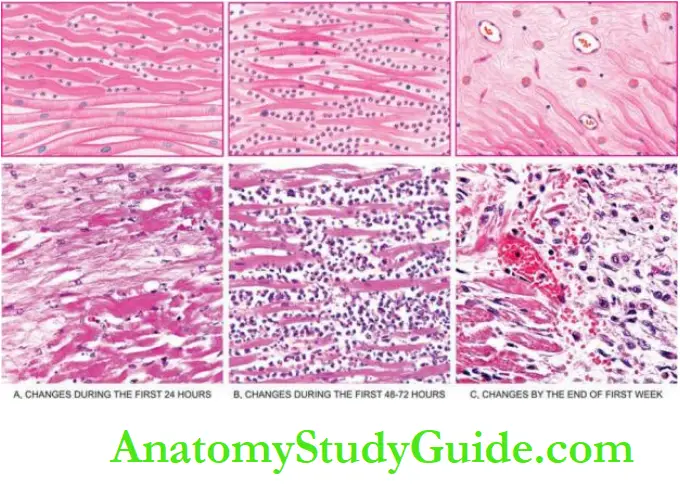
In the first 6 hours after infarction, usually no detectable histologic change is observed in routine light microscopy. However, some investigators have described stretching and waviness of the myocardial fibres within one hour of the onset of ischaemia.
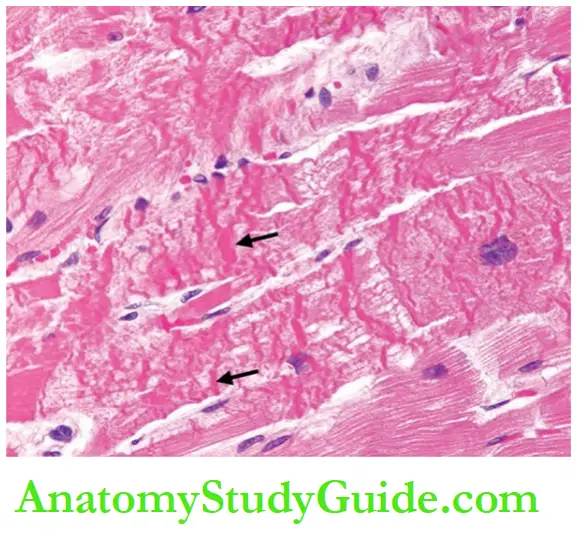
After 6 hours, there is the appearance of some oedema fluid between the myocardial fibres. The muscle fibres at the margin of the infarct show vacuolar degeneration called myocytolysis.
By 12 hours, coagulative necrosis of the myocardial fibres sets in and neutrophils begin to appear at the margin of the infarct.
Coagulative necrosis of fibres is characterised by loss of striations and intense eosinophilic, hyaline appearance and may show nuclear changes like karyolysis, pyknosis and karyorrhexis.
Haemorrhages and oedema are present in the interstitium.
During the first 24 hours, coagulative necrosis progresses further as evidenced by shrunken eosinophilic cytoplasm and pyknosis of the nuclei. The neutrophilic infiltrate at the margins of the infarct is slight.
During the first 48 to 72 hours, coagulative necrosis is complete with loss of nuclei. The neutrophilic infiltrate is well developed and extends centrally into the interstitium.
In 3-7 days, neutrophils are necrosed and gradually disappear. The process of resorption of necrosed muscle fibres by macrophages begins.
Simultaneously, there is the onset of the proliferation of capillaries and fibroblasts from the margins of the infarct.
2. Second week The changes are as under:
By the 10th day, most of the necrosed muscle at the periphery of the infarct is removed.
The fibrovascular reaction at the margin of the infarct is more prominent.
Many pigmented macrophages containing yellow-brown lipofuscin (derived from the breakdown of myocardial cells) and golden-brown haemosiderin (derived from lysed erythrocytes in haemorrhagic areas) are seen.
Also present are a few other inflammatory cells like eosinophils, lymphocytes and plasma cells.
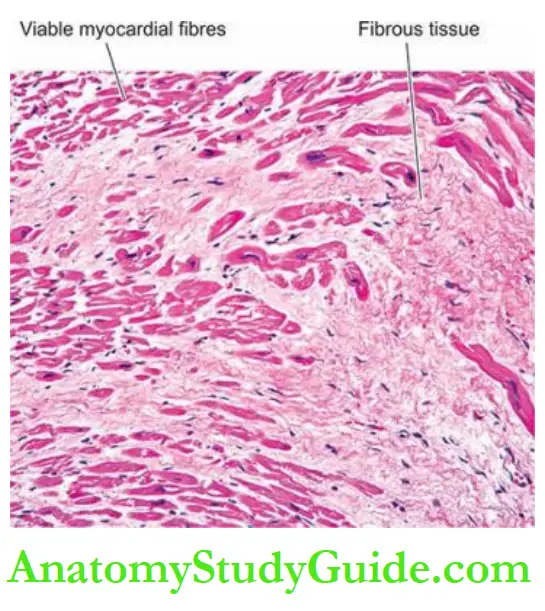
By the end of the 2nd week, most of the necrosed muscle in small infarcts is removed, neutrophils have almost disappeared, and newly laid collagen fibres replace the periphery of the infarct.
3. Third week Necrosed muscle fibres from larger infarcts continue to be removed and replaced by ingrowth of newly formed collagen fibres.
Pigmented macrophages as well as lymphocytes and plasma cells are prominent while eosinophils gradually disappear.
4. Fourth to sixth week With further removal of necrotic tissue, there is an increase in collagenous connective tissue, decreased vascularity and fewer pigmented macrophages,
lymphocytes and plasma cells.
Thus, at the end of 6 weeks, a contracted fibro collagenic scar with diminished vascularity is formed.
The pigmented macrophages may persist for a long duration in the scar, sometimes for years.
A summary of the sequence of gross and microscopic changes in myocardial infarction of varying duration.
Salvage In Early Infarcts And Reperfusion Injury
In the vast majority of cases of acute MI, occlusive coronary artery thrombosis has been demonstrated superimposed on the fibrofatty plaque.
The ischaemic injury to the myocardium is reversible if perfusion is restored within the first 30 minutes of the onset of infarction failing which irreversible ischaemic necrosis
of myocardium sets in.
The salvage in early infarcts can be achieved by the following interventions:
- Institution of thrombolytic therapy with thrombolytic agents such as streptokinase and
tissue plasminogen activator (door-to-needle time ≤30 minutes). - Percutaneous transluminal coronary angioplasty (PTCA).
- Coronary artery stenting.
- Coronary artery bypass surgery.
However, a late attempt at reperfusion is fraught with the risk of ischaemic reperfusion injury.
Further myonecrosis during reperfusion occurs due to the rapid influx of calcium ions and the generation of toxic oxygen free radicals.
Grossly, the myocardial infarct following reperfusion injury appears haemorrhagic rather than pale.
Microscopically, myofibres show contraction band necrosis which is transverse and thick eosinophilic bands.
Changes In Early Infarcts
Through special techniques like electron microscopy, and chemical and histochemical studies, changes can be demonstrated in early infarcts before detectable light microscopic alterations appear.
1. Electron microscopic changes Changes: by EM examination are evident in less than half an hour on the onset of infarction.
These changes are as under:
- The disappearance of perinuclear glycogen granules within 5 minutes of ischaemia.
- Swelling of mitochondria in 20 to 30 minutes.
- Disruption of the sarcolemma.
- Nuclear alterations like peripheral clumping of nuclear chromatin.
2. Chemical and histochemical changes: Analysis of tissues from early infarcts by chemical and histochemical techniques has shown a number of findings.
These are as follows:
- Glycogen depletion in myocardial fibres within 30 to 60 minutes of infarction.
- Increase in lactic acid in the myocardial fibres.
- Loss of K+ from the ischaemic fibres.
- Increase of Na+ in the ischaemic cells.
- The influx of Ca++ into the cells causes irreversible cell injury.
Based on the above observations and on leakage of enzymes from the ischaemic myocardium, alterations in the concentrations of various enzymes are detected in the blood of these patients.
Diagnosis
The diagnosis of acute MI is made on the observations of 3 types of features—
clinical features, ECG changes, and serum enzyme determinations.
1. Clinical features Typically, acute MI has a sudden onset.
The following clinical features usually characterise a case of acute MI.
Pain: Usually sudden, severe, crushing and prolonged, substernal or precordial in location, unrelieved by rest or nitroglycerin, often radiating to one or both the arms, neck and back.
Indigestion: Pain is often accompanied by epigastric or substernal discomfort interpreted as ‘heartburn’ with nausea and vomiting.
Apprehension: The patient is often terrified, restless and apprehensive due to great fear of death.
Shock: Systolic blood pressure is below 80 mmHg; lethargy, cold clammy limbs, peripheral cyanosis, weak pulse, tachycardia or bradycardia are often present.
Oliguria: Urine flow is usually less than 20 ml per hour.
Low-grade fever: Mild rise in temperature occurs within 24 hours and lasts up to one week, accompanied by leucocytosis and elevated ESR.
Acute pulmonary oedema: Some cases develop severe pulmonary congestion due to left ventricular failure and develop suffocation, dyspnoea, orthopnoea and bubbling respiration.
2. ECG changes: The ECG changes are one of the most important parameters.
The most characteristic ECG change is ST segment elevation in acute MI (termed as STEMI); other changes include T wave inversion and the appearance of wide deep Q waves.
3. Serum cardiac biomarkers Certain proteins and enzymes are released into the blood from necrotic heart muscle after acute MI.
Measurement of their levels in serum is helpful in making a diagnosis and plan management.
Rapid assay of some more specific cardiac proteins is available rendering the estimation of non-specific estimation of SGOT of historical importance only in current practice.
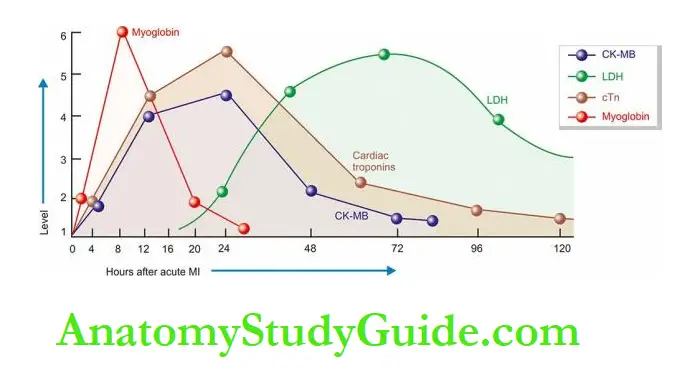
Important myocardial markers in use nowadays are as under:
- Creatine phosphokinase (CK) and CK-MB CK has three forms—
-
- CK-MM derived from skeletal muscle;
- CK-BB is derived from the brain and lungs; and
- CK-MB, mainly from cardiac muscles and an insignificant amount from extracardiac tissue.
Thus, total CK estimation lacks specificity while elevation of CK-MB isoenzyme is considerably specific for myocardial damage.
CK-MB has further 2 forms—CK-MB2 is the myocardial form while CK-MB1 is the extracardiac form.
A ratio of CK-MB2: CK-MB1 more than 1.5 is highly sensitive for the diagnosis of acute MI after 4-6 hours of the onset of myocardial ischaemia.
CK-MB disappears from blood within 48 hours.
Lactate dehydrogenase (LDH) Total LDH estimation also lacks specificity since this enzyme is present in various tissues besides the myocardium such as in skeletal muscle, kidneys, liver, lungs and red blood cells.
However, like CK, LDH too has two isoforms of which LDH-1 is myocardial-specific.
Estimation of the ratio of LDH-1: LDH-2 above 1 is reasonably helpful in making a diagnosis.
LDH levels begin to rise after 24 hours, reach a peak in 3 to 6 days and return to normal in 14 days.
Cardiac-specific troponins (cTn) Immunoassay of cTn as a serum cardiac marker has rendered LDH estimation obsolete.
Troponins are contractile muscle proteins present in human cardiac and skeletal muscle but cardiac troponins are specific for the myocardium.
There are two types of cTn:
- cardiac troponin T (cTnT); and
- cardiac troponin I (cTnI).
Both cTnT and cTnI are not found in the blood normally, but after myocardial injury, their levels rise very high around the same time when CK-MB is elevated (i.e. after 4-6 hours).
Both troponin levels remain high for much longer duration; cTnI for 7-10 days and cTnT for 10-14 days.
Myoglobin Though myoglobin is the first cardiac marker to become elevated after myocardial infarction, it lacks cardiac specificity and is excreted in the urine rapidly.
Its levels, thus, return to normal within 24 hours of the attack of acute MI.
4. Other supportive laboratory tests: In addition to serum cardiac biomarkers, a few other laboratory tests either indicate myocardial ischaemic injury or are supportive of susceptibility to acute MI in a patient.
Polymorphonuclear leucocytosis is a nonspecific reaction to myocardial damage, appearing within a few hours after coronary ischaemia and persists for up to one week.
- ESR rises slowly after myocardial injury and peaks a week after ischaemia.
- Lipid profile with measurements of various lipid fractions in the blood.
- Elevated CRP in serum is a sensitive and independent risk marker for IHD.
- Urine analysis including microalbuminuria for evidence of diabetes mellitus and renal disease.
- Serum creatinine for renal disease.
- Glycosylated haemoglobin estimation for control diabetes mellitus.
Complications
Following an attack of acute MI, only 10-20% of cases do not develop major complications and recover.
The remainder 80-90% of cases develop one or more major complications, some of which are fatal.
The immediate mortality from acute MI (sudden cardiac death) is about 25%.
The important complications which may develop following acute MI are as follows:
1. Arrhythmias
Arrhythmias (or abnormalities in the normal heart rhythm) are the most common complication in acute MI.
These occur due to ischaemic injury or irritation to the conduction system, resulting in an abnormal rhythm.
Other causes of arrhythmias include leakage of K+ from ischaemic muscle cells and increased concentration of lactate and free fatty acids in the tissue fluid.
Arrhythmias may be in the form of sinus tachycardia or sinus bradycardia, atrial fibrillation, premature systoles, and the most serious ventricular fibrillation responsible for many sudden cardiac deaths.
2. Congestive heart failure: About half the patients with MI develop CHF which may be in the form of right ventricular failure, left ventricular failure or both.
CHF is responsible for about 40% of deaths from acute MI. If the patient survives, healing may restore normal cardiac function but in some CHF may persist and require regular treatment later.
3. Cardiogenic shock: About 10% of patients with acute MI develop cardiogenic shock characterised by hypotension with systolic blood pressure of 80 mmHg or less for many days.
Shock may be accompanied by peripheral circulatory failure, oliguria and mental confusion.
4. Mural thrombosis and thromboembolism: The incidence of thromboembolism from intracardiac thrombi and from thrombosis in the leg veins is 15-45% in cases of acute MI and is the major cause of death in 12% of cases.
Mural thrombosis in the heart develops due to the involvement of the endocardium and subendocardium in the infarct and due to the slowing of the heart rate.
Mural thrombi often form thromboembolic. Another source of thromboembolic is venous thrombosis in the leg veins due to prolonged bed rest.
Thromboemboli from either source may cause occlusion of the pulmonary, renal, mesenteric, splenic, pancreatic or cerebral arteries and cause infarcts in these organs.
5. Rupture: Rupture of the heart occurs in up to 5% of cases of acute MI causing death.
Rupture occurs most often from the infarcted ventricular wall into the pericardial cavity-causing haemopericardium and tamponade.
Other sites of rupture are through the interventricular septum and the rupture of a papillary muscle in the infarct of the left ventricle.
Rupture at any of these sites occurs usually in the first week and is often fatal.
6. Cardiac aneurysm: Another 5% of patients of acute MI develop aneurysm, often of the left ventricle.
It occurs in healed infarcts through thin, fibrous, non-elastic scar tissue.
Cardiac aneurysms impair the function of the heart and are the common sites for mural thrombi.
Rarely, calcification of the wall of aneurysm may occur.
7. Pericarditis: Sterile pericarditis appearing on about the second day is common over transmural infarcts.
It is characterised by fibrinous pericarditis and may be associated with pericardial effusion. Often, it is of no functional significance and resolves spontaneously.
8. Postmyocardial infarction syndrome: About 3 to 4% of patients who suffered from acute MI develop postmyocardial infarction syndrome or Dressler’s syndrome subsequently.
It usually occurs 1 to 6 weeks after the attack of MI. It is characterised by pneumonitis.
The symptoms are usually mild and disappear in a few weeks. The exact pathogenesis of this syndrome is not known.
It may be due to an autoimmune reaction as evidenced by circulating anti-heart antibodies in the serum of these patients.
But these antibodies are also present in some patients with acute MI who do not develop this syndrome.
Chronic Ischaemic Heart Disease
Chronic ischaemic heart disease, ischaemic cardiomyopathy or myocardial fibrosis, are the terms used for focal or diffuse fibrosis in the myocardium characteristically found in elderly patients of progressive IHD.
Such small areas of fibrous scarring are commonly found in the heart of patients who have a history of episodes of angina and attacks of MI some years back.
The patients generally have gradually developed CHF due to decompensation over a period of years.
Occasionally, serious cardiac arrhythmias or infarctions may supervene and cause death.
Etiopathogenesis: In the majority of cases, coronary atherosclerosis causes progressive ischaemic myocardial damage and replacement by myocardial fibrosis.
A small percentage of cases may result from other causes such as emboli, coronary arteritis and myocarditis.
The mechanism of development of myocardial fibrosis can be explained by one of the following concepts:
Myocardial fibrosis represents the healing of minute infarcts involving small scattered groups of myocardial fibres.
An alternate concept of the development of myocardial fibrosis is the healing of minute areas of focal myocytolysis—the myocardial fibres in a small area undergo slow degeneration due to myocardial ischaemia.
These fibres lose their myofibrils but nuclei remain intact.
These foci are infiltrated by macrophages and eventually are replaced by proliferating fibroblasts and collagen.
Morphologic Features:
Grossly, the heart may be normal in size or hypertrophy. The left ventricular wall generally shows foci of grey-white fibrosis in the brown myocardium.
Healed scars of previous MI may be present. Valves of the left heart may be distorted, thickened and show calcification.
Coronary arteries invariably show moderate to severe atherosclerosis.
Microscopically, the characteristic features are as follows:
There are scattered areas of diffuse myocardial fibrosis, especially around the small blood vessels in the interstitial tissue of the myocardium.
Intervening single fibres and groups of myocardial fibres show variation in fibre size and foci of myocytolysis.
Areas of brown atrophy of the myocardium may also be present.
Coronary arteries show atherosclerotic plaques and may have complicated lesions in the form of superimposed thrombosis.
Sudden Cardiac Death
Sudden cardiac death is defined as sudden death within 24 hours of the onset of cardiac symptoms.
The most important cause is coronary atherosclerosis; less commonly it may be due to coronary vasospasm and other non-ischaemic causes.
These include calcific aortic stenosis, myocarditis of various types, hypertrophic cardiomyopathy, mitral valve prolapse, endocarditis, and hereditary and acquired defects of the conduction system.
The mechanism of sudden death by myocardial ischaemia is almost always by fatal arrhythmias, chiefly ventricular asystole or fibrillation.
Morphologic Features:
At autopsy, such cases reveal most commonly critical atherosclerotic coronary narrowing (more than 75% compromised lumen) in one or more of the three major coronary arterial trunks with superimposed thrombosis or plaque haemorrhage.
Healed and new myocardial infarcts are found in many cases.
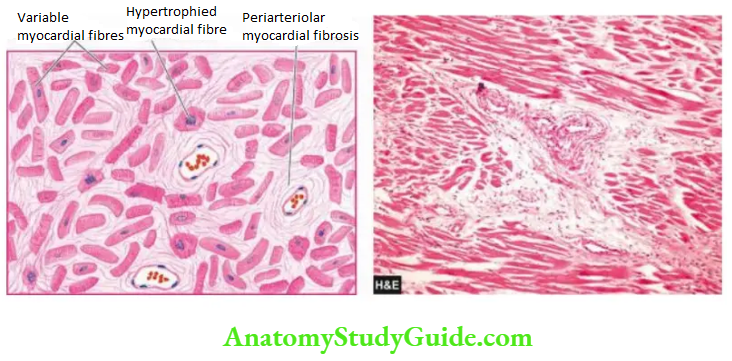
Myocardial Infarction And Stem Cells:
Permanent loss of damaged myocardial tissue in MI and its replacement with scar tissue results in loss of function by healed myocardium.
Traditional therapies are directed at lowering mortality, preventing additional damage to the myocardium and reducing the risk of subsequent heart attack.
Futuristic therapeutic options are directed at cardiac tissue regeneration by stem cell therapy for both acute and chronic myocardial ischaemic damage to the myocardium.
Currently, stem cell therapy for patients of IHD is undergoing clinical trials by use of human pluripotent stem cells, and human adult and foetal stem cells (such as bone marrow-derived stem cells) that can transdifferentiate into cardiac myocytes.
Fundamental research is also going on for the use of certain biomaterials and bioengineering technology to stimulate resident myocardial stem cells.
Preliminary studies on the transplantation of stem cells in IHD have yielded encouraging results in clinical improvement and reduction in infarct size and hold promise for the future.
Ischaemic Heart Disease:
Ischaemic heart disease (IHD) is an acute or chronic cardiac disability arising from an imbalance between the myocardial supply and demand for oxygenated blood.
Atherosclerotic coronary artery disease (CAD) is the most common cause of IHD, most commonly of LAD, others are RCA and CXA. Often, there are superimposed changes on the plaque.
Acute coronary syndromes include a triad of acute myocardial infarction, unstable angina and sudden cardiac death
Angina pectoris results from transient myocardial ischaemia and is characterised by paroxysmal pain in the substernal or precordial region Acute myocardial infarction (MI) is the most important and feared consequence of coronary artery disease.
Early thrombolytic therapy within 30 minutes of occurrence may help in the restoration of blood supply.
Gross and microscopic changes in myocardial infarction, most often in the left ventricle, vary according to the age of the infarct.
Diagnosis of acute MI is made by clinical features, ECG changes, serum cardiac biomarkers, and some supportive laboratory tests.
Chronic ischaemic heart disease is focal or diffuse fibrosis in the myocardium characteristically found in elderly patients with progressive IHD.
Sudden death by myocardial ischaemia is almost always by fatal arrhythmias, chiefly ventricular asystole or fibrillation.
Stem cell therapy in IHD holds promise for the future since it stimulates the regeneration of damaged myocardial tissue instead of replacement with a fibrous scar which does not
perform a cardiac function.
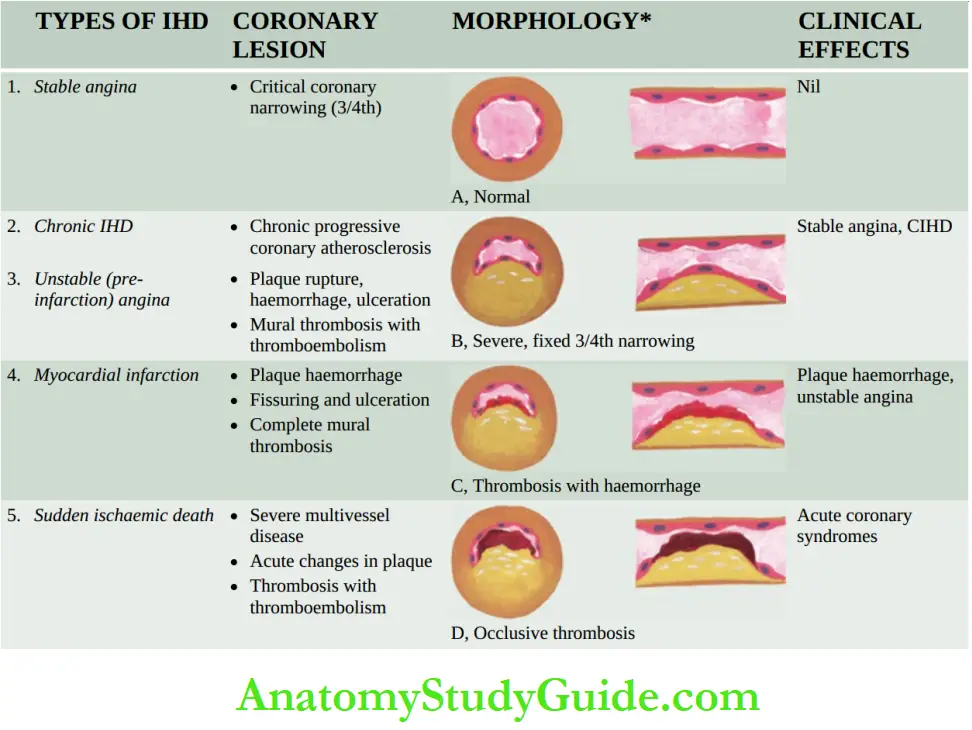
Rheumatic Fever And Rheumatic Heart Disease
Rheumatic fever (RF) is a systemic, post-streptococcal, non-suppurative inflammatory disease, principally affecting the heart, joints, central nervous system, skin and subcutaneous tissues.
The chronic stage of RF involves all the layers of the heart (pancarditis) causing major cardiac sequelae referred to as rheumatic heart disease (RHD).
In spite of its name suggesting acute arthritis migrating from joint to joint, it is well known that it is the heart rather than the joints which is the first and major organ affected.
Decades ago, William Boyd gave the dictum ‘rheumatism licks the joint, but bites the whole heart’.
Incidence
The disease appears most commonly in children between the age of 5 to 15 years when the streptococcal infection is most frequent and intense.
Both sexes are affected equally, though some investigators have noted a slight female preponderance.
The geographic distribution, incidence and severity of RF and RHD are generally related to the frequency and severity of streptococcal pharyngeal infection.
The disease is seen more commonly in poor socioeconomic strata of society living in damp and overcrowded places which promotes the interpersonal spread of streptococcal infection.
Its incidence has declined in developed countries as a result of improved living conditions and the early use of antibiotics for streptococcal infection.
But it is still common in the developing countries of the world, particularly prevalent in the Indian subcontinent (India, Pakistan, Bangladesh, Nepal, Afghanistan), some Pacific countries, sub-Saharan Africa and Latin America.
In India, RHD and RF continue to be major public health problems.
In a multicentric survey of school-going children by the Indian Council of Medical Research, an incidence of 1 to 5.5 per 1000 children has been reported.
Etiopathogenesis
After a long controversy, the etiologic role of preceding throat infection with group A β-haemolytic streptococci (GAS) in RF is now well accepted.
However, the mechanism of lesions in the heart, joints and other tissues is not by direct infection but by induction of hypersensitivity or autoimmunity in a susceptible host.
Thus, there are 3 groups of factors in the aetiology and pathogenesis of RF and RHD: environmental factors, host susceptibility and immunologic evidence.
1. Environmental Factors:
There is sufficient clinical and epidemiological evidence to support the concept that RF occurs following infection of the throat and upper respiratory tract with β-haemolytic streptococci of Lancefield group A.
These evidences are as under:
1. There is often a history of infection of the pharynx, and upper respiratory tract with this microorganism about 2 to 3 weeks prior to the attack of RF.
This period is usually the latent period required for sensitisation to the bacteria.
2. Subsequent or ongoing attacks of streptococcal infection are generally associated with recurrent episodes of acute RF.
3. A higher incidence of RF has been observed after outbreaks and epidemics of streptococcal infection of the throat in children from schools or in young men from training camps.
4. Administration of antibiotics leads to a lowering of the incidence as well as the severity of RF and its recurrences.
5. Cardiac lesions similar to those seen in RHD have been produced in experimental animals by induction of repeated infection with β-haemolytic streptococci of group A.
6. Socioeconomic factors like poverty, poor nutrition, the density of the population, overcrowding in quarters for sleeping etc are associated with the spread of infection, lack of proper medical attention, and hence the higher incidence of RF.
7. The geographic distribution of the disease, as already pointed out, shows a higher frequency and severity of the disease in the developing countries of the world where the living conditions in underprivileged populations are substandard and medical facilities are insufficient.
children in these regions develop recurrent throat infections which remain untreated and have a higher incidence of RF.
8. The role of climate in the development of RF has been reported by some workers.
The incidence of the disease is higher in subtropical and tropical regions with cold, damp climates near the rivers and waterways which favour the spread of infection.
Despite all this evidence, only a small proportion of patients with streptococcal pharyngeal infection develop RF—the attack rate is less than 3%.
There is a suggestion that a concomitant virus enhances the effect of streptococci in individuals who develop RF.
2. Host Susceptibility:
Since all individuals with streptococcal infections do not develop RF, role of inherited characteristics for the disease has been reported:
- Clustering of disease in families.
- Occurrence in identical twins.
- Individuals with HLA class II alleles (particularly HLA-DR7 and HLA-DR4) have strong association with RF.
- First-degree relatives of patients with RF and RHD have increased expression of a particular alloantigen, D8-17, on B cells, which may act as a marker for inherited susceptibility to the disease.
3. Immunologic Evidence:
It has been observed that though the throat of patients during acute RF may contain streptococci, the clinical symptoms of RF appear after a delay of 2-3 weeks and the organisms cannot be grown from the lesions in the target tissues.
This has led to the concept that lesions have immune pathogenesis. Evidence in support is as under:
1. Patients with RF have elevated titres of antibodies to the antigens of β-haemolytic streptococci of group A such as anti-streptolysin O (ASO) and S, anti-streptokinase, antistreptohyaluronidase and anti-DNAase B.
2. Cell wall polysaccharide of group A Streptococcus forms antibodies which are reactive against cardiac valves.
This is supported by the observation of persistently elevated corresponding autoantibodies in patients who have cardiac valvular involvement than those without cardiac valve involvement.
3. Hyaluronate capsule of group A Streptococcus is identical to human hyaluronate present in joint tissues and thus these tissues are the target of attack.
4. Membrane antigens of group A Streptococcus react with sarcolemma of smooth and cardiac muscle, dermal fibroblasts and neurons of caudate nucleus.
Summary Of Organism-Host Susceptibility-Immunity Hypothesis:
Combining the three types of pieces of evidence given above, the pathogenesis of RF-RHD can be summed up as under:
1. A susceptible host, on being encountered with group A Streptococcus infection, mounts an autoimmune reaction by the formation of autoantibodies against bacteria.
2. These autoantibodies cause damage to human tissues due to cross-reactivity between epitopes in the components of bacteria and the host.
3. Streptococcal epitopes present on the bacterial cell wall, cell membrane and the streptococcal M protein, are immunologically identical to human molecules on myosin, keratin, actin, laminin, vimentin and N-acetylglucosamine.
4. Molecular mimicry and cross-reactivity between streptococcal M protein in particular and the human molecules forms the basis of autoimmune damage to human target tissues in RHD i.e. cardiac muscle, valves, joints, skin, neurons etc.
Morphologic Features:
RF is generally regarded as an autoimmune focal inflammatory disorder of the connective tissues throughout the body.
The cardiac lesions of RF in the form of pancarditis, particularly the valvular lesions, are its major manifestations.
However, supportive connective tissues at other sites like the synovial membrane, periarticular tissue, skin and subcutaneous tissue, arterial wall, lungs, pleura and the CNS are all affected (extracardiac lesions).
A. Cardiac Lesions:
The cardiac manifestations of RF are in the form of focal inflammatory involvement of the interstitial tissue of all three layers of the heart, the so-called pancarditis.
The pathognomonic feature of pancarditis in RF is the presence of distinctive Aschoff nodules or Aschoff bodies.
The Aschoff Nodules Or Bodies:
The Aschoff nodules or the Aschoff bodies are spheroidal or fusiform distinct tiny structures, 1-2 mm in size, occurring in the interstitium of the heart in RF and may be visible to the naked eye.
They are especially found in the vicinity of small blood vessels in the myocardium and endocardium and occasionally in the pericardium and the adventitia of the proximal part of the aorta.
Lesions similar to the Aschoff nodules may be found in the extracardiac tissues.
The evolution of fully-developed Aschoff bodies occurs through 3 stages all of which may be found in the same heart at different stages of development.
These are as follows:
1. Early (exudative or degenerative) stage The earliest sign of injury in the heart in RF is apparent by about 4th week of illness.
Initially, there is oedema of the connective tissue and an increase in acid mucopolysaccharide in the ground substance.
This results in the separation of the collagen fibres by accumulating ground substance.
Eventually, the collagen fibres are fragmented and disintegrated and the affected focus takes the appearance and staining characteristics of fibrin. This change is referred to as fibrinoid degeneration.
2. Intermediate (proliferative or granulomatous) stage: It is this stage of the Aschoff body which is pathognomonic of rheumatic conditions. This stage is apparent in 4th to 13th week of illness.
The early stage of fibrinoid change is followed by the proliferation of cells that includes infiltration by lymphocytes (mostly T cells), plasma cells, a few neutrophils and the characteristic cardiac histiocytes (Anitschkow cells) at the margin of the lesion.
Cardiac histiocytes or Anitschkow cells are present in small numbers in normal hearts but their number is increased in the Aschoff bodies; therefore they are not characteristic of RHD.
These are large mononuclear cells having central round nuclei and contain a moderate amount of amphophilic cytoplasm.
The nuclei are vesicular and contain a prominent central chromatin mass which in the longitudinal section appears serrated or caterpillar-like, while in cross-section the chromatin mass appears as a small rounded body in the centre of the vesicular nucleus, just
like an owl’s eye.
Some of these modified cardiac histiocytes become multinucleate cells containing 1 to 4 nuclei and are called Aschoff cells and are pathognomonic of RHD.
3. Late (healing or fibrous) stage: The stage of healing by fibrosis of the Aschoff nodule occurs in about 12 to 16 weeks after the illness.
The nodule becomes oval or fusiform in shape, about 200 µm wide and 600 µm long.
The Anitschkow cells in the nodule become spindle-shaped with diminished cytoplasm and the nuclei stain solidly rather than showing vesicular character.
These cells tend to be arranged in a palisaded manner. With the passage of months and years, the Aschoff body becomes less cellular and the collagenous tissue is increased.
Eventually, it is replaced by a small fibro collagenous scar with little cellularity, frequently in perivascular location.
Rheumatic Pancarditis: Although all three layers of the heart are affected by RF, the intensity of their involvement is variable.
1. Rheumatic Endocarditis: Endocardial lesions of RF may involve the valvular and mural endocardium, causing rheumatic valvulitis and mural endocarditis, respectively.
Rheumatic valvulitis is chiefly responsible for the major cardiac manifestations in chronic RHD.
Rheumatic Valvulitis:
Grossly, the valves in acute RF show thickening and loss of the translucency of the valve leaflets or cusps.
This is followed by the formation of characteristic, small (1 to 3 mm in diameter), multiple, warty vegetations or verrucae, chiefly along the line of closure of the leaflets and cusps.
These tiny vegetations are almost continuous so that the free margin of the cusps or leaflets appears as a rough and irregular ridge.
The vegetations in RF appear grey-brown, and translucent and are firmly attached so that they are not likely to get detached to form emboli, unlike the friable vegetations of infective endocarditis.
Though all four heart valves are affected, their frequency and severity of involvement vary the mitral valve alone is the most common site, followed in decreasing order of frequency, by combined mitral and aortic valves.
The tricuspid and pulmonary valves usually show infrequent and slight involvement.
The higher incidence of vegetation on the left side of the heart is possible because of the greater mechanical stresses on the valves of the left heart, especially along the line of closure of the valve cusps.
The occurrence of vegetations on the atrial surfaces of the atrioventricular valves (mitral and tricuspid) and on the ventricular surface of the semilunar valves (aortic and pulmonary) further lends support to the role of mechanical pressure on the valves in the pathogenesis of vegetations.
The chronic stage of RHD is characterised by a permanent deformity of one or more valves, especially the mitral (in 98% of cases alone or along with other valves) and aortic.
The approximate frequency of deformity of various valves is as under:
- Mitral alone = 37% of cases.
- Mitral + aortic = 27% of cases.
- Mitral + aortic + tricuspid = 22% cases.
- Mitral + tricuspid = 11% cases.
- Aortic alone = 2%.
- Mitral + aortic + tricuspid + pulmonary = less than 1% of cases.
Thus, the mitral valve is almost always involved in RHD.
The gross appearance of the chronic healed mitral valve in RHD is characteristic of ‘fish mouth’ or ‘button hole’ stenosis.
Mitral stenosis and insufficiency are commonly combined in chronic RHD; calcific aortic stenosis may also be found.
These healed chronic valvular lesions in RHD occur due to diffuse fibro collagenous thickening and calcification of the valve cusps or leaflets which cause adhesions between the lateral portions, especially in the region of the commissures.
Thickening, shortening and fusion of the chordae tendineae further contribute to chronic valvular lesions.
Microscopically, the inflammatory changes begin in the region of the valve rings (where the leaflets are attached to the fibrous annulus) and then extend throughout the entire leaflet, whereas vegetations are usually located on the free margin of the leaflets and cusps.
In the early (acute) stage, the histological changes are oedema of the valve leaflet, the presence of an increased number of capillaries and infiltration with lymphocytes, plasma cells, histiocytes with many Anitschkow cells and a few polymorphs.
Occasionally, Aschoff bodies with central foci of fibrinoid necrosis and surrounded by a palisade of cardiac histiocytes are seen, but more often the cellular infiltration is diffuse in the acute stage of RF.
Vegetations present at the free margins of cusps appear as eosinophilic, tiny structures mainly consisting of fibrin with superimposed platelet thrombi and do not contain bacteria.
In the healed (chronic) stage, the vegetation has undergone organisation.
The valves show diffuse thickening as a result of fibrous tissue with hyalinisation, and often calcification.
Vascularisation of the valve cusps may still be evident in the form of thick-walled blood vessels with narrowed lumina.
Typical Aschoff bodies are rarely seen in the valves at this stage.
Rheumatic Mural Endocarditis:
The mural endocardium may also show features of rheumatic carditis though the changes are less conspicuous as compared to valvular changes.
Grossly, the lesions are seen most commonly as MacCallum’s patch which is the region of the endocardial surface in the posterior wall of the left atrium just above the posterior leaflet of the mitral valve.
Maccallum’s patch appears as a map-like area of thickened, roughened and wrinkled part of the endocardium.
Microscopically, the appearance of MacCallum’s patch is similar to that seen in rheumatic valvulitis.
The affected area shows oedema, fibrinoid change in the collagen, and a cellular infiltrate of lymphocytes, plasma cells and macrophages with many Anitschkow cells.
Typical Aschoff bodies may sometimes be found.
2. Rheumatic Myocarditis:
Grossly, in the early (acute)stage, the myocardium, especially of the left ventricle, is soft and flabby. In the intermediate stage, the interstitial tissue of the myocardium shows small foci of necrosis.
Later, tiny pale foci of the Aschoff bodies may be visible throughout the myocardium.
Microscopically, the most characteristic feature of rheumatic myocarditis is the presence of distinctive Aschoff bodies.
These diagnostic nodules are scattered throughout the interstitial tissue of the myocardium and are most frequent in the interventricular septum, left ventricle and left atrium.
Derangements of the conduction system may, thus, be present.
The Aschoff bodies are best identified in the intermediate stage when they appear as granulomas with central fibrinoid necrosis and are surrounded by a palisade of Anitschkow cells and multinucleate Aschoff cells.
There is infiltration by lymphocytes, plasma cells and some neutrophils.
In the late stage, the Aschoff bodies are gradually replaced by small fibrous scars in the vicinity of blood vessels and the inflammatory infiltrate subsides.
The presence of active Aschoff bodies along with old healed lesions is indicative of rheumatic
activity.
3. Rheumatic Pericarditis:
Inflammatory involvement of the pericardium commonly accompanies RHD.
Grossly, the usual finding is fibrinous pericarditis in which there is loss of normal shiny pericardial surface due to deposition of fibrin on its surface and accumulation of slight amount of fibrinous exudate in the pericardial sac.
If the parietal pericardium is pulled off from the visceral pericardium, the two separated surfaces are shaggy due to thick fibrin covering them.
This appearance is often likened to ‘bread and butter appearance’ i.e. resembling the buttered surfaces of two slices in a sandwich when they are gently pulled apart.
If fibrinous pericarditis fails to resolve and, instead, undergoes organisation, the two layers of the pericardium form fibrous adhesions resulting in chronic adhesive pericarditis.
Microscopically, fibrin is identified on the surfaces. The subserosal connective tissue is infiltrated by lymphocytes, plasma cells, histiocytes and a few neutrophils.
Characteristic Aschoff bodies may be seen which later undergo organisation and fibrosis.
The organisation of the exudate causes fibrous adhesions between the visceral and parietal surfaces of the pericardial sac and obliterates the pericardial cavity.
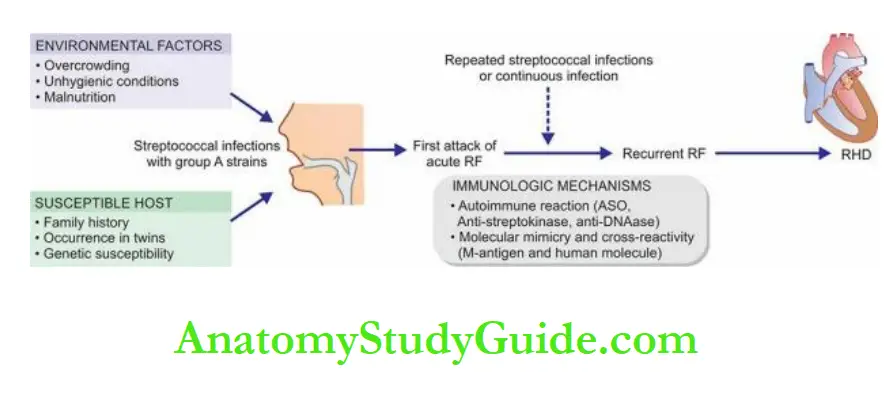
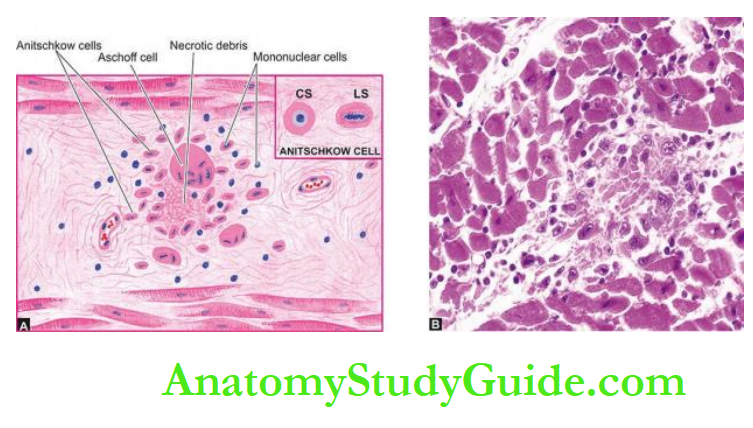
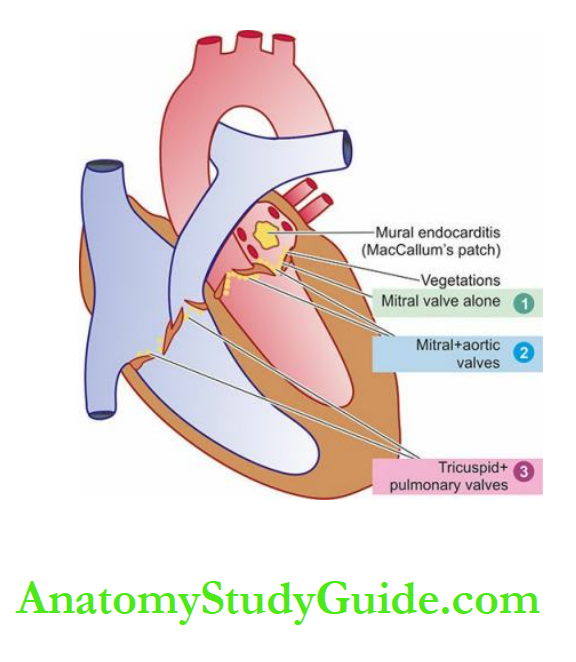
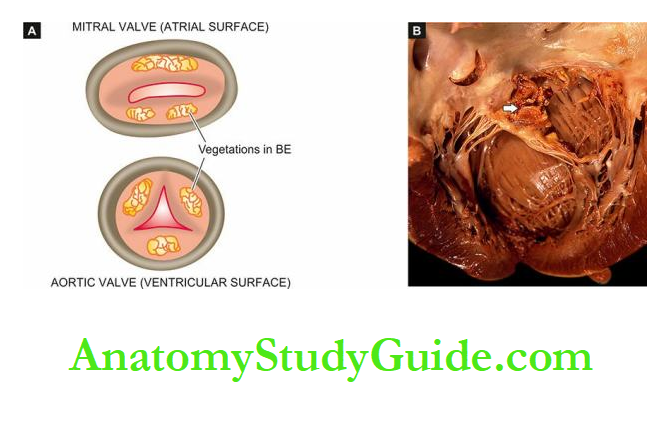
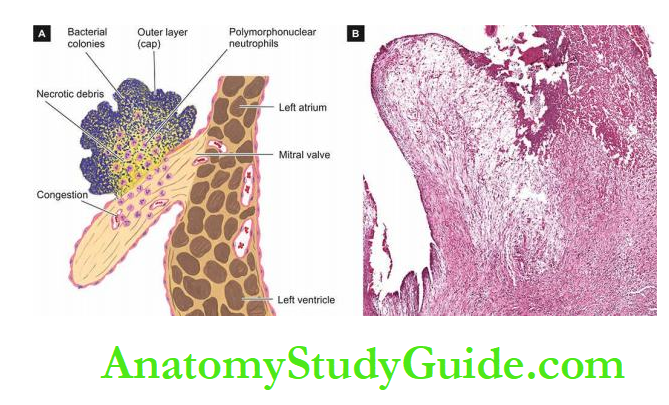 .
.
2. Extracardiac Lesions:
Patients of the syndrome of acute rheumatism develop lesions in connective tissue elsewhere in the body, chiefly the joints, subcutaneous tissue, arteries, brain and lungs.
1. Polyarthritis: Acute and painful inflammation of the synovial membranes of some of the joints, especially the larger joints of the limbs, is seen in about 90% of cases of RF in adults and
less often in children.
As pain and swelling subside in one joint, others tend to get involved, producing the characteristic ‘migratory polyarthritis’ involving two or more joints at a time.
Histologically, the changes are transitory. The synovial membrane and the periarticular connective tissue show hyperaemia, oedema, fibrinoid change and neutrophilic infiltration.
Sometimes, focal lesions resembling Aschoff bodies are observed. A serous effusion into the joint cavity is commonly present.
2. Subcutaneous Nodules:
The subcutaneous nodules of RF occur more often in children than in adults. These nodules are small (0.5 to 2 cm in diameter), spherical or ovoid and painless.
They are attached to deeper structures like tendons, ligaments, fascia or periosteum and therefore often remain unnoticed by the patient.
Characteristic locations are extensor surfaces of the wrists, elbows, ankles and knees.
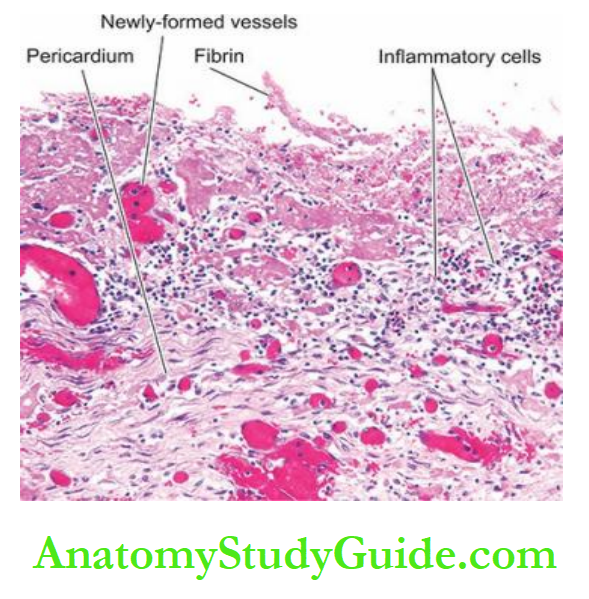
Histologically, the subcutaneous nodules of RF are representative of giant Aschoff bodies of the heart.
They consist of 3 distinct zones:
a central area with fibrinoid changes, surrounded by a zone of histiocytes and fibroblasts forming a palisade arrangement,
And the outermost zone of connective tissue which is infiltrated by non-specific chronic inflammatory cells and proliferating blood vessels.
It may be mentioned here that histologically similar but clinically different subcutaneous lesions appear in rheumatoid arthritis; they are larger, painful and tender and persist for months to years.
3. Erythema Marginatum: This non-pruritic erythematous rash is characteristic of RF.
The lesions occur mainly on the trunk and proximal parts of the extremities.
The erythematous area develops central clearing and has slightly elevated red margins. The erythema is transient and migratory.
4. Rheumatic Arteritis: Arteritis in RF involves not only the coronary arteries and the aorta but also occurs in arteries of various other organs such as renal, mesenteric and cerebral
arteries.
The lesions in the coronaries are seen mainly in the small intramyocardial branches.
Histologically, the lesions may be like those of hypersensitivity angiitis or sometimes may resemble polyarteritis nodosa.
Occasionally, foci of fibrinoid necrosis or ill-formed Aschoff bodies may be present close to the vessel wall.
5. Chorea Minor: Chorea minor or Sydenham’s chorea or Saint Vitus dance is a delayed manifestation of RF as a result of the involvement of the central nervous system.
The condition is characterised by disordered and involuntary jerky movements of the trunk and the extremities accompanied by some degree of emotional instability.
The condition occurs more often at younger ages, particularly in girls.
Histologically, the lesions are located in the cerebral hemispheres, brainstem and basal ganglia.
They consist of small haemorrhages, oedema and perivascular infiltration of lymphocytes. There may be endarteritis obliterans and thrombosis of cortical and meningeal vessels.
6. Rheumatic Pneumonitis And Pleuritis: Involvement of the lungs and pleura occurs rarely in RF.
Pleuritis is often accompanied by serofibrinous pleural effusion but definite Aschoff bodies are not present. In rheumatic pneumonitis, the lungs are large, firm and rubbery.
Histologically, the changes are oedema, capillary haemorrhages and focal areas of fibrinous exudate in the alveoli. Aschoff bodies are generally not found.
Clinical Features, Diagnosis And Prognosis
The first attack of acute RF generally appears 2 to 3 weeks after streptococcal pharyngitis, most often in children between the age of 5 to 15 years.
With subsequent streptococcal pharyngitis, there is reactivation of the disease and similar clinical manifestations appear with each recurrent attack.
The disease generally presents with migratory polyarthritis and fever.
However, RF has widespread systemic involvement and no single specific laboratory diagnostic test is available.
As per revised WHO criteria (2004) based on revised Jones’criteria (first described by Dr TD Jones in 1944, and last revised in 1992),
Following major and minor criteria and some supportive evidence of preceding infection are included for diagnosis:
1. Major criteria:
- Carditis (50-60% cases)
- Polyarthritis (60-75% cases)
- Chorea (Sydenham’s chorea) (2-30% cases)
- Erythema marginatum (<5%)
- Subcutaneous nodules (<5%)
2. Minor criteria:
- Fever
- Polyarthralgia
- Previous history of RF
- Laboratory findings: elevated ESR, raised C-reactive protein, and leucocytosis
- ECG finding of prolonged PR interval.
3. Supportive evidence of group A streptococcal infection in the preceding 45 days
- Positive throat culture for group A streptococci
- Raised titres of streptococcal antibodies (antistreptolysin O and S, anti streptokinase, antistreptohyaluronidase and anti-DNAase B).
Clinical diagnosis of RF and RHD is made in a case with antecedent laboratory evidence of streptococcal throat infection in the presence of any two of the major criteria, or the occurrence of one major and two minor criteria.
If the heart is spared in a case of acute RF, the patient may have complete recovery without any sequelae.
However, once the heart is involved, it is often associated with reactivation and recurrences of the disease.
Myocarditis, in particular, is the most life-threatening due to the involvement of the conduction system of the heart and results in serious arrhythmias.
The long-term sequelae or stigmata are chronic valvular deformities, especially mitral stenosis, as already just explained.
Initially, a state of compensation occurs, while later decompensation of the heart leads to full-blown cardiac failure.
Currently, surgical replacement of the damaged valves can alter the clinical course of the disease.
The major causes of death in RHD are cardiac failure, bacterial endocarditis and embolism:
Cardiac failure is the most common cause of death from RHD.
In young patients, cardiac failure occurs due to chronic valvular deformities, while in older patients coronary artery disease may be superimposed on the old RHD.
Bacterial endocarditis of both acute and subacute types may supervene due to inadequate use of antibiotics.
Embolism in RHD originates most commonly from mural thrombi in the left atrium and its appendages, in association with mitral stenosis.
The organs most frequently affected are the brain, kidneys, spleen and lungs.
Sudden death may occur in RHD as a result of a ball thrombus in the left atrium or due to acute coronary insufficiency in association with aortic stenosis.
Rheumatic Fever and Rheumatic Heart Disease
Rheumatic fever is a systemic, post-streptococcal, non-suppurative inflammatory disease, principally affecting the heart, joints, central nervous system, skin and subcutaneous tissues.
Chronic stage of RF involves all the layers of the heart (pancarditis) causing major cardiac sequelae referred to as rheumatic heart disease (RHD).
The disease is more common in children between the age of 5 to 15 years.
There are 3 groups of factors in the aetiology and pathogenesis of RF and RHD:
environmental factors, host susceptibility and immunologic evidence.
Pathognomonic feature of pancarditis in RF is the presence of distinctive Aschoff nodules or Aschoff bodies.
The chronic stage of RHD is characterised by a permanent deformity of one or more valves, especially the mitral (alone or with other valves) and aortic.
Patients of RF-RHD may develop extra-cardiac lesions in connective tissue elsewhere in the body, chiefly the joints, subcutaneous tissue, arteries, brain and lungs.
Diagnosis of RHD is made by WHO criteria based on revised Jones criteria consisting of some major and some minor clinical and laboratory features; major criteria are carditis, polyarthritis, chorea, erythema marginatum and subcutaneous nodules.
Non-Rheumatic Endocarditis
Inflammatory involvement of the endocardial layer of the heart is called endocarditis.
Though in common usage, if not specified endocarditis would mean inflammation of the valvular endocardium, several workers designate endocarditis on the basis of the anatomic area of the involved endocardium such as:
valvular for the valvular endocardium, mural for the inner lining of the lumina of cardiac chambers, chordal for the endocardium of the chordae tendineae, trabecular for the endocardium of trabeculae carnage, and papillary for the endocardium covering the papillary muscles.
Endocarditis can be broadly grouped into non-infective and infective types-
Most types of endocarditis are characterised by the presence of ‘vegetations’ or ‘verrucae’ which have distinct features.
Atypical Verrucous (Libman-Sacks) Endocarditis
Libman and Sacks, two American physicians, described a form of endocarditis in 1924 that is characterised by sterile endocardial vegetations which are distinguishable from the vegetations of RHD and bacterial endocarditis.
1. Non-Infective:
- Rheumatic endocarditis
- Atypical verrucous (Libman-Sacks) endocarditis
- Non-bacterial thrombotic (cachectic, marantic) endocarditis
2. Infective:
- Bacterial endocarditis
- Other infective types (tuberculous, syphilitic, fungal, viral, rickettsial)
Etiopathogenesis:
Atypical verrucous endocarditis is one of the manifestations of ‘collagen diseases.
Characteristic lesions of Libman-Sacks endocarditis are seen in 50% of cases of acute systemic lupus erythematosus (SLE); other diseases associated with this form of
endocarditis is systemic sclerosis, thrombotic thrombocytopenic purpura (TTP) and other collagen diseases.
Morphologic Features:
Grossly, characteristic vegetations occur most frequently on the mitral and tricuspid valves.
The vegetations of atypical verrucous endocarditis are small (1 to 4 mm in diameter), granular, and multiple and tend to occur on both surfaces of affected valves, in the valve pockets and on the adjoining ventricular and atrial endocardium.
The vegetations are sterile unless superimposed by bacterial endocarditis.
Unlike vegetations of RHD, the healed vegetations of Libman-Sacks endocarditis do not produce any significant valvular deformity.
Frequently, fibrinous or serofibrinous pericarditis with pericardial effusion is associated.
Microscopically, the verrucae of Libman-Sacks endocarditis are composed of fibrinoid material with superimposed fibrin and platelet thrombi.
The endocardium underlying the verrucae shows characteristic histological changes which include fibrinoid necrosis, proliferation of capillaries and infiltration by histiocytes, plasma cells, lymphocytes, neutrophils and the pathognomonic haematoxylin bodies of Gross which are counterparts of LE cells of the blood.
Similar inflammatory changes may be found in the interstitial connective tissue of the myocardium.
The Aschoff bodies are never found in the endocardium or myocardium.
Non-Bacterial Thrombotic (Cachectic, Marantic) Endocarditis:
Non-bacterial thrombotic, cachectic, marantic or terminal endocarditis or endocarditis simplex is an involvement of the heart valves by sterile thrombotic vegetations.
Etiopathogenesis:
The exact pathogenesis of lesions in non-bacterial thrombotic endocarditis (NBTE) is not clear.
Vegetations are found at autopsy in 0.5 to 5% of cases.
Following diseases and conditions are frequently associated with their presence:
1. In patients having hypercoagulable state from various etiologies e.g. advanced cancer (in 50% cases of NBTE), especially mucinous adenocarcinomas, chronic tuberculosis, renal failure and chronic sepsis.
In view of its association with chronic debilitating and wasting diseases, alternate names for NBTE such as ‘cachectic’, ‘marantic’ and ‘terminal’ endocarditis are used
synonymously.
2. Occurrence of these lesions in young and well-nourished patients is explained on the basis of alternative hypotheses such as allergy,
vitamin C deficiency, deep vein thrombosis, and endocardial trauma (e.g. due to a catheter in the pulmonary artery and haemodynamic trauma to the valves).
Morphologic Features:
Grossly, the verrucae of NBTE are located on cardiac valves, chiefly mitral, and less often aortic and tricuspid valves.
These verrucae are usually small (1 to 5 mm in diameter), single or multiple, brownish and occur along the line of closure of the leaflets but are more friable than the vegetations of RHD.
Organised and healed vegetations appear as fibrous nodules.
The normal age-related appearance of tag-like appendage at the margin of the valve cusps known as ‘Lambl’s excrescences’ is an example of such healed lesions.
Microscopically, the vegetations in NBTE are composed of fibrin along with entangled RBCs, WBCs and platelets.
Vegetations in NBTE are sterile, and bland and do not cause tissue destruction.
The underlying valve shows swollen collagen, fibrinoid change and capillary proliferation but does not show any inflammatory infiltrate.
The embolic phenomenon is seen in many cases of NBTE and results in infarcts in the brain, lungs, spleen and kidneys. The bland vegetations of NBTE on infection may produce infective endocarditis.
Infective (Bacterial) Endocarditis:
Definition:
Infective or bacterial endocarditis (IE or BE) is a serious infection of the valvular and mural endocardium caused by different forms of microorganisms and is characterised by
typical infected and friable vegetation.
A few specific forms of IE are named by the microbial etiologic agent causing them e.g. tubercle bacilli, fungi etc.
Depending upon the severity of the infection, BE is subdivided into 2 clinical forms:
1. Acute bacterial endocarditis (ABE): is a fulminant and destructive acute infection of the endocardium by highly virulent bacteria in a previously normal heart and almost invariably runs a rapidly fatal course in a period of 2-6 weeks.
2. Subacute bacterial endocarditis (SABE) or endocarditis lenta: (lenta = slow) is caused by less virulent bacteria in a previously diseased heart and has a gradual downhill course in a period of 6 weeks to a few months and sometimes years.
Although the classification of bacterial endocarditis into acute and subacute forms has been largely discarded because the clinical course is altered by antibiotic treatment, still a few important distinguishing features are worth noting.
However, features of the vegetation in the two forms of BE are difficult to distinguish.
Incidence:
The introduction of antibiotic drugs has helped greatly in lowering the incidence of BE as compared with its incidence in the pre-antibiotic era.
Though BE may occur at any age, most cases of ABE as well as SABE occur over 50 years of age.
Males are affected more ten than females.
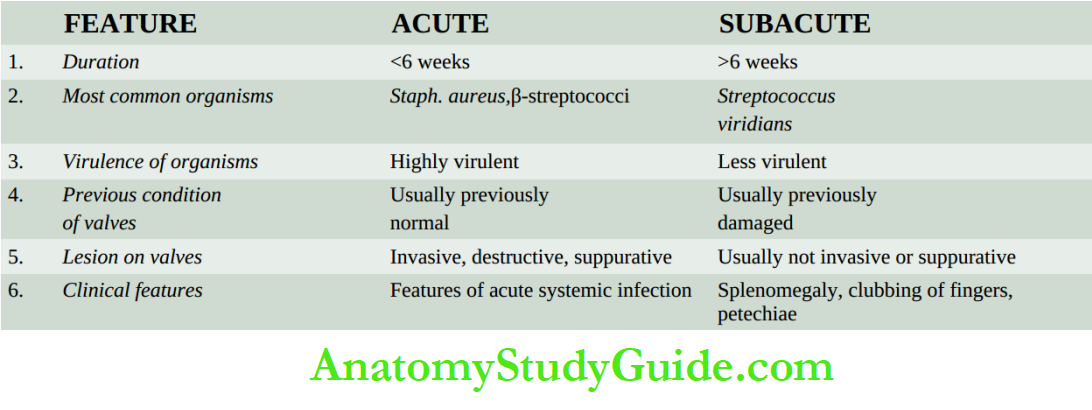
Etiology:
All cases of BE are caused by infection with microorganisms in patients having certain predisposing factors.
1. Infective agents: About 90% of cases of BE are caused by streptococci and staphylococci.
In ABE, the most common causative organisms are virulent strains of staphylococci, chiefly Staphylococcus aureus.
Others are pneumococci, gonococci, β-streptococci and enterococci.
In SABE, the commonest causative organisms are the streptococci with low virulence, predominantly Streptococcus viridans, which form part of the normal flora of the mouth and pharynx.
Other less common etiologic agents include other strains of streptococci and staphylococci (e.g. Streptococcus bovis which is the normal inhabitant of the gastrointestinal tract,
Streptococcus pneumoniae, and Staphylococcus epidermidis which is a commensal of the skin), gram-negative enteric bacilli (e.g. E. coli, Klebsiella, Pseudomonas and Salmonella), pneumococci, gonococci and Haemophilus influenzae.
2. Predisposing factors: There are 3 main types of factors which predispose to the development of both forms of BE:
- Conditions initiating transient bacteraemia, septicaemia and pyaemia.
- Underlying heart disease.
- Impaired host defences.
1. Bacteraemia, septicaemia and pyaemia Bacteria gain entry to the bloodstream causing transient and clinically silent bacteraemia in a variety of day-to-day procedures as well as from other sources of infection. Some of the common examples are:
- Periodontal infections such as trauma from vigorous brushing of teeth, hard chewing, tooth extraction and other dental procedures.
- Infections of the genitourinary tract such as in catheterisation, cystoscopy, and obstetrical procedures including normal delivery and abortions.
- Infections of the gastrointestinal and biliary tract.
- Surgery of the bowel, biliary tract and genitourinary tracts.
- Skin infections such as boils, carbuncles and abscesses.
- Upper and lower respiratory tract infections including bacterial pneumonia.
- Intravenous drug abuse.
Cardiac catheterisation and cardiac surgery for implantation of prosthetic valves.
2. Underlying heart disease SABE occurs much more frequently in previously diseased heart valves, whereas ABE is common in the previously normal heart.
Amongst the commonly associated underlying heart diseases are the following:
- Chronic rheumatic valvular disease in about 50% of cases.
- Congenital heart disease in about 20% of cases. These include VSD, subaortic stenosis, pulmonary stenosis, bicuspid aortic valve, coarctation of the aorta, and PDA.
- Other causes are syphilitic aortic valve disease, atherosclerotic valvular disease, floppy mitral valve, and prosthetic heart valves.
3. Impaired host defences All conditions in which there is depression of specific immunity, deficiency of complement and defective phagocytic function, predispose to BE.
Following are some examples of such conditions:
- Impaired specific immunity in lymphomas.
- Leukaemias.
- Cytotoxic therapy for various forms of cancers and transplant patients.
- Deficient functions of neutrophils and macrophages.
Pathogenesis:
Bacteria causing BE on entering the bloodstream from any of the abovementioned routes are implanted on the cardiac valves or mural endocardium because they have surface adhesion molecules which mediate their adherence to the injured endocardium.
There are several predisposing conditions which explain the development of bacterial implants on the valves:
1. The circulating bacteria are lodged much more frequently on previously damaged valves from diseases, chiefly RHD, congenital heart diseases and prosthetic valves, than on healthy valves.
2. Conditions producing haemodynamic stress on the valves are liable to cause damage to the endothelium, favouring the formation of platelet-fibrin thrombi which get infected from circulating bacteria.
3. Another alternative hypothesis is the occurrence of non-bacterial thrombotic endocarditis from prolonged stress which is followed by bacterial contamination.
Morphologic Features:
The characteristic pathologic feature in both ABE and SABE is the presence of typical vegetations or verrucae on the valve cusps or leaflets, and less often, on the mural endocardium, which is quite distinct from other types.
A summary of the distinguishing features of the principal types of vegetation.
Grossly, the lesions are found commonly on the valves of the left heart, most frequently on the mitral, followed in descending frequency, by the aortic, simultaneous involvement of both mitral and aortic valves, and quite rarely on the valves of the right heart.
The vegetations in SABE are more often seen on previously diseased valves, whereas the vegetation of ABE is often found on previously normal valves.
Like in RHD, the vegetations are often located on the atrial surface of atrioventricular valves and the ventricular surface of the semilunar valves.
They begin from the contact areas of the valve and may extend along the surface of the valves and onto the adjacent endocardium.
The vegetations of BE vary in size from a few millimetres to several centimetres, greytawny to greenish, irregular, single or multiple, and typically friable.
They may appear flat, filiform, fungating or polypoid. The vegetations in ABE tend to be bulkier and more globular than those of SABE and are located more often on previously normal valves, may cause ulceration or perforation of the underlying valve leaflet, or may produce myocardial abscesses.
Microscopically, the vegetation of BE consists of 3 zones:
- The outer layer or cap consists of eosinophilic material composed of fibrin and platelets.
- Underneath this layer is the basophilic zone containing colonies of bacteria.
- However, a bacterial component of the vegetation may be lacking in treated cases.
- The deeper zone consists of non-specific inflammatory reactions in the cusp itself, and in the case of SABE, there may be evidence of repair.
In the acute fulminant form of the disease, the inflammatory cell infiltrate chiefly consists of neutrophils and is accompanied by tissue necrosis and abscesses in the valve rings and in the myocardium.
In the subacute form, there is healing by granulation tissue, mononuclear inflammatory cell infiltration and fibroblastic proliferation.
Histological evidence of pre-existing valvular disease such as RHD may be present in SABE.
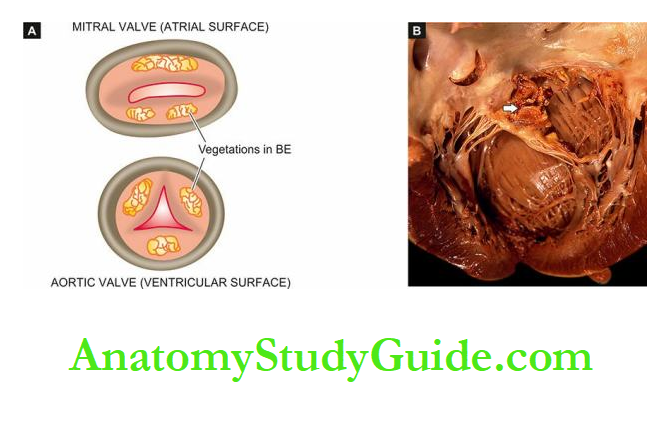
Clinical Features And Complications: Most cases of BE present with fever.
The acute form of BE is characterised by high-grade fever, chills, weakness and malaise while the subacute form of the disease has non-specific manifestations like slight fever, fatigue, loss of weight and flu-like symptoms.
Although the diagnosis of BE is confirmed by vegetation, criteria for clinical diagnosis have been proposed called modified Duke criteria.
1. Modified Duke criteria:
It includes major and minor criteria; the presence of two major and three minor criteria establishes the diagnosis of BE.
1. Major criteria These are:
Blood culture: from two separate blood cultures positive for typical microorganisms of BE (Streptococcus viridans, Streptococcus bovis, Staphylococcus aureus, or community-acquired enterococci, or persistently positive blood culture taken 12 hours apart.
Evidence of endocardial involvement: by positive ECG changes for IE, intracardiac mass on valve or on supportive tissue, abscess or new partial dehiscence of the prosthetic valve, or new valvular regurgitation.
2. Minor criteria These include:
- Predisposing heart condition
- Fever >100.4oF
- The major vascular embolic phenomenon
- Immunologic phenomenon (e.g. glomerulonephritis, Osler’s nodes)
- Microbial evidence by positive blood culture but meeting major criteria
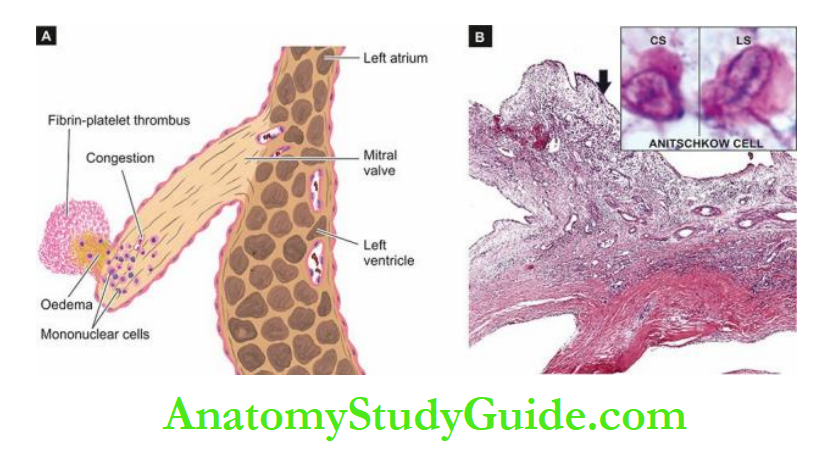
2. Complications: In the early stage, the lesions are confined to the heart, while the subsequent progression of the disease leads to the involvement of extra-cardiac organs.
In general, severe complications develop early in ABE than in SABE.
Complications and sequelae of BE are divided into cardiac and extracardiac:
1. Cardiac complications: These include the following:
- Valvular stenosis or insufficiency
- Perforation, rupture, and aneurysm of valve leaflets
- Abscesses in the valve ring
- Myocardial abscesses
- Suppurative pericarditis
- Cardiac failure from one or more of the foregoing complications.
2. Extracardiac complications Since the vegetations in BE are typically friable, they tend to get dislodged due to the rapid stream of blood and give rise to embolism which is responsible for very common and serious extra-cardiac complications.
These are as follows:
Emboli originating from the left side of the heart and entering the systemic circulation affect organs like the spleen, kidneys, and brain causing infarcts, abscesses and mycotic aneurysms.
Emboli arising from the right side of the heart enter the pulmonary circulation and produce pulmonary abscesses.
Petechiae may be seen in the skin and conjunctiva due to either emboli or toxic damage to the capillaries.
In SABE, there are painful, tender nodules on the fingertips of hands and feet called Osler’s nodes, while in ABE there is the appearance of painless, non-tender subcutaneous maculopapular lesions on the pulp of the fingers called Janeway’s lesions.
In either case, their origin is due to toxic or allergic inflammation of the vessel wall.
Focal necrotising glomerulonephritis is seen more commonly in SABE than in ABE.
Occasionally diffuse glomerulonephritis may occur. Both these have their pathogenesis in circulating immune complexes (hypersensitivity phenomenon).
Treatment of BE with antibiotics in adequate dosage kills the bacteria but complications and sequelae of healed endocardial lesions may occur even after successful therapy.
The causes of death are cardiac failure, persistent infection, embolism to vital organs, renal failure and rupture of mycotic aneurysm of cerebral arteries.
Specific Types Of Infective Endocarditis
Besides BE, various other types of microbes may occasionally produce infective endocarditis which is named according to the etiologic agent causing it.
These include the following:
1. Tuberculous endocarditis: Though tubercle bacilli are bacteria, tuberculous endocarditis is described as separate from bacterial endocarditis due to specific granulomatous inflammation found in tuberculosis.
It is characterised by the presence of typical tubercles on the valvular as well as mural endocardium and may form tuberculous thromboembolic.
2. Syphilitic endocarditis: The endocardial lesions in syphilis have already been described in relation to syphilitic aortitis.
The severest manifestation of cardiovascular syphilis is aortic valvular incompetence.
3. Fungal endocarditis: Rarely, endocardium may be infected with fungi such as Candida albicans, Histoplasma capsulatum, Aspergillus, Mucor, coccidioidomycosis, cryptococcosis, blastomycosis and actinomycosis.
Opportunistic fungal infections like candidiasis and aspergillosis are seen more commonly in patients receiving long-term antibiotic therapy, intravenous drug abusers and after prosthetic valve replacement.
Fungal endocarditis produces an appearance similar to that in BE but the vegetation is bulkier in fungal endocarditis.
4. Viral endocarditis: There is only experimental evidence of the existence of this entity.
5. Rickettsial endocarditis: Another rare cause of endocarditis is infection with rickettsiae in Q fever.
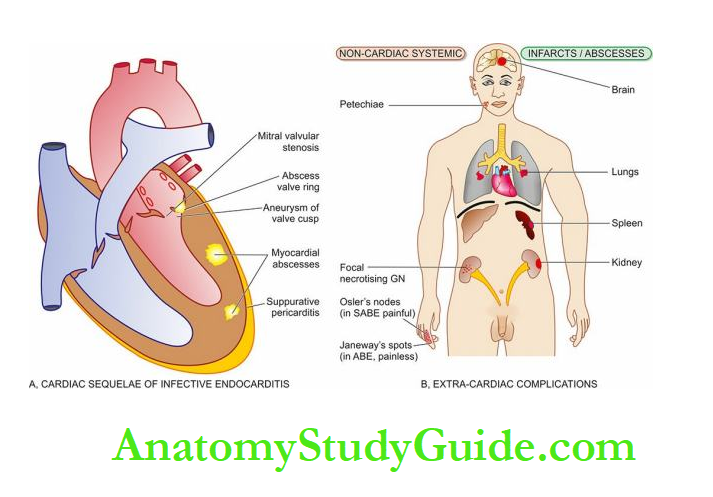
Non-rheumatic Endocarditis
Endocarditis can be non-infective and infective types; most types of endocarditis are characterised by the presence of ‘vegetations’ or ‘verrucae’.
Atypical verrucous (Libman-Sacks) endocarditis is a manifestation of collagen diseases.
Vegetations are small, granular and multiple. Non-bacterial thrombotic endocarditis (NBTE) is an involvement of the heart valves by sterile thrombotic vegetations, often preceded by a hypercoagulable state.
Infective or bacterial endocarditis (IE or BE) occurs following conditions initiating transient bacteraemia, septicaemia and pyaemia, underlying heart disease and impaired host defences.
It is characterised by typical infected, large and friable vegetation.
Since vegetations in BE are typically friable, they tend to get dislodged due to the rapid stream of blood and give rise to embolism and cause serious extra-cardiac complications.
Clinical diagnosis of IE is made by modified Duke criteria:
major criteria are positive blood culture for organisms of IE and evidence in support of endocardial damage, while minor criteria include predisposing conditions, fever and evidence of vascular, immunologic and microbial infection.
A few other uncommon forms of IE are tuberculous, syphilitic, fungal and viral endocarditis
Valvular Diseases And Deformities
Valvular diseases are various forms of congenital and acquired diseases which cause valvular deformities.
Many of them result in cardiac failure. Rheumatic heart disease is the most common form of acquired valvular disease.
Valves of the left side of the heart are involved much more frequently than those of the right side of the heart.
The mitral valve is affected most often, followed in descending frequency, by the aortic valve, and combined mitral and aortic valves.
The valvular deformities may be of 2 types: stenosis and insufficiency:
- Stenosis is the narrowing of the valvular opening from whatever cause. Resultantly, it fails to open fully and obstructs the forward flow of the blood.
- Insufficiency or incompetence or regurgitation is due to damaged valvular cusps or its ring. The result is the failure of the valve to close tightly and thus blood flows backwards.
Congenital valvular diseases have already been described.
Various acquired valvular diseases that may deform the heart valves are listed below:
- RHD, the commonest cause
- Infective endocarditis
- Non-bacterial thrombotic endocarditis
- Libman-Sacks endocarditis
- Syphilitic valvulitis
- Calcific aortic valve stenosis
- Calcification of mitral annulus
- Myxomatous degeneration (floppy valve syndrome)
- Carcinoid heart disease.
Out of this list, major forms of vegetative endocarditis involving the valves have already been described.
Others along with the consequences of these valvular diseases in the form of stenosis and insufficiency of the heart valves are described below.
Mitral Stenosis
Mitral stenosis occurs in approximately 40% of all patients with RHD.
About 70% of the patients are women. The latent period between rheumatic carditis and the development of symptomatic mitral stenosis is about two decades.
Etiology
The most common causes are:
- Rheumatic origin, the most common
- Congenital parachute mitral valve
- Sever mitral annular calcification
- Libman-Sacks endocarditis
Morphologic Features:
The appearance of the mitral valve in stenosis varies according to the extent of involvement.
Generally, the valve leaflets are diffusely thickened by fibrous tissue and/or calcific deposits, especially towards the closing margin.
There are fibrous adhesions of mitral commissures and fusion and shortening of chordae tendineae.
In less extensive involvement, the bases of the leaflets of the mitral valve are mobile while the free margins have puckered and thickened tissue with narrowed orifice; this is called ‘pursestring puckering’.
The more advanced cases have rigid, fixed and immobile diaphragm-like valve leaflets with narrow, slit-like or oval mitral openings, commonly referred to as ‘buttonhole’ or ‘fish-mouth’ mitral orifice.
Effects: In normal adults, the mitral orifice is about 5 cm2.
Symptomatic mitral stenosis develops if the valve opening is reduced to 1 cm2 resulting in significant elevation of left atrial pressure from the normal of 12 mmHg to about 25 mmHg leading to dilatation of the left atrium.
Elevated left atrial pressure, in turn, raises the pressure in the pulmonary veins and capillaries, reducing pulmonary function and causing exertional dyspnoea which is the chief symptom of mitral stenosis.
The effects of mitral stenosis can thus be summarised as under:
- Dilatation and hypertrophy of the left atrium.
- Normal-sized or atrophic left ventricle due to reduced inflow of blood.
- Pulmonary hypertension resulting from the passive backward transmission of elevated left partial pressure which causes:
-
- chronic passive congestion of the lungs,
- hypertrophy and dilatation of the right ventricle, and
- dilatation of the right atrium when right heart failure supervenes.
Mitral Insufficiency
Mitral insufficiency is caused by RHD in about 50% of patients but in contrast to mitral stenosis, pure mitral insufficiency occurs more often in men (75%).
Subsequently, mitral insufficiency is associated with some degree of mitral stenosis.
Etiology: All the causes of mitral stenosis may produce mitral insufficiency; besides there are other causes.
- RHD, a most common cause
- Non-inflammatory calcification of mitral valve annulus (in the elderly)
- Myxomatous transformation of the mitral valve (floppy valve syndrome)
- Healed endocarditis rupture
- Congenital
- Rupture of a leaflet or papillary muscle post-MI
Morphologic Features:
The appearance of the mitral valve in insufficiency varies according to the underlying cause.
The rheumatic process produces rigidity, deformity and retraction of the valve leaflets and fusion of commissures as well as shortening and fusion of chordae tendineae.
- In myxomatous degeneration of the mitral valve leaflets (floppy valve syndrome) which is described, there is prolapse of one or both leaflets into the left atrium during
systole. - In non-inflammatory calcification of the mitral annulus seen in the aged, there is irregular, stony-hard, bead-like thickening in the region of the mitral annulus without any associated inflammatory changes. It is thought to reflect degenerative changes of ageing.
Effects: Regurgitant mitral orifice produces a progressive increase in left ventricular end-diastolic volume as well as pressure since the left ventricle cannot empty completely.
This results in a rise in left atrial pressure and dilatation.
As a consequence of left atrial hypertension, pulmonary hypertension occurs resulting in pulmonary oedema and right heart failure.
In symptomatic cases of mitral insufficiency, the major symptoms are related to decreased cardiac output
(e.g. fatigue and weakness) and due to pulmonary congestion (e.g. exertional dyspnoea and orthopnoea) but the features are less well-marked than in mitral stenosis.
The effects of mitral insufficiency are summarised as under:
- Dilatation and hypertrophy of the left ventricle.
- Marked dilatation of the left atrium.
- Features of pulmonary hypertension such as:
- Chronic passive congestion of the lungs,
- hypertrophy and dilatation of the right ventricle, and
- dilatation of the right atrium when right heart failure supervenes.
Aortic Stenosis
Aortic stenosis comprises about one-fourth of all patients with chronic valvular heart disease.
About 80% patients of with symptomatic aortic stenosis are males.
Etiology:
It is of 2 main types: non-calcific and calcific type, the latter being more common.
1. Non-calcific aortic stenosis: The most common cause of non-calcific aortic stenosis is chronic RHD.
Other causes are congenital valvular and subaortic stenosis, congenitally bicuspid aortic valve and radiation exposure.
2. Calcific aortic stenosis: Calcific aortic stenosis is a more common type.
Its various causes are the healing of the valve by scarring followed by calcification of the aortic valve such as in RHD, bacterial endocarditis,
Brucella endocarditis, Mönckeberg’s calcific aortic stenosis, and familial hypercholesterolaemic xanthomatosis.
Morphologic Features: The aortic cusps show characteristic fibrous thickening and
calcific nodularity of the closing edges. Calcified nodules are often found in the sinuses of
Valsalva.
In rheumatic aortic stenosis, the commissures are fused and calcified, while in nonrheumatic aortic stenosis, there is no commissural fusion.
Effects:
Aortic stenosis becomes symptomatic when the valve orifice is reduced to 1 cm2 from its normal 3 cm2. The symptoms appear many years later when the heart cannot compensate and the stenosis is quite severe.
The major effect of aortic stenosis is an obstruction to the outflow resulting in concentric hypertrophy of the left ventricle.
Later, when cardiac failure supervenes, there is dilatation as well as hypertrophy of the left ventricle (eccentric hypertrophy).
The three cardinal symptoms of aortic stenosis are exertional dyspnoea, angina pectoris and syncope.
Exertional dyspnoea results from an elevation of pulmonary capillary pressure.
Angina pectoris usually results from an elevation of pulmonary capillary pressure and usually develops due to increased demand for hypertrophied myocardial mass.
Syncope results from accompanying coronary insufficiency. Sudden death may also occur in an occasional case of aortic stenosis.
Aortic Insufficiency
About three-fourths of all patients with aortic insufficiency are males with some having a family history of Marfan’s syndrome.
Etiology Major causes are:
- Chronic RHD is most common, seen in 75% of cases (along with other valvular lesions)
- Congenital bicuspid valve
- Myxomatous degeneration of the aortic valve (floppy valve syndrome)
- Infective endocarditis (healed)
- Syphilitic endocarditis (healed)
- Marfan’s syndrome
- Cystic medial degeneration
- Traumatic rupture of the valve cusps
- Ankylosing spondylitis
- Hypertension
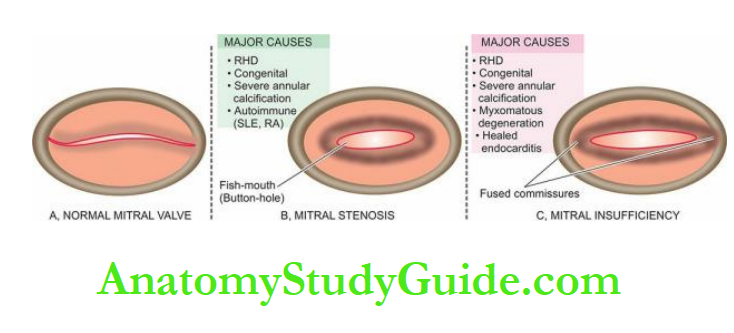
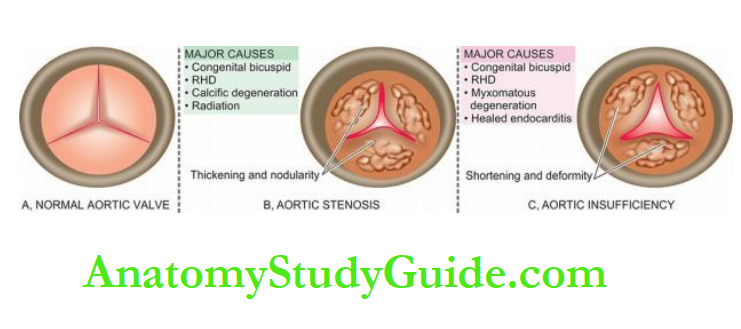
Morphologic Features: The aortic valve cusps are thickened, deformed and shortened
and fail to close. There is generally distension and distortion of the ring.
Effects:
As a result of the regurgitant aortic orifice, there is an increase in the left ventricular end-diastolic volume.
This leads to hypertrophy and dilatation of the left ventricle producing massive cardiac enlargement so that the heart may weigh as much as 1000 gm.
Failure of the left ventricle increases the pressure in the left atrium and eventually pulmonary hypertension and right heart failure occur.
The characteristic physical findings in a patient of aortic insufficiency are awareness of the beatings of the heart, poundings in the head with each heartbeat, low diastolic and high pulse pressure, rapidly rising and collapsing water hammer pulse (Corrigan’s pulse), booming ‘pistol
shot’ sound over the femoral artery and systolic and diastolic murmurs were heard over the femoral artery when it is lightly compressed (Durozier’s sign).
Sometimes, angina pectoris occurs due to increased myocardial demand or due to coronary insufficiency.
Carcinoid Heart Disease
Etilogy: Carcinoid syndrome developing in patients with extensive hepatic metastases from a carcinoid tumour is characterised by cardiac manifestations in about half the cases.
The lesions are characteristically located in the valves and endocardium of the right side of the heart. The pathogenesis of the cardiac lesions is not certain.
But in carcinoid tumours with hepatic metastasis, there is an increased blood level of serotonin secreted by the tumour.
The increased concentration of serotonin reaches the right side of the heart and causes the lesions but serotonin is inactivated on passage of the blood through the lungs and hence the left heart is relatively spared.
In addition, high levels of bradykinin may play a contributory role in carcinoid heart disease.
However, chronic infusion of serotonin or bradykinin in experimental animals has not succeeded in producing cardiac lesions; hence the exact pathogenesis of carcinoid heart disease remains obscure.
Morphologic Features:
In the majority of cases, the lesions are limited to the right side of the heart.
Both pulmonary and tricuspid valves as well as the endocardium of the right chambers show characteristic cartilage-like fibrous plaques.
Similar plaques may occur on the intima of the great veins, the coronary sinus and the great arteries.
Occasionally, the lesions may be found on the left side of the heart.
Effects: Thickening and contraction of the cusps and leaflets of the valves of the outflow tracts of the right heart result mainly in pulmonary stenosis and tricuspid regurgitation, and to a lesser extent, pulmonary regurgitation and tricuspid stenosis.
Myxomatous Degeneration Of Mitral Valve (Mitral Valve Prolapse)
Myxomatous or mucoid degeneration of the valves of the heart is a peculiar condition occurring in young patients between the age of 20 and 40 years and is more common in women.
The condition is common and seen in 5% of the general adult population.
The condition is also known by other synonyms like ‘floppy valve syndrome’ or ‘mitral valve prolapse’.
Etiology: The cause of the condition is not known but in some cases, it may be genetically determined collagen disorder.
Association with Marfan’s syndrome has been observed in 90% of patients.
Others have noted myxomatous degeneration in cases of Ehlers-Danlos syndrome and in myotonic dystrophy.
However, the myxomatous valvular changes seen in aged patients are not related to this entity.
Morphologic Features:
Any cardiac valve may be involved but the mitral valve is affected most frequently.
The disease is usually most severe and most common in the posterior leaflet of the mitral valve.
The affected leaflet shows either excessive or redundant leaflet tissue, which is opaque white, soft and floppy.
The cut section of the valve reveals a mucoid or myxoid appearance.
A significant feature is the ballooning or aneurysmal protrusion of the affected leaflet and hence the name ‘mitral valve prolapse’ and ‘floppy valve syndrome’.
Microscopically, the enlarged cusp shows loose connective tissue with abundant mucoid or myxoid material due to the abundance of mucopolysaccharide.
Effects: Usually, the condition does not produce any symptoms or significant valvular dysfunction.
The condition is recognised during life by the characteristic mid-systolic click followed by a systolic murmur due to a mildly incompetent mitral valve caused by the mitral valve prolapse.
Occasionally, complications may develop such as superimposed infective endocarditis, mitral insufficiency and arrhythmias.
Rarely, sudden death from serious ventricular arrhythmias may occur.
Valvular Diseases and Deformities:
Valvular diseases may be congenital or acquired and cause valvular deformities.
RHD is the most common form of acquired valvular disease. Valves of the left side of the heart, particularly the mitral valve, are involved much more often.
Mitral stenosis, often from RHD, is accompanied by dilatation and hypertrophy of the left atrium.
Mitral insufficiency causes dilatation and hypertrophy of the left ventricle.
In aortic stenosis, aortic cusps show fibrous thickening and calcific nodularity of the closing edges, while in aortic insufficiency aortic valve cusps are thickened, deformed and shortened and fail to close.
In carcinoid heart disease, the lesions are limited to the right side of the heart, i.e. both pulmonary and tricuspid valves.
Myxomatous degeneration of the mitral valve is associated with Marfan’s syndrome and causes mitral valve prolapse.
Other Myocardial Diseases
Involvement of the myocardium occurs in three major forms of diseases already discussed ischaemic heart disease, hypertensive heart disease and rheumatic heart disease.
In addition, there are two other broad groups of isolated myocardial diseases:
- Myocarditis i.e. inflammatory involvement of the myocardium.
- Cardiomyopathy i.e. a non-inflammatory myocardial involvement due to several causes, largely genetic causes.
Myocarditis
Inflammation of the heart muscle is called myocarditis. It is a rather common form of heart disease that can occur at any age.
Its exact incidence is difficult to ascertain as the histological examination has been largely confined to autopsy material.
Reports from different studies have estimated the incidence of myocarditis in 1 to 4% of all autopsies.
A number of classifications of myocarditis have been proposed in the past as follows:
- Interstitial and parenchymatous type, depending upon whether the inflammation is confined to interstitial tissue or the parenchyma.
- Specific and non-specific types, depending upon whether the inflammation is granulomatous or non-specific type.
- Acute, subacute and chronic type, depending upon the duration of the inflammatory response. However, currently most commonly used is etiologic.
According to this classification, myocarditis is divided into 4 main etiologic types described below.
1. Infective Myocarditis:
A number of infectious agents such as bacteria, viruses, protozoa, parasites, fungi, rickettsiae and spirochaetes may cause myocarditis by direct invasion or by their toxins.
Some of the common forms are described below.
1. Infective Myocarditis
- Viral myocarditis
- Bacterial myocarditis
- Toxic myocarditis
- Infective granulomatous myocarditis
- Syphilitic myocarditis
- Rickettsial myocarditis
- Protozoal myocarditis
- Helminthic myocarditis
- Fungal myocarditis
2. Idiopathic (Fiedler’S) Myocarditis
- Diffuse type
- Giant cell (idiopathic granulomatous) type
3. Myocarditis In Connective Tissue Diseases
- Rheumatoid arthritis
- Lupus erythematosus
- Polyarteritis nodosa
- Dermatomyositis
- Scleroderma
4. Miscellaneous Types Of Myocarditis
- Physical agents
- Chemical agents
- Drugs
- Immunologic agents
- Metabolic derangements
1. Viral Myocarditis: A number of viral infections are associated with myocarditis.
Some of the common examples are influenza, poliomyelitis, infectious mononucleosis, hepatitis, smallpox, chickenpox, measles, mumps, rubella, viral pneumonia, coxsackievirus and HIV infections.
Cardiac involvement occurs in about 5% of viral infections.
Viral myocarditis usually appears after a few days to a few weeks of viral infections elsewhere in the body.
The damage to the myocardium is caused either by direct viral cytotoxicity or by a cell-mediated immune reaction.
Regardless of the type of virus, the pathologic changes are similar.
Grossly, the myocardium is pale and flabby with dilatation of the chambers. There may be focal or patchy areas of necrosis.
Histologically, there are changes of acute myocarditis. Initially, there is oedema and infiltration of the interstitial tissue by neutrophils and lymphocytes.
Later, there is necrosis of individual myocardial fibres and the infiltrate consists of lymphocytes and macrophages.
2. Bacterial Myocarditis: Pyogenic bacteria, chiefly Staphylococcus aureus or Streptococcus pyogenes, which cause septicaemia and pyaemia may produce suppurative
myocarditis.
As already pointed out, acute bacterial endocarditis may sometimes cause bacterial myocarditis.
Grossly, there are either abscesses in the myocardium or there is diffuse myocardial involvement.
Microscopically, the exudate chiefly consists of neutrophils, admixed with lymphocytes, plasma cells and macrophages.
There may be foci of myocardial degeneration and necrosis; later there may be areas of healing by fibrosis.
3. Toxic Myocarditis: A number of acute bacterial infections produce myocarditis by toxins e.g. in diphtheria, typhoid fever and pneumococcal pneumonia.
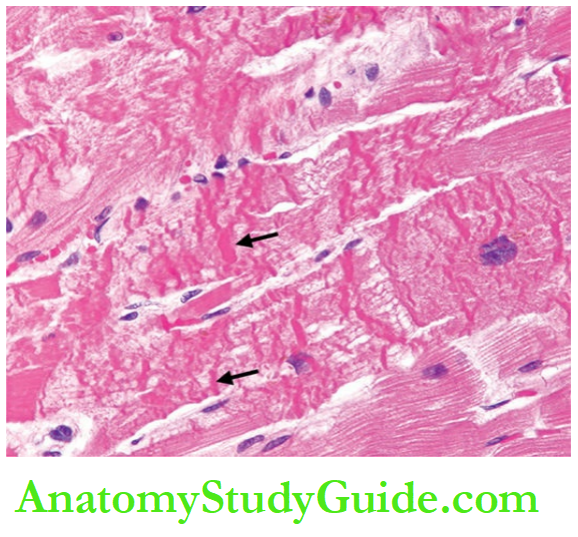
Grossly, the appearance is similar to that seen in viral myocarditis.
Histologically, there are small foci of coagulative necrosis in the muscle which are surrounded
by nonspecific acute and chronic inflammatory infiltrate.
Toxic myocarditis manifests clinically by cardiac arrhythmias or acute cardiac failure due to
the involvement of the conduction system. It may cause sudden death.
4. Granulomatous Myocarditis: Tuberculosis, brucellosis and tularaemia are some examples of bacterial infections characterised by granulomatous inflammation in the
myocardium.
Sarcoidosis, though not a bacterial infection, has a histological resemblance to other granulomatous myocarditis.
Tuberculous myocarditis is rare and occurs either by haematogenous spread or by extension from tuberculous pericarditis.
The condition must be distinguished from idiopathic granulomatous (giant cell) myocarditis (described later).
5. Syphilitic Myocarditis: Syphilitic involvement of the myocardium may occur in 2 forms—a gummatous lesion consisting of granulomatous inflammation which is more common, and primary non-specific myocarditis which is rare.
The syphilitic gummas in the myocardium may be single or multiple and may be grossly discernible.
The gummas may affect the conduction system of the heart.
6. Rickettsial Myocarditis: Myocarditis occurs quite frequently in scrub typhus (R.tsutsugamushi) and Rocky Mountain typhus fever caused by spotted rickettsia.
Microscopically, there is interstitial oedema and focal or patchy infiltration by inflammatory cells which include lymphocytes, plasma cells, macrophages, mast cells and eosinophils but necrosis and degeneration are generally not present.
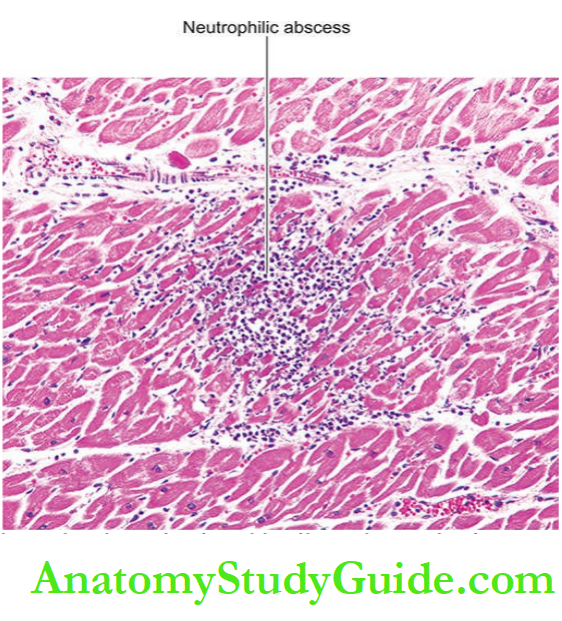
7. Protozoal Myocarditis: Chagas’ disease and toxoplasmosis are the two protozoal diseases causing myocarditis.
Chagas’ disease caused by Trypanosoma cruzi frequently attacks the myocardium besides involving the skeletal muscle and the central nervous system.
Toxoplasmosis caused by the intracellular protozoan, Toxoplasma gondii, sometimes causes myocarditis in children and adults.
Microscopically, both these conditions show focal degeneration and necrosis of the myocardium, oedema and cellular infiltrate consisting of histiocytes, plasma cells, lymphocytes and a few polymorphs.
The organisms are found in the muscle fibres.
8. Helminthic Myocarditis: Echinococcus granulosus and Trichinella spiralis are the two intestinal helminths which may cause myocarditis.
Echinococcus rarely produces hydatid cysts in the myocardium while the larvae of Trichinella in trichinosis cause heavy inflammation in the myocardium as well as in the interstitial tissue.
9. Fungal Myocarditis: Patients with immunodeficiency, cancer and other chronic debilitating diseases are more prone to develop fungal myocarditis.
These include candidiasis, aspergillosis, blastomycosis, actinomycosis, cryptococcosis, coccidioidomycosis and histoplasmosis.
2. Idiopathic (Fiedler’s) Myocarditis:
Idiopathic or Fiedler’s myocarditis is an isolated myocarditis unaccompanied by inflammatory changes in the endocardium or pericardium and occurs without the usual apparent causes.
The condition is rapidly progressive and causes sudden severe cardiac failure or sudden death.
Grossly, the heart is soft and flabby. The cardiac chambers are generally dilated and sometimes show hypertrophy.
There are yellow-grey focal lesions throughout the myocardium. Mural thrombi are commonly present.
Histologically, two forms of idiopathic myocarditis are described: diffuse type and giant cell (idiopathic granulomatous) type.
Diffuse type is more common of the two. It is characterised by diffuse non-specific inflammatory infiltrate consisting of lymphocytes, plasma cells, macrophages, eosinophils and a few polymorphs in the interstitial tissue without the formation of granulomas. Late stage shows healing by fibrosis.
Giant cell type or idiopathic granulomatous type is characterised by the formation of noncaseating granulomas consisting of macrophages, lymphocytes, plasma cells and multinucleate giant cells.
The giant cells are of a foreign body or Langhans’ type or of myogenic origin. The granulomas do not show the presence of acid-fast bacilli or spirochaetes.
Some have suggested the relationship of this condition with sarcoidosis but sarcoid granulomas are known to occur in the myocardium secondary to generalised sarcoidosis.
3. Myocarditis In Connective Tissue Diseases:
Inflammatory involvement of the myocardium occurs in a number of connective tissue diseases such as rheumatoid arthritis, lupus erythematosus, polyarteritis nodosa, dermatomyositis and scleroderma.
The pathologic changes in the heart muscle are similar to the changes seen in other organs in these conditions as described elsewhere in relevant chapters.
4. Miscellaneous Types Of Myocarditis:
Apart from the above forms of myocarditis, the miscellaneous group consists of myocarditis caused by a variety of agents—physical and chemical agents, drugs and metabolic derangements.
1. Physical agents: Physical agents like contusion of the myocardium, heat stroke, cardiac surgery and irradiation can initiate non-specific myocarditis.
The features consist of an infiltrate of neutrophils, eosinophils and mononuclear cells and show contraction-band necrosis of the myocardial fibres.
2. Chemical agents: Toxic chemicals such as arsenic, phosphorus and carbon monoxide cause focal areas of degeneration and necrosis of myocardial fibres and non-specific inflammatory reactions, chiefly consisting of lymphocytes and macrophages.
3. Drugs: Changes similar to those induced by chemical poisons are produced by certain drugs such as phenothiazine compounds, sulfonamides, catecholamines and cytotoxic compounds.
4. Immunologic agents: Myasthenia gravis, Friedreich’s ataxia, and progressive muscular dystrophies initiate a state of auto-immunisation against the myocardium resulting in focal myocardial degeneration and necrosis with secondary inflammatory reaction. Later, there may be myocardial fibrosis.
5. Metabolic derangements: Uraemia, hypokalaemia and shock are associated with degeneration and necrosis of the myocardial fibres, oedema of the interstitial tissue and nonspecific inflammatory reaction.
Cardiomyopathy
Definition And Classification
Cardiomyopathy (CMP) literally means disease of the heart muscle but the term was originally coined to restrict its usage to myocardial disease of unknown cause.
However, the term cardiomyopathy has been loosely used by various workers for myocardial diseases of known etiology as well e.g. alcoholic CMP, amyloid CMP, ischaemic CMP etc.
By definition, CMPs are a heterogeneous group of myocardial diseases which are:
- associated with mechanical and/or electrical dysfunction,
- have morphological features in the myocardium, and
- are due to several causes which are frequently genetic.
Based on this definition, the classification of CMPs and their subtypes.
There are three pathophysiologic categories of CMPs:
- Dilated (congestive) cardiomyopathy
- Hypertrophic cardiomyopathy
- Restrictive or obliterative or infiltrative cardiomyopathy
Dilated (Congestive) Cmp:
This type of CMP is characterised by gradually progressive cardiac failure along with dilatation of all four chambers of the heart.
The condition occurs more often in adults and the average survival from onset to death is less than 5 years.
Etiology A few hypotheses based on associations with the following conditions have been proposed:
A possible association of viral myocarditis (especially coxsackievirus B) with dilated CMP to the presence of viral nucleic acids in the myocardium, has been noted.
Association with toxic damage from cobalt and chemotherapy with doxorubicin and other anthracyclines is implicated in some cases.
Inherited mutations have been implicated due to the occurrence of disease in families.
Mutations in certain sarcomere proteins such as cardiac troponin-T and I, β-and α-myosin, and α-cardiac actin have been observed.
Abnormality in protein due to mutations causes contractile dysfunction.
Another mutation in the cytoskeletal protein dystrophin gene on X-chromosome has been held responsible for muscular dystrophy as well as dilated cardiomyopathy.
Chronic alcoholism has been found associated with dilated cardiomyopathy.
It may be due to thiamine deficiency induced by alcohol and resulting in beriberi heart disease.
This is referred to as ‘alcoholic CMP’. However, moderate consumption of alcohol provides some cardioprotection against IHD by raising HDL cholesterol.
Another form of alcoholic CMP is associated with the consumption of large quantities of beer (beer drinkers’ myocarditis).
Cobalt added to the beer so as to improve the appearance of foam is thought to cause direct toxic injury to the heart in this condition.
Peripartum association has been observed in some cases. Poorly-nourished women may develop this form of CMP within a month before or after delivery (peripartum CMP).
1. Dilated (Or Congestive) Cardiomyopathy
2. Hypertrophic Cardiomyopathy
- Obstructive type
- Non-obstructive type
3. Restrictive (Or Obliterative Or Infiltrative)
- Cardiomyopathy
- Cardiac amyloidosis
- Endocardial fibroelastosis
- Endomyocardial fibrosis
- Loeffler’s endocarditis (fibroblastic parietal endocarditis with peripheral blood eosinophilia)
- Other forms
Morphologic Features: Grossly, the heart is enlarged and increased in weight (up to 1000 gm).
The most characteristic feature is the prominent dilatation of all four chambers giving the heart a typical globular appearance.
Thickening of the ventricular walls even if present is masked by the ventricular dilatation. The endocardium is thickened and mural thrombi are often found in the ventricles and atria. The cardiac valves are usually normal.
Microscopically, the endomyocardial biopsies or autopsy examination of the heart reveal nonspecific and variable changes.
There may be hypertrophy of some myocardial fibres and atrophy of others.
Sometimes degenerative changes and small areas of interstitial fibrosis are found with focal mononuclear inflammatory cell infiltrate.
Hypertrophic Cmp This form of CMP is known by various synonyms like asymmetrical hypertrophy, hypertrophic subaortic stenosis and Teare’s disease.
The disease occurs more frequently between the age of 25 and 50 years.
It is often asymptomatic but becomes symptomatic due to heavy physical activity causing dyspnoea, angina, congestive heart failure and even sudden death.
Etiology Following factors have been implicated:
Autosomal dominant inheritance of the disease is available in about half the cases suggesting genetic factors in its causation.
Inherited mutations in genes encoding for sarcomere proteins have been reported in a much larger number of cases of hypertrophic CMP than those of dilated CMP.
Particularly implicated are the mutations in heavy and light chains of cardiac β-myosin, troponin-I and troponin-T.
The condition may result from myocardial ischaemia resulting in fibrosis of the intramyocardial arteries and compensatory hypertrophy.
Other contributory factors are: increased circulating levels of catecholamines, myocardial ischaemia as a result of thickened vasculature of the myocardium and abnormally increased fibrous tissue in the myocardium due to hypertrophy.
Morphologic Features: Grossly, the characteristic features are cardiac enlargement, increase in weight, normal or small ventricular cavities and myocardial hypertrophy.
The hypertrophy of the myocardium is typically asymmetrical and affects the interventricular septum more than the free walls of the ventricles.
This asymmetric septal hypertrophy may be confined to the apical region of the septum (non-obstructive type) or may extend up to the level of the mitral valve causing obstruction to left ventricular outflow in the form of subaortic stenosis (obstructive type).
The designation of rhabdomyoma of the septum has also been applied to this form of CMP.
Microscopically, the classical feature is the myocardial cell disorganisation in the ventricular
septum.
The bundles of myocardial fibres are irregularly and haphazardly arranged rather than the usual parallel pattern and are separated by bands of interstitial fibrous tissue.
The individual muscle cells show hypertrophy and large prominent nuclei.
Restrictive (Obliterative Or Infiltrative) CMP
This form of CMP is characterised by restriction in ventricular filling due to a reduction in the
the volume of the ventricle.
The common feature in this heterogeneous group of conditions producing restrictive cardiomyopathy is abnormal diastolic function.
Restrictive CMP includes the following entities:
Cardiac amyloidosis
Endocardial fibroelastosis
Endomyocardial fibrosis
Loeffler’s endocarditis (Fibroblastic parietal endocarditis with peripheral blood eosinophilia)
Other forms of restrictive cardiomyopathy.
1. Cardiac Amyloidosis:
Amyloidosis of the heart may occur in any form of systemic amyloidosis or may occur as isolated organ amyloidosis in amyloid of ageing and result in subendocardial deposits.
The deposit of amyloid occurs in the interstitium and replaces and constricts the myocardial fibres.
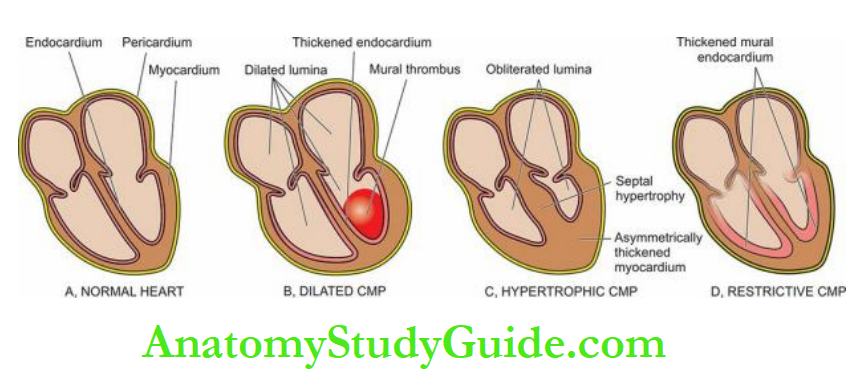
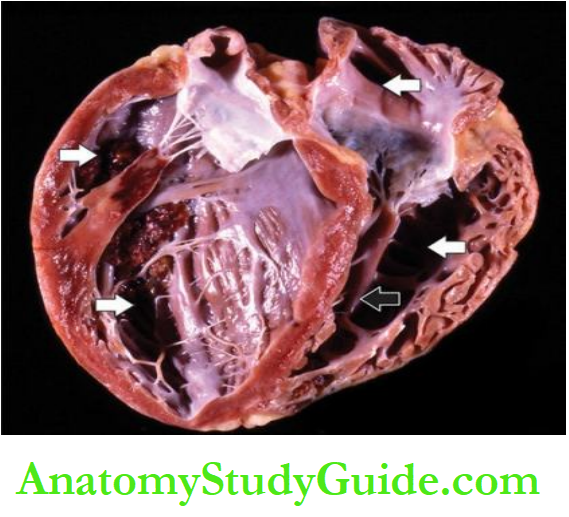
2. Endocardial Fibroelastosis:
This is an unusual and uncommon form of heart disease occurring predominantly in infants and children under 2 years of age and less often in adults.
The infantile form is clinically characterised by sudden breathlessness, cyanosis, cardiac failure and death whereas the symptoms in the adult form last for a longer duration.
Etiology: The etiology of the condition remains obscure. However, a number of theories have been proposed.
These are as under:
The infantile form is believed to be congenital in origin occurring due to the effect of intrauterine endocardial anoxia.
The adult form may also be induced by anoxia-causing lesions such as anomalous coronary arteries, metabolic derangements influencing myocardial function etc.
It may occur due to haemodynamic pressure overload such as in congenital septal defects and coarctation of the aorta.
It may be an expression of a genetic disorder as noticed in twins, triplets and siblings.
The association of endocardial fibroelastosis with various congenital malformations in the heart or elsewhere further supports the genetic theory.
Some workers consider this disease a form of connective tissue disorder.
Certain factors causing myocardial injury may initiate the endocardial disease such as thiamine deficiency (beriberi heart disease) or preceding idiopathic myocarditis.
Lymphatic obstruction of the heart has been suggested by some as the causative mechanism.
Morphologic Features: Grossly, the characteristic feature is the diffuse or patchy, rigid, pearly-white thickening of the mural endocardium.
The left ventricle is predominantly involved, followed in decreasing frequency by the left atrium, right ventricle and right atrium.
Quite often, the valves, especially of the left heart, are affected. Some cases contain mural thrombi.
Enlargement of the heart is present and is mainly due to left ventricular hypertrophy but the volume of the chamber is decreased.
Microscopically, the typical finding is the proliferation of the collagen and elastic tissue (fibroelastosis) comprising the thickened endocardium.
The fibroelastosis generally does not extend into the subjacent myocardium.
The lesion is devoid of inflammatory cells.
3. Endomyocardial Fibrosis:
This form of restrictive cardiomyopathy is a tropical condition prevalent in Africa, especially in Uganda and Nigeria, but some cases occur in South India, Sri Lanka, Malaysia and tropical South America.
It is seen in children and young adults. The clinical manifestations consist of congestive heart failure of unknown cause just as in the adult variety of endocardial fibroelastosis.
Etiology:
The aetiology of the condition remains obscure but the geographic distribution suggests the role of certain factors like malnutrition, viral infections and heavy consumption of bananas (rich in serotonin).
Morphologic Features:
Grossly, endomyocardial fibrosis is characterised by fibrous scarring of the ventricular endocardium that extends to involve the inner third of the myocardium.
The atrioventricular valve leaflets are often affected but the semilunar valves are uninvolved.
Mural thrombi may be present. The heart may be normal-sized or hypertrophied but the volume of the affected chambers is diminished due to fibrous scarring.
Microscopically, the endocardium and parts of the inner third of the myocardium show the destruction of normal tissue and replacement by fibrous tissue.
The condition differs from endocardial fibroelastosis in having mononuclear inflammatory cell infiltrate and lacking in elastic tissue.
The superficial layer may show dense hyalinised connective tissue and even calcification.
4. Loeffler’s Endocarditis
Also known by the more descriptive term of ‘fibro plastic parietal endocarditis with peripheral blood eosinophilia’, the condition is considered by some as a variant of the entity described above, endomyocardial fibrosis.
However, it differs from the latter in the following respects:
There is generally a peripheral blood eosinophilic leucocytosis.
The inflammatory infiltrate in the endocardium and in the part of the affected myocardium chiefly consists of eosinophils. The condition has a worse prognosis.
5. Other Forms of Restrictive Cardiomyopathy:
Besides the above, restrictive cardiomyopathy may result from various diverse causes as:
Haemochromatosis due to iron overload from multiple transfusions, haemoglobinopathies
- Myocardial sarcoidosis
- Carcinoid syndrome
- Scleroderma
- Neoplastic infiltration in the heart
Other Myocardial Diseases:
Myocardial diseases are myocarditis (i.e. inflammatory involvement of the myocardium) and cardiomyopathy (i.e. a non-inflammatory myocardial involvement from various
causes, often genetic).
Based on the etiologic classification, myocarditis is divided into: infective (viral, bacterial, toxic, granulomatous, syphilitic, rickettsial, protozoal and helminthic), idiopathic (Fiedler’s), in connective tissue disorders and caused by miscellaneous agents.
Cardiomyopathies (CMPs) are heart diseases associated with mechanical and/or electrical dysfunction, having morphological features in the myocardium, and are due to several causes which are frequently genetic.
CMPs are subdivided into 3 pathophysiologic types: dilated (congestive), hypertrophic, and restrictive (or obliterative or infiltrative).
Dilated CMP is characterised by four-chamber dilatation of the heart.
Hypertrophic CMP has cardiac enlargement. Restrictive CMP has restrictions to ventricular filling and reduced volume of ventricles.
Pericardial Diseases
Diseases of the pericardium are usually secondary to, or associated with other cardiac and systemic diseases.
These are discussed under 2 headings:
- Pericardial fluid accumulations
- Pericarditis
Pericardial Fluid Accumulations
Accumulation of fluid in the pericardial sac may be watery or pure blood.
Accordingly, it is of 2 types: hydropericardium (pericardial effusion) and haemopericardium.
1. Hydropericardium (Pericardial Effusion)
Accumulation of fluid in the pericardial cavity due to non-inflammatory causes is called hydropericardium or pericardial effusion.
Normally, the pericardial cavity contains 30 to 50 ml of clear watery fluid.
Considerable quantities of fluid (up to 1000 ml) can be accommodated in the pericardial cavity without seriously affecting the cardiac function if the accumulation is slow.
But the sudden accumulation of a smaller volume (up to 250 ml) may produce deficient diastolic filling of the cardiac chambers (cardiac tamponade).
Pericardial effusion is detected by cardiac enlargement in the X-rays and by faint apex beat.
The various types of effusions and their causes are as follows:
1. Serous effusions: This is the most common type occurring in conditions in which there is generalised oedema e.g. in cardiac (in CHF), renal, nutritional and hepatic causes.
The serum effusion is clear, watery, and straw-coloured with a specific gravity of less than 1.015 (transudate). The serosal surface is smooth and glistening.
2. Serosanguineous effusion: This type is found following blunt trauma to the chest and cardiopulmonary resuscitation.
3. Chylous effusion: Milky or chylous fluid accumulates in conditions causing lymphatic obstruction.
4. Cholesterol effusion: This is a rare type of fluid accumulation characterised by the presence of cholesterol crystals such as in myxoedema.
2. Haemopericardium: Accumulation of pure blood in the pericardial sac is termed haemopericardium.
The condition must be distinguished from haemorrhagic pericarditis in which there is escape of small quantities of blood into the pericardial cavity.
Massive and sudden bleeding into the sac causes compression of the heart leading to cardiac tamponade.
The causes of haemopericardium are as under:
- Rupture of the heart through a myocardial infarct.
- Rupture of dissecting aneurysm.
- Bleeding diatheses such as scurvy, acute leukaemias, and thrombocytopenia.
- Trauma following cardiopulmonary resuscitation or by laceration of a coronary artery.
Pericarditis
Pericarditis is the inflammation of the pericardial layers and is generally secondary to diseases in the heart or caused by systemic diseases.
Primary or idiopathic pericarditis is quite rare. Based on the morphologic appearance, pericarditis is classified into acute and chronic types, each of which may have several etiologies.
Acute and chronic pericarditis has further subtypes based on the character of the exudate.
1. Acute Pericarditis
Acute bacterial and non-bacterial pericarditis are the most frequently encountered forms of pericarditis. These may have the following subtypes:
1. Serous Pericarditis: Acute pericarditis may be accompanied by accumulation of serous effusion which differs from the transudate of hydropericardium in having increased protein content and higher specific gravity. Its various causes are as under:
- Viral infection e.g. coxsackie A or B viruses, influenza virus, mumps virus, adenovirus and infectious mononucleosis.
- Rheumatic fever.
- Rheumatoid arthritis.
- Systemic lupus erythematosus.
- Involvement of the pericardium by malignant tumours in the vicinity e.g. carcinoma lung, mesothelioma and mediastinal tumours.
- Tuberculous pericarditis in the early stage. The fluid accumulation is generally not much and ranges from 50 to 200 ml but may rarely be large enough to cause cardiac tamponade.
1. Acute Pericarditis
- Serous pericarditis
- Fibrinous or serofibrinous pericarditis
- Purulent or fibrinopurulent pericarditis
- Haemorrhagic pericarditis
2. Chronic Pericarditis
- Tuberculous pericarditis
- Chronic adhesive pericarditis
- Chronic constrictive pericarditis
- Pericardial plaques (milk spots, soldiers’ spots)
Microscopically, the epicardial and pericardial surfaces show infiltration by some neutrophils, lymphocytes and histiocytes.
The fluid usually resorbs with the resolution of the underlying disease.
2. Fibrinous And Serofibrinous Pericarditis: The response of the pericardium by fibrinous exudate is the most common type of pericarditis. Quite often, there is an admixture of fibrinous exudate with serous fluid.
The various causes of this type of pericarditis are as follows:
- Uraemia
- Myocardial infarction
- Rheumatic fever
- Trauma such as in cardiac surgery
- Acute bacterial infections.
The amount of fluid accumulation is variable.
The cardiac surface is characteristically covered by dry or moist, shaggy, fibrinous exudate which gives a ‘bread and butter’ appearance.
Clinically, these cases manifest by friction rub. In less extensive cases of fibrinous or serofibrinous pericarditis, there is complete resorption of the exudate.
In cases with advanced fibrinous exudate, pericarditis heals by the organisation and develops fibrous adhesions resulting in adhesive pericarditis.
3. Purulent Or Fibrinopurulent Pericarditis: Purulent or fibrinopurulent pericarditis is mainly caused by pyogenic bacteria (e.g. staphylococci, streptococci and pneumococci) and less frequently by fungi and parasites.
The infection may spread to the pericardium by the following routes:
- By direct extension from neighbouring inflammation e.g. in empyema of the pleural cavity, lobar pneumonia, infective endocarditis and mediastinal infections.
- By haematogenous spread.
- By lymphatic permeation.
- Direct implantation during cardiac surgery.
Generally, fibrinous or serofibrinous pericarditis precedes the development of purulent pericarditis.
The amount of exudate is variable and is generally thick, creamy pus, coating the pericardial surfaces.
Microscopically, besides the purulent exudate on the pericardial surfaces, the serosal layers show dense infiltration by neutrophils.
Purulent exudate generally does not resolve completely but instead heals by organisation resulting in adhesive or chronic constrictive pericarditis
4. Haemorrhagic Pericarditis: Haemorrhagic pericarditis is the one in which the exudate consists of admixture of an inflammatory effusion of one of the foregoing types along
with blood.
The causes are as under:
- Neoplastic involvement of the pericardium
- Haemorrhagic diathesis with effusion
- Tuberculosis
- Severe acute infections
The outcome of haemorrhagic pericarditis is generally similar to that of purulent pericarditis.
2. Chronic Pericarditis: Chronic pericarditis is the term used for tuberculous pericarditis and the healed stage of one of the various forms of acute pericarditis already described.
Included under this are: tuberculous pericarditis, chronic adhesive pericarditis, chronic constrictive pericarditis, and the pericardial plaques.
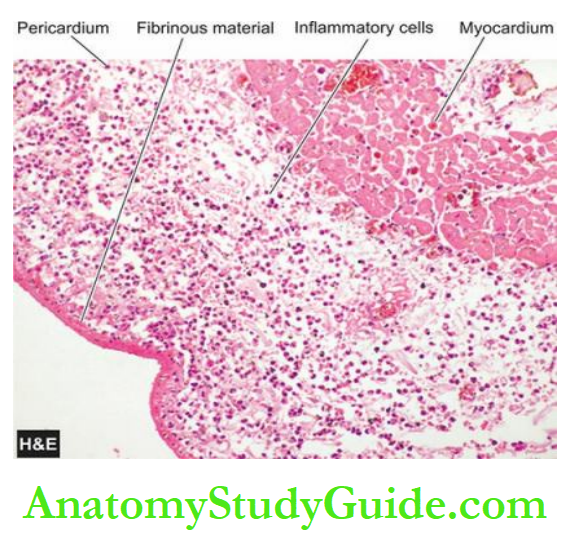
The space between the layers of the pericardium contains numerous mixed inflammatory cells, extending into the subjacent myocardium.
1. Tuberculous Pericarditis:
Tuberculous pericarditis is the most frequent form of granulomatous inflammation of the pericardium.
The lesions may occur by one of the following mechanisms:
Direct extension from an adjacent focus of tuberculosis.
By lymphatic spread e.g. from tracheobronchial lymph nodes, chronic pulmonary tuberculosis or infected pleura.
The exudate is slightly turbid, caseous or blood-stained with sufficient fibrin.
Tubercles are generally visible on the pericardial surfaces and sometimes caseous areas are also visible to the naked eye.
Microscopically, typical tuberculous granulomas with caseation necrosis are seen in the pericardial wall.
The lesions generally do not resolve but heal by fibrosis and calcification resulting in chronic constrictive pericarditis.
2. Chronic Adhesive Pericarditis:
Chronic adhesive pericarditis is the stage of organisation and healing by the formation of fibrous adhesions in the pericardium following preceding fibrinous, suppurative or haemorrhagic pericarditis.
The process begins with the formation of granulation tissue and neovascularization.
Subsequently, fibrous adhesions develop between the parietal and the visceral layers of the pericardium and obliterate the pericardial space.
Sometimes, fibrous adhesions develop between the parietal pericardium and the adjacent mediastinum and are termed adhesive mediastinopericarditis.
Chronic adhesive pericarditis differs from chronic constrictive pericarditis in not embarrassing the function of the heart.
However, cardiac hypertrophy and dilatation may occur in severe cases due to increased workload.
3. Chronic Constrictive Pericarditis:
This is a rare condition characterised by dense fibrous or fibrocalcific thickening of the pericardium resulting in mechanical interference with the function of the heart and reduced cardiac output.
The condition usually results from long-standing preceding causes, e.g.
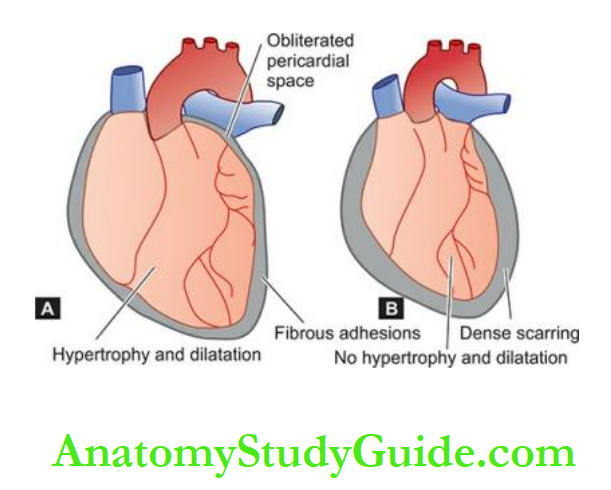
- Tuberculous pericarditis
- Purulent pericarditis
- Haemopericardium
- Concato’s disease (polyserositis)
- Rarely, acute non-specific and viral pericarditis.
The heart is encased in 0.5 to 1 cm thick and dense collagenous scar which may be calcified.
As a result, the heart fails to dilate during diastole.
The dense fibro-collagenous tissue may cause narrowing of the openings of the vena cavae, resulting in obstruction to the venous return to the right heart and consequent right heart failure.
In contrast to chronic adhesive pericarditis, hypertrophy and dilatation do not occur due to dense fibrous scarring.
Instead, the heart size is normal or smaller.
4. Pericardial Plaques (Milk Spots, Soldiers’ Spots): These are opaque, white, shining and well-circumscribed areas of the organisation with fibrosis in the pericardium measuring 1 to 3 cm in diameter.
They are seen most frequently on the anterior surface of the right ventricle.
The exact cause is not known but they are generally believed to arise from healing of preceding pericarditis.
The plaque-like lesions of pericardial thickenings are also termed milk spots or soldiers’ spots as they were often found at autopsy in the soldiers in World War I who carried their shoulder bags causing pressure against the chest wall by the straps which produced chronic irritation of the pericardium.
Pericardial Disease:
Pericardial effusion is an accumulation of fluid in the pericardial sac.
Hydropericardium is a watery collection (serous, serosanguineous, chylous) while haemopericardium is a collection of pure blood.
Pericarditis is the inflammation of the pericardial layers; it may be acute or chronic types.
Depending upon the character of accumulated fluid, acute pericarditis may be serous, fibrinous, purulent, and haemorrhagic.
Chronic pericarditis is due to fibrous healing by the organisation and may be chronic adhesive, chronic constrictive and plaque formation.
Tumours Of The Heart
Tumours of the heart are classified into primary and secondary, the latter being more common than the former.
Primary Tumours
Primary tumours of the heart are quite rare, found in 0.04% of autopsies.
In decreasing order of frequency, the benign tumours encountered in the heart are myxoma, lipoma, fibroelastoma, rhabdomyoma, haemangioma and lymphangioma.
The malignant tumours are still rarer, the important ones are rhabdomyosarcoma, angiosarcoma and malignant mesothelioma.
Out of all these, only myxoma of the heart requires elaboration.
Myxoma:
This is the most common primary tumour of the heart comprising about 50% of all primary cardiac tumours.
The majority of them occur in the age range of 30 to 60 years. Myxomas may be located in any cardiac chamber or the valves, but 90% of them are situated in the left atrium.
Grossly, they are often single but may be multiple. They range in size from less than 1 to 10 cm, polypoid, pedunculated, spherical, soft and haemorrhagic masses resembling mulling an original thrombus.
Some investigators actually consider myxomas to be organising mural thrombi rather than true neoplasms.
Microscopically, the tumour shows the following features:
There is abundant myxoid or mucoid intercellular stroma positive for mucin.
The cellularity is sparse. The tumour cells are generally stellate-shaped, spindled and polyhedral, scattered in the stroma.
Occasional multinucleate tumour giant cells are present.
Numerous capillary-sized blood vessels are found and the tumour cells may be aggregated around them.
A few lymphocytes, plasma cells and macrophages are seen.
Foci of haemorrhage and deposits of haemosiderin granules are often present.
Secondary Tumours:
Metastatic tumours of the heart are more common than primary tumours.
About 10% of cases with disseminated cancer have metastases in the heart.
Most of these result from the haematogenous or lymphatic spread. In descending order of frequency, primary sites of origin are carcinoma of the lung, breast, malignant lymphoma, leukaemia and malignant melanoma.
Occasionally, there may be a direct extension of a primary intrathoracic tumour such as carcinoma of the lung into the pericardium and into the cardiac chambers.
Tumours of the Heart
Tumours of the heart are classified into primary and secondary.
Myxoma of the heart is the most common benign primary tumour occurring most often in the left atrium.
Secondary tumours are more common than primary tumours and are metastases from disseminated cancers
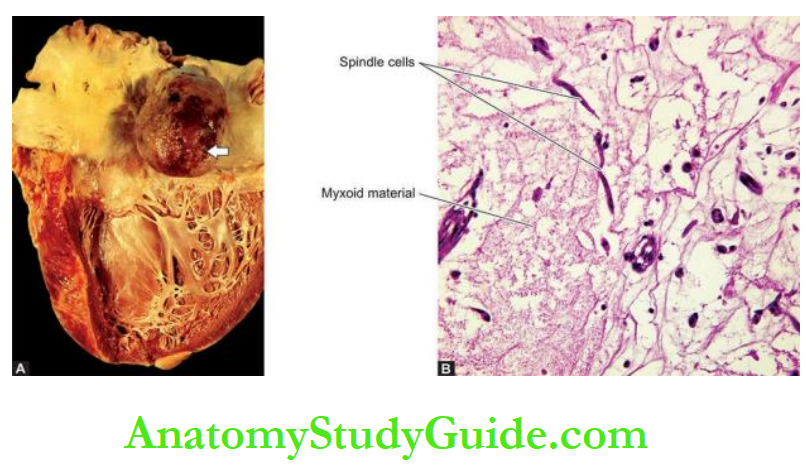
Pathology Of Cardiovascular Interventions
With the development of surgical and non-surgical coronary revascularisation procedures in coronary artery disease, it has become possible to study the pathology of native as well as grafted vessels.
Besides, the myocardial tissue by endomyocardial biopsy is also accessible for histopathologic study.
Endomyocardial Biopsy:
Endomyocardial biopsy (EMB) is done for making a final histopathologic diagnosis in certain cardiac diseases.
The main indications for EMB are myocarditis, cardiac transplant cases, restrictive heart disease, infiltrative heart diseases such as amyloidosis, storage disorders etc.
EMB is done by biopsy forceps introduced via a cardiac catheter into either of the ventricles but preferably right ventricle is biopsied for its relative ease and safety.
The route for the catheter may be through the internal jugular vein or femoral vein for accessing the right ventricle.
Balloon Angioplasty:
Balloon angioplasty or percutaneous coronary intervention (PCI) is a non-surgical procedure that employs percutaneous insertion and manipulation of a balloon catheter into the occluded coronary artery.
The balloon is inflated to dilate the stenotic after which causes endothelial damage, plaque fracture, medial dissection and haemorrhage in the affected arterial wall.
PCI is accompanied by the insertion of coronary stents in the blocked coronaries with a success rate of symptoms in over 95% of cases.
However, case selection for PCI is important and major indications are 2 or 3 vessel block but blockage of the left main coronary is a contraindication for PCI.
Unstable angioplasty may be associated with acute coronary syndromes.
PCI is followed by the administration of anti-platelet (oral aspirin) and antithrombin therapy to avoid the occurrence of coronary thrombosis.
Recurrent stenosis after metal stenting in PCI may occur within 6 months in about 20% of patients, more often in patients with diabetes mellitus.
Restenosis is multifactorial in aetiology that includes smooth muscle cell proliferation, extracellular matrix and local thrombosis.
However, the widespread use of drug-delivering stents has made it possible to overcome several long-term complications of coronary stenting.
Currently, stents with anti-proliferative, anti-inflammatory, cytotoxic and cytostatic agents are commercially available.
Coronary Artery Bypass Grafting:
Coronary artery bypass grafting (CABG) employs the use of autologous grafts to replace or bypass blocked coronary arteries.
The most frequently used is an autologous graft of a saphenous vein which is reversed (due to valves in the vein) transplanted, or a left internal mammary artery may be used which is available nearest to the operative area of the heart.
Long-term follow-up of CABG surgery has yielded the following observations on the pathology of a grafted vessel:
1. In a reversed saphenous vein graft, long-term luminal patency is 50% after 10 years.
Pathologic changes which develop in grafted veins include thrombosis in the early stage, intimal thickening and graft atherosclerosis with or without complicated lesions.
2. Internal mammary artery graft, however, has a patency of more than 90% after 10 years.
3. Atherosclerosis with superimposed complications may develop in the native coronary artery distal to the grafted vessel as well as in the grafted vessel.
Cardiac Transplantation:
Since the first human-to-human cardiac transplant was carried out successfully by South African surgeon Dr Christian Barnard in 1967,
cardiac transplantation and prolonged assisted circulation are being done in many countries for end-stage cardiac diseases, most often in idiopathic dilated cardiomyopathy, heart failure and IHD.
Major complications of heart transplant are:
transplant rejection reaction, infections (particularly with Toxoplasma gondii and cytomegaloviruses), graft coronary atherosclerosis, and higher incidence of malignancy due to long-term administration of immunosuppressive therapy.
Worldwide, about 3,000 cardiac transplants are performed annually.
The survival following heart transplants is reported as 1 year in 85%, 5 years in 65% and 10 years in 45% of cases.
One of the main problems in cardiac transplant centres is the availability of donors.
Pathology of Cardiovascular Interventions
Endomyocardial biopsy (EMB) from the right ventricle is done for making a final histopathologic diagnosis in certain cardiac diseases.
Balloon angioplasty is the percutaneous insertion and manipulation of a balloon catheter into the occluded coronary artery.
Coronary artery bypass grafting (CABG) employs the use of autologous grafts to replace or bypass blocked coronary arteries.
The most frequently used is an autologous graft of the saphenous vein.
Following CABG, atherosclerosis with superimposed complications may develop in the native coronary artery distal to the grafted vessel as well as in the grafted vessel.
Heart transplants are performed infrequently due to the non-availability of the donor’s heart, and the risk of complications.
Leave a Reply