Haemostatic System, Bleeding Disorders and Transfusion Biology
Haemostatic System
Haemostasis is the process of the formation of a plug to halt bleeding and restore normal blood flow. There are three major components of a haemostatic system: platelets, coagulation cascade, and the vessel wall. Pathophysiology of haemostasis is quite linked to that of thrombosis discussed in thus discussion of haemostasis and thrombosis is likely to overlap.
Table of Contents
Read And Learn More: General Pathology Notes
Discussion on haemostasis involves the following topics:
- Thrombopoiesis
- Mechanism of haemostasis
- Tests for haemostatic function
Thrombopoiesis:
As outlined earlier in the trilineage myeloid stem cells in the bone marrow differentiate into erythroid progenitor, granulocyte-monocyte progenitor, and megakaryocyte progenitor cells.
- Platelets are formed in the bone marrow by a process of fragmentation of the cytoplasm of megakaryocytes.
- Platelet production is under the regulatory control of thrombopoietin (TPO), a glycoprotein produced by the liver and kidney. Synthesis of TPO is increased in inflammation, especially in response to IL-6.
The stages in platelet production are:
Megakaryoblast, promegakaryocyte, megakaryocyte, and discoid platelets.
Megakaryoblast:
The earliest precursor of platelets in the bone marrow is megakaryoblast. It arises from haematopoietic stem cells by a process of differentiation.
Promegakaryocyte:
- A megakaryoblast undergoes endoreduplication of nuclear chromatin i.e. nuclear chromatin replicates repeatedly in multiples of two without division of the cell.
- Ultimately, a large cell containing up to 32 times the normal diploid content of nuclear DNA (polyploidy) is formed when further nuclear replication ceases and the cytoplasm becomes granular.
Megakaryocyte:
- A mature megakaryocyte is a large cell, 30-90 µm in diameter, and contains 4-16 nuclear lobes having coarsely clumped chromatin. The cytoplasm is abundant, light blue and contains red-purple granules.
- Platelets are formed from pseudopods of megakaryocyte cytoplasm which get detached into the blood stream. Each megakaryocyte may form up to 4000 platelets. The formation of platelets from the stem cell takes about 10 days.
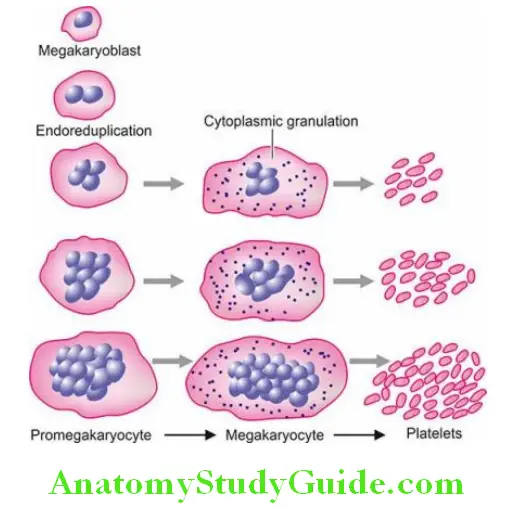
Platelets:
Platelets are small (1-4 µm in diameter), discoid, non-nucleate structures containing red-purple granules. The normal platelet count ranges from 150,000-400,000/µl and the lifespan of platelets is 7-10 days.
- About 70% of platelets are in circulation while the remaining 30% lie sequestered in the spleen.
- Newly-formed platelets spend 24-36 hours in the spleen before being released into circulation but splenic stasis does not cause any injury to the platelets normally. Factors such as stress, epinephrine and exercise stimulate platelet production.
Mechanism Of Haemostasis:
The role of the three components involved in haemostasis is as under:
1. The Vessel Wall:
Normal vascular endothelium has a metabolically active surface.
- An intact endothelium provides an antithrombotic surface by inhibiting platelet function and thus contributes to preventing thrombosis.
- Normally, endothelium-derived vasoconstrictor, endothelin, promotes thrombosis and activates platelets, while endothelium-derived vasodilators such as nitric oxide platelet inhibitors.
- Thus, the net effect of two opposing forces-vasoconstriction and platelet activation (prothrombotic forces), and vasodilatation and platelet inhibition (antithrombotic forces), is a fluidity of the blood flow and haemostasis.
- Injured endothelium activates platelets as well as coagulation cascade, and inhibits fibrinolysis.
2. The Platelets:
On vascular injury, platelets undergo activation that involves platelet adhesion, platelet granule release and platelet aggregation, which are regulated by changes in membrane phospholipids, and calcium. At the molecular level.
These important events are depicted diagrammatically and briefly outlined below:
1. Adhesion:
Platelets adhere to collagen in the subendothelium due to the presence of a receptor on the platelet surface, glycoprotein (Gp) Ia-IIa which is an integrin.
- The adhesion to the vessel wall is further stabilised by von Willebrand factor, an adhesion glycoprotein.
- This is achieved by the formation of a link between the von Willebrand factor and another platelet receptor, the GpIb-IX complex.
2. Release reaction:
- After adhesion, platelets become activated and release three types of granules from their cytoplasm: dense granules, α-granules and lysosomal vesicles.
- Important products released from these granules are ADP, ATP, calcium, serotonin, platelet factor 4, factor V, factor VIII, thrombospondin, platelet-derived growth factor (PDGF), von Willebrand factor (vWF), fibronectin, fibrinogen, plasminogen activator inhibitor –1 (PAI-1) and thromboxane A2.
3. Aggregation:
This process is mediated by fibrinogen which forms a bridge between adjacent platelets via glycoprotein receptors on platelets, GpIIb-IIIa.
- The term primary haemostasis is used for the initial platelet plug formed at the site of vascular injury.
- It is an immediate phenomenon appearing within seconds of injury and is responsible for the cessation of bleeding from microvas culture.
3. Coagulation Cascade:
Vascular damage exposes blood to tissue factor (cofactor of VIIa, formerly called factor III) triggers a coagulation cascade
- This involves a plasma coagulation system consisting of several plasma proteins—13 coagulation factors (intrinsic and extrinsic and common pathway), fibrinolytic factors, and inhibitors, culminating in the generation of fibrin.
- The fibrinolytic system is a physiological mechanism of the dissolution of fibrin. Integration of fibrin clot in the initial platelet plug is termed secondary haemostasis.
Tests For Haemostatic Function:
In general, the haemostatic mechanisms have 2 primary functions:
- To promote local haemostasis at the site of injured blood vessels.
- To ensure that the circulating blood remains in a fluid state while in the vascular bed i.e. to prevent the occurrence of generalised thrombosis.
The formation of a haemostatic plug is a complex mechanism and involves the maintenance of a delicate balance among at least 5 components. Accordingly, there are specific tests for assessing each of these components:
- Blood vessel wall: Tests for disordered vascular haemostasis
- Platelets: Tests for platelets and their functions
- Plasma coagulation factors: Tests for blood coagulation
- Fibrinolytic system: Tests or fibrinolysis
- Inhibitors: Tests for coagulation inhibitors
Anything that interferes with any of these components results in defective haemostasis with abnormal bleeding. In general, to establish a definite diagnosis in any case suspected to have 

Comprehensive clinical evaluation, including the patient’s history, family history and details of the site, frequency and character of haemostatic defect.
- Series of screening tests for assessing the abnormalities in various components involved in maintaining haemostatic balance.
- Specific tests to pinpoint the cause. A brief review of general principles of tests used to investigate haemostatic abnormalities is presented below and summarised in Table.
1. Investigation Of Disordered Vascular Haemostasis:
Disorders of vascular haemostasis may be due to increased vascular permeability, reduced capillary strength and failure to contract after injury.
Tests of defective vascular function are as under:
1. Bleeding time: This simple test is based on the principle of formation of a haemostatic plug following a standard incision on the volar surface of the forearm and the time the incision takes to stop bleeding is measured.
The test is dependent upon capillary function as well as on platelet number and the ability of platelets to adhere to form aggregates. The normal range is 3-8 minutes.
A prolonged bleeding time may be due to the following causes:
- Thrombocytopenia
- Disorders of platelet function
- von Willebrand’s disease
- Vascular abnormalities (e.g. in Ehlers-Danlos syndrome)
- Severe deficiency of factor V and XI
Hess Capillary Resistance Test (Tourniquet Test):
This test is done by tying the sphygmomanometer cuff to the upper arm and raising the pressure in it between diastolic and systolic for 5 minutes.
- After deflation, the number of petechiae appearing in the next 5 minutes in a 3 cm2 area over the cubital fossa is counted.
- The presence of more than 20 petechiae is considered a positive test. The test is positive for increased capillary fragility as well as thrombocytopenia.
Investigation Of Platelets And Platelet Function:
Haemostatic disorders are commonly due to abnormalities in platelet number, morphology or function.

1. Screening Tests:
The screening tests carried out for assessing platelet-related causes are as under:
- Peripheral blood platelet count
- Skin bleeding time
- Examination of fresh blood film to see the morphologic abnormalities of platelets
2. Special Tests:
If these screening tests suggest a disorder of platelet function, the following platelet function tests may be carried out:
- Platelet adhesion tests such as retention in a glass bead column, and other sophisticated techniques.
- Aggregation tests are turbidimetric techniques using ADP, collagen or ristocetin.
- The granular content of the platelets and their release can be assessed by electron microscopy or by measuring the substances released.
- Platelet coagulant activity is measured indirectly by the prothrombin consumption index.
3. Investigation Of Blood Coagulation:
The normal blood coagulation system consists of a cascade of activation of 13 coagulation factors. These form intrinsic, extrinsic and common pathways which culminate in the formation of thrombin that acts on fibrinogen to produce fibrin.
- The Fibrin clot so formed is strengthened by factor XIII which itself gets activated by thrombin.
- The process of fibrinolysis or clot dissolution and the role of platelets in the activation of cascade and formation of the haemostatic plug is illustrated.
Coagulation tests include screening and confirmatory special tests as under:
Screening Tests:
Tests for the blood coagulation system include a battery of screening tests.
These are as under:

1. Whole blood coagulation time The estimation of whole blood coagulation time done by various capillary and tube methods is of limited value since it is an insensitive and nonspecific test. The normal range is 4-9 minutes at 37°C.
2. Activated partial thromboplastin time (APTT) or partial thromboplastin time with kaolin (PTTK) This test is used to measure the intrinsic system factors (VIII, IX, XI and XII)* as well as factors common to both intrinsic and extrinsic systems (factors X, V, prothrombin and fibrinogen). The test consists of the addition of 3 substances to the plasma—calcium, phospholipid and a surface activator such as kaolin.
The normal range is 30-40 seconds. The common causes of a prolonged PTTK (or APTT) are as follows:
- Parenteral administration of heparin
- Disseminated intravascular coagulation
- Liver disease
- Circulating anticoagulants
3. One-stage prothrombin time (PT) PT measures the extrinsic system factor VII as well as factors in the common pathway. In this test, tissue thromboplastin (for example., brain extract) and calcium are added to the test. The normal PT in this test is 10-14 seconds.
The common causes of prolonged one-stage PT are as under:
- Patients on oral anticoagulant drug therapy e.g. warfarin
- Liver disease, especially obstructive liver disease
- Vitamin K deficiency
- Disseminated intravascular coagulation
4. International normalised ratio (INR) Patients on oral anticoagulants are tested and monitored more often by INR; it is the ratio of PT test results of patients with test results from other laboratories.
- In normal persons, INR is <1.1.
- In patients on oral blood-thinning drugs (e.g. warfarin), acceptable INR is 2.0 to 3.0.
- Higher INR means blood clots slower and lower INR means blood clots more quickly than desired.
5. Measurement of fibrinogen The screening tests for fibrinogen deficiency are semiquantitative fibrinogen titre and thrombin time (TT). The normal value of thrombin time is under 20 seconds, while a fibrinogen titre in plasma dilution up to 32 is considered normal.
The following are the common causes for higher values in both these tests:
- Hypofibrinogenaemia (e.g. in DIC)
- Raised concentration of FDP
- Presence of heparin
Special Tests:
In the presence of an abnormality in screening tests, detailed investigations for the possible cause are carried out.
These include the following:
1. Coagulation factor assays These bioassays are based on the results of PTTK or PT tests and employ the use of substrate plasma that contains all other coagulation factors except the one to be measured. The unknown level of the factor activity is compared with a standard control plasma with a known level of activity. Results are expressed as a percentage of normal activity.
2. Quantitative assays The coagulation factors can be quantitatively assayed by immunological and other chemical methods.
3. Thromboelastography (TEG) This is a viscoelastic haemostatic assay used in-house more often by ICUs and surgeons. TEG provides a measure of the interaction of platelets with the coagulation cascade i.e. aggregation, clot strength, fibrin cross-linking and fibrinolysis. However, TEG does not always correlate with other tests such as platelet count, PT/INR, and APTT.
Investigation Of Fibrinolytic System:
Increased levels of circulating plasminogen activator are present in patients with hyperfibrinolysis.
Following screening tests are done to assess these abnormalities in the fibrinolytic system:
- Estimation of fibrinogen
- Fibrin degradation products (FDP) in the serum
- Ethanol gelation test
- Euglobulin or whole blood lysis time
More specific tests include functional assays, immunological assays by ELISA, and chromogenic assays of plasminogen activators, plasminogen, plasminogen activator inhibitors, and FDP.
Investigation Of Coagulation Inhibitors:
There is an inbuilt system in the body by which coagulation remains confined as per need and does not become generalised.
Important inhibitors are as under:
1. Antithrombin III This binds to thrombin and forms thrombin-antithrombin complex which does not permit coagulation.
2. Protein C and S Activated protein C acts as an anticoagulant by cleaving and inactivating activated factors V and VIII. This reaction is further augmented by a cofactor, protein S. Deficiency of protein C and S, or failure of action of activated protein C on activated factor V due to mutation of the target site on factor V (Leiden factor) leads to hypercoagulability
Haemostatic System:
- The haemostatic system consists of three closely interlinked components: the platelets, the coagulation cascade and the vessel wall.
- Thrombopoiesis is under the regulatory control of thrombopoietin. The stages in platelet production are megakaryoblast, promegakaryocyte, megakaryocyte, and finally discoid platelets are formed.
- Primary haemostasis is the formation of platelet plugs after vascular injury. Platelets adhere to the subendothelium, are activated to release biologically active compounds and undergo aggregation.
- Secondary haemostasis is the involvement of fibrin clots in the platelet plug. This occurs by activation of coagulation cascade and formation of fibrin.
- Haemostatic balance is maintained by 5 components: blood vessel wall, platelets, coagulation factors, fibrinolysis and inhibitors. For assessing haemostatic function, there are screening tests and special or confirmatory tests.
- Tests for vessel walls are bleeding time and Hess capillary test.
- Platelet tests are counts, and tests for adhesion, aggregation and release.
- Screening tests for coagulation factors are coagulation test, activated partial thromboplastin time, prothrombin time and thrombin time. Special tests are assays of various coagulation factors.
- Tests for fibrinolysis are the euglobulin lysis test and measurement of FDPs.
- Tests for inhibitors are for antithrombin and proteins C and S
Bleeding Disorders
Bleeding disorders or haemorrhagic diatheses are a group of disorders characterised by defective haemostasis with abnormal bleeding. The tendency to bleed may be spontaneous in the form of small haemorrhages into the skin and mucous membranes (for example., petechiae, purpura, ecchymoses), or there may be excessive external or internal bleeding following trivial trauma and surgical procedure (for example., haematoma, haemarthrosis etc).
Since bleeding disorder may be the result of the predominant involvement of any one or more of the main components in the haemostatic system, etiologically they are divided into the following main groups.
- Vascular bleeding disorders
- Bleeding due to platelet disorders
- Bleeding due to coagulation disorders
- Disseminated intravascular coagulation (DIC) due to a combination of factors
Vascular Bleeding Disorders:
- Vascular bleeding disorders, also called non-thrombocytopenic purpuras or vascular purpuras, are normally mild and characterised by petechiae, purpuras or ecchymoses confined to the skin and mucous membranes.
- Pathogenesis of bleeding is poorly understood since a majority of the standard screening tests of haemostasis including the bleeding time, coagulation time, platelet count and platelet function, are usually normal.
- Vascular purpuras arise from damage to the capillary endothelium, abnormalities in the subendothelial matrix or extravascular connective tissue that supports the blood vessels, or from formation of abnormal blood vessels. Vascular bleeding disorders may be inherited or acquired.
Inherited Vascular Bleeding Disorders:
A few examples of hereditary vascular disorders are given below:
Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu disease):
- This is an uncommon inherited autosomal dominant disorder. The condition begins in childhood and is characterised by abnormally telangiectatic (dilated) capillaries due to vascular malformations in the skin and mucosal surfaces, especially on lips, tongue, nose palms and soles.
- Clinically, patients present with bleeding from the stomach, epistaxis or from other mucosal surfaces.
Inherited disorders of connective tissue matrix:
These include Marfan’s syndrome, EhlersDanlos syndrome and pseudoxanthoma elasticum, all of which have inherited defects in the connective tissue matrix and, thus, have fragile skin vessels and easy bruising.
Acquired Vascular Bleeding Disorders:
Several acquired conditions are associated with vascular purpuras. These are as under:
1. Henoch-Schönlein purpura The salient features of Henoch-Schönlein or anaphylactoid purpura are as under:
- It is a self-limited type of hypersensitivity vasculitis occurring in children and young adults.
- The hypersensitivity vasculitis produces a purpuric rash on the extensor surfaces of arms, legs and buttocks, as well as haematuria, colicky abdominal pain due to bleeding into the GIT, polyarthralgia and acute nephritis.
- Despite these haemorrhagic features, all coagulation tests are normal.
- The vessel wall shows leucocytoclastic vasculitis as seen by viable and necrotic neutrophils.
- Circulating immune complexes are deposited in the vessel wall consisting of IgA, C3 and fibrin, and in some cases, properdin suggesting activation of alternate complement pathway as the trigger event.
2. Haemolytic-uraemic syndrome:
- The haemolytic-uraemic syndrome is a disease of infancy and early childhood in which there is a bleeding tendency and varying degrees of acute renal failure.
- The disorder remains confined to the kidney where hyaline thrombi are seen in the glomerular capillaries.
3. Simple easy bruising (Devil’s pinches):
Easy bruising of unknown cause is a common phenomenon in women of child-bearing age group.
4. Infection:
Many infections cause vascular haemorrhages either by causing toxic damage to the endothelium or by DIC. These are especially prone to occur in septicaemia and severe measles.
5. Drug reactions:
Certain drugs form antibodies and produce hypersensitivity (or leucocytoclastic) vasculitis responsible for abnormal bleeding.
6. Steroid purpura:
Long-term steroid therapy or Cushing’s syndrome may be associated with vascular purpura due to defective vascular support.
7. Senile purpura:
Atrophy of the supportive tissue of cutaneous blood vessels in old age may cause senile atrophy, especially in the dorsum of the forearm and hand.
8. Scurvy:
Deficiency of vitamin C causes defective collagen synthesis which causes skin bleeding as well as bleeding into muscles, and occasionally into the gastrointestinal and genitourinary tracts.
Vascular Bleeding Disorders:
These are commonly called vascular purpuras because the cause lies in the vascular wall—damaged endothelium, abnormality in a subendothelial matrix, or abnormally formed blood vessels.
- Examples of inherited vascular purpuras are hereditary haemorrhagic telangiectasia and connective tissue matrix disorders.
- Acquired causes are Henoch-Schönlein purpura, haemolytic-uraemic syndrome, simple easy bruising, certain bacterial and viral infections, intake of certain drugs and steroids, senility, scurvy et
Bleeding Due To Platelet Disorders:
Platelet disorders producing bleeding may fall into one of the following 3 categories:
- Due to a reduction in the number of platelets i.e. various forms of thrombocytopenias
- Due to a rise in platelet count i.e. thrombocytosis
- Due to defective platelet functions.
Thrombocytopenias:
Thrombocytopenia is defined as a reduction in the peripheral blood platelet count below the lower limit of normal i.e. below 150,000/µl.
- Thrombocytopenia is associated with abnormal bleeding that includes spontaneous cutaneous purpura and mucosal haemorrhages as well as prolonged bleeding after trauma.
- However, the spontaneous haemorrhagic tendency may become clinically evident only after severe depletion of the platelet count to a level below 20,000/µl.
Thrombocytopenia may result from the following groups of causes:
- Impaired platelet production
- Accelerated platelet destruction
- Splenic sequestration
- Dilutional loss
- Inherited
Some of the common and important causes of thrombocytopenia—drug-induced, heparin-induced, immune (or idiopathic), and thrombotic type, are discussed below.
Drug-induced Thrombocytopenia:
- Many commonly used drugs cause thrombocytopenia by depressing megakaryocyte production.
- In most cases, an immune mechanism by the formation of drug-antibody complexes is implicated in which the platelet is damaged as an ‘innocent bystander’.
Drug-induced thrombocytopenia is associated with many commonly used drugs and includes:
Chemotherapeutic agents (alkylating agents, anthracyclines, antimetabolites), certain antibiotics (sulfonamides, PAS, rifampicin, penicillins), drugs used in cardiovascular diseases (digitoxin, thiazide diuretics), diclofenac, acyclovir, heparin and excessive consumption of ethanol.

- Clinically, the patient presents with acute purpura. The platelet count is markedly lowered, often below 10,000/µl and the bone marrow shows a normal or increased number of megakaryocytes.
- The immediate treatment is to stop or replace the suspected drug with instruction to the patient to avoid taking the offending drug in future.
- Occasional patients may require temporary support with glucocorticoids, plasmapheresis or platelet transfusions.
Heparin-induced Thrombocytopenia:
Thrombocytopenia due to the administration of heparin is distinct from that caused by other drugs in the following ways:
- Thrombocytopenia is generally not so severe to fall to a level below 20,000/µl.
- Unlike drug-induced thrombocytopenia, heparin-induced thrombocytopenia is not associated with bleeding but instead, these patients are more prone to develop thrombosis.
The underlying mechanism of heparin-induced thrombocytopenia is the formation of antibodies against platelet factor 4 (PF-4)-heparin complex.
This specific antibody activates the endothelial cells and initiates thrombus formation. It occurs in a small proportion of cases after the patient has received heparin for 5-10 days.
Diagnosis is made by a combination of laboratory and clinical features with 4 Ts:
thrombocytopenia, thrombosis, time of fall of platelet count, absence of other causes of thrombocytopenia.
Immune Thrombocytopenic Purpura (ITP):
Idiopathic (or immune) thrombocytopenic purpura (ITP), is characterised by immunologic destruction of platelets and normal or increased megakaryocytes in the bone marrow.
Pathogenesis:
Based on the duration of illness, ITP is classified into acute and chronic forms, both of which have different pathogenesis.
Acute ITP This is a self-limited disorder, seen most frequently in children following recovery from a viral illness (for example., hepatitis C, infectious mononucleosis, CMV infection, HIV infection) or an upper respiratory illness.
- The onset of acute ITP is sudden and severe thrombocytopenia but recovery occurs within a few weeks to 6 months.
- The mechanism of acute ITP is by the formation of immune complexes containing viral antigens, and by the formation of antibodies against viral antigens which crossreact with platelets and lead to their immunologic destruction.
Chronic ITP Chronic ITP occurs more commonly in adults, particularly in women of childbearing age (20-40 years). The disorder develops insidiously and persists for several years.
- Though chronic ITP is idiopathic, similar immunologic thrombocytopenia may be seen in association with SLE, AIDS and autoimmune thyroiditis.
- The pathogenesis of chronic ITP is explained by the formation of anti-platelet autoantibodies, usually by platelet-associated IgG humoral antibodies synthesised mainly in the spleen.
- These antibodies are directed against target antigens on the platelet glycoproteins, GpIIb-IIIa and GpIb-IX complex.
- Some of the antibodies directed against the platelet surface also interfere in their function. The mechanism of platelet destruction is similar to that seen in autoimmune haemolytic anaemias.
- Sensitised platelets are destroyed mainly in the spleen and rendered susceptible to phagocytosis by cells of the reticuloendothelial system.
Clinical Features:
- The clinical manifestation of ITP may develop abruptly in cases of acute ITP, or the onset may be insidious as occurs in a majority of cases of chronic ITP.
- The usual manifestations are petechial haemorrhages, easy bruising, and mucosal bleeding such as menorrhagia in women, nasal bleeding, bleeding from gums, melaena and haematuria. Intracranial haemorrhage is, however, rare.
- Splenomegaly and hepatomegaly may occur in cases with chronic ITP but lymphadenopathy is quite uncommon in either type of ITP.
Laboratory Findings:
The diagnosis of ITP can be suspected on clinical features after excluding the known causes of thrombocytopenia and is supported by the following haematologic findings:
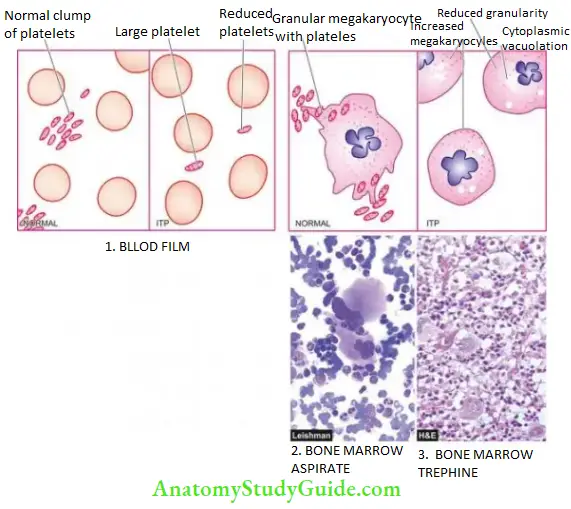
- Platelet count is markedly reduced, usually in the range of 10,000-50,000/µl.
- Blood film shows only occasional platelets which are often large in size.
- Bone marrow shows an increased number of megakaryocytes which have large non-lobulated single nuclei and may have reduced cytoplasmic granularity and the presence of vacuoles
- With sensitive techniques, anti-platelet IgG antibodies can be demonstrated on platelet surfaces or in the serum of patients.
- Platelet survival studies reveal markedly reduced platelet lifespan, sometimes less than one hour, as compared with a normal lifespan of 7-10 days.
Principles Of Treatment:
Spontaneous recovery occurs in 90% of cases of acute ITP, while only less than 10% of cases of chronic ITP recover spontaneously.
- Treatment is directed at reducing the level and source of autoantibodies and reducing the rate of destruction of sensitised platelets.
- This is possible by corticosteroid therapy, immunosuppressive drugs (for example., vincristine, cyclophosphamide and azathioprine) and splenectomy.
- The beneficial effects of splenectomy in chronic ITP are due to both the removal of the major site of platelet destruction and the major source of autoantibody synthesis.
- Platelet transfusions are helpful as a palliative measure only in patients with severe haemorrhage.
Thrombotic Thrombocytopenic Purpura (TTP) and Haemolytic-Uraemic Syndrome (HUS):
- Thrombotic thrombocytopenic purpura (TTP) and haemolytic-uraemic syndrome (HUS) are a group of thrombotic microangiopathies which are essentially characterised by a triad of thrombocytopenia, microangiopathic haemolytic anaemia and formation of hyaline fibrin microthrombi within the microvasculature throughout the body.
- These are often fulminant and lethal disorders occurring in young adults. The intravascular microthrombi are composed predominantly of platelets and fibrin.
- The widespread presence of this platelet microthrombi is responsible for thrombocytopenia due to increased consumption of platelets, microangiopathic haemolytic anaemia and protean clinical manifestations involving different organs and tissues throughout the body.
Pathogenesis:
Unlike DIC, a clinicopathologically related condition, activation of the clotting system is not the primary event in the formation of microthrombi.
- TTP is initiated by endothelial injury followed by the release of von Willebrand factor and other procoagulant material from endothelial cells, leading to the formation of microthrombi.
- The trigger for endothelial injury comes from immunologic damage by diverse conditions such as pregnancy, metastatic cancer, high-dose chemotherapy, HIV infection, and mitomycin C.
Pathogenesis Clinical Features:
The clinical manifestations of TTP are due to microthrombi in the arterioles, capillaries and venules throughout the body. Besides features of thrombocytopenia and microangiopathic haemolytic anaemia, characteristic findings include fever, transient neurologic deficits and renal failure. The spleen may be palpable.
Laboratory Findings:
The diagnosis can be made from the following findings:
- Thrombocytopenia
- Microangiopathic haemolytic anaemia with negative Coombs’ test
- Leucocytosis, sometimes with leukemoid reaction
- Bone marrow examination reveals normal or slightly increased megakaryocytes accompanied by some myeloid hyperplasia.
- Diagnosis is, however, established by examination of biopsy (e.g. from gingiva) which demonstrates typical microthrombi in arterioles, capillaries and venules, unassociated with any inflammatory changes in the vessel wall.
Thrombocytosis:
- Thrombocytosis is defined as a platelet count of more than 4,00,000/µl. While essential or primary thrombocytosis or thrombocythaemia is discussed under myeloproliferative disorders, secondary or reactive thrombocytosis can occur following massive haemorrhage, iron deficiency, severe sepsis, marked inflammation, disseminated cancers, haemolysis, or following splenectomy.
- Thrombocytosis causes bleeding or thrombosis but how it produces is not clearly known. As such, transitory and secondary thrombocytosis does not require any separate treatment other than treating the cause.
Disorders Of Platelet Functions:
Defective platelet function is suspected in patients who show skin and mucosal haemorrhages and have prolonged bleeding time but a normal platelet count. These disorders may be hereditary or acquired.
Hereditary Disorders:
Depending upon the predominant functional abnormality, inherited disorders of platelet functions are classified into the following 3 groups:
1. Defective Platelet Adhesion:
These are as under:
- Bernard-Soulier syndrome is an autosomal recessive disorder with inherited deficiency of a platelet membrane glycoprotein which is essential for adhesion of platelets to vessel wall.
- In von Willebrand’s disease, there is defective platelet adhesion as well as deficiency of factor VII.
2. Defective platelet adhesion:
In thrombasthenia (Glanzmann’s disease), there is failure of primary platelet aggregation with ADP or collagen due to inherited deficiency of two of platelet membrane glycoproteins.
3. Disorders Of Platelet Release Reaction:
These disorders are characterised by normal initial aggregation of platelets with ADP or collagen but the subsequent release of ADP, prostaglandins and 5-HT is defective due to complex intrinsic deficiencies.
Acquired Disorders:
Acquired defects of platelet functions include the following clinically significant examples:
1. Aspirin Therapy:
- Prolonged use of aspirin leads to easy bruising and abnormal bleeding time. This is because aspirin inhibits the enzyme cyclooxygenase, and thereby suppresses the synthesis of prostaglandins which are involved in platelet aggregation as well as release reaction.
- The anti-platelet effect of aspirin is clinically applied in preventing major thromboembolic disease in recurrent myocardial infarction.
2. Others:
Several other acquired disorders are associated with various abnormalities in platelet functions at different levels. These include uraemia, liver disease, multiple myeloma, Waldenström’s macroglobulinaemia and various myeloproliferative disorders.
Bleeding due to Platelet Disorders:
- Platelet disorders may be due to abnormality in count (reduction or increased), or due to defective functions.
- Thrombocytopenia may result from impaired production, accelerated destruction, splenic sequestration, and dilutional loss and is rarely inherited.
- Many drugs and heparin may induce thrombocytopenia that is reversed on withdrawal of the offending drug.
- Idiopathic (or immune) thrombocytopenic purpura (ITP), is characterised by immunologic destruction of platelets and normal or increased megakaryocytes in the bone marrow. It may be acute or chronic.
- Thrombotic thrombocytopenic purpura (TTP) and haemolytic-uraemic syndrome (HUS) cases have thrombocytopenia, microangiopathic haemolytic anaemia and hyaline fibrin microthrombi within the microvasculature throughout the body.
- Defective platelet functions may be hereditary (platelet adhesion, aggregation, release reaction) or acquired (aspirin, uraemia, liver disease etc)
Bleeding Due To Coagulation Disorders:
The physiology of normal coagulation is described in Chapter 6 along with relatively more common coagulation disorders of arterial and venous thrombosis and embolism.
- Hypercoagulability or thrombophilia is an increased risk of thrombosis due to abnormality in haemostatic equilibrium (i.e. the reverse of abnormal bleeding).
- Here, coagulation disorders as a cause of bleeding are discussed. These are much less common causes of bleeding as compared with other bleeding disorders.
- The type of bleeding in coagulation disorders is different from that seen in vascular and platelet abnormalities.
- Instead of the spontaneous appearance of petechiae and purpuras, the plasma coagulation defects manifest more often in the form of large ecchymoses, haematomas and bleeding into muscles, joints, body cavities, GIT and urinary tract.
- A deficiency of each of the thirteen known plasma coagulation factors has been reported.
For establishing the diagnosis, screening tests for coagulation (whole blood coagulation time, bleeding time, activated partial thromboplastin time and prothrombin time) are carried out, followed by coagulation factor assays.
Disorders of plasma coagulation factors may have hereditary or acquired origin:
Hereditary coagulation disorders Most of the inherited plasma coagulation disorders are due to qualitative or quantitative defects in a single coagulation factor.
- Out of defects in various coagulation factors, two of the most common inherited coagulation disorders are the sex-(X)- linked disorders—classic haemophilia or haemophilia A (due to inherited deficiency of factor VIII), and Christmas disease or haemophilia B (due to inherited deficiency of factor IX).
- A much more common and related coagulation disorder is von Willebrand’s disease (due to an inherited defect of von Willebrand’s factor) discussed here.
Acquired coagulation disorders Acquired coagulation disorders, on the other hand, are usually characterised by deficiencies of multiple coagulation factors.
The most common acquired clotting abnormalities are:
vitamin K deficiency, coagulation disorder in liver diseases, fibrinolytic defects and disseminated intravascular coagulation (DIC). Common hereditary and acquired coagulation disorders are as under.
1. Classic Haemophilia (Haemophilia A):
Classic haemophilia or haemophilia A is the most common hereditary coagulation disorder occurring due to deficiency or reduced activity of factor VIII (anti-haemophilic factor).
- The disorder is inherited as a sex-(X-) linked recessive trait and, therefore, manifests clinically in males, while females are usually the carriers.
- However, rarely there may be true female haemophilics (homozygous state) arising from consanguinity within the family for example.,daughter born to a haemophiliac father and carrier mother.
- The chances of a proven carrier mother passing on the abnormality to her children are 50:50 for each son and 50:50 for each daughter.
- A haemophilic father cannot transmit the disorder to his sons (as they inherit his Y chromosome only that does not carry the genetic abnormality) while all his daughters will be asymptomatic carriers.
- However, cases without a family history of haemophilia are due to spontaneous somatic mutations of the gene coding for factor VIII.
The disease has been known since ancient times but Schönlein in 1839 gave this bleeder’s disease its present name haemophilia.
- In 1952, it was found that haemophilia was not always due to deficiency of factor VIII as was previously considered but instead, the blood of some patients was deficient in factor IX (Christmas factor or plasma thromboplastin component).
- Currently, haemophilia A (classic haemophilia) is an inherited deficiency of factor VIII due to a mutation in the F8 gene, and haemophilia B (Christmas disease) is an inherited deficiency of factor IX due to a mutation in the F9 gene.
- The frequency of haemophilia varies in different races, the highest incidence being in populations of Britain, Northern Europe and Australia.
- Haemophilia A comprises about 80% of cases. Western literature reports give an overall incidence of haemophilia A in 1 in 10,000 male births.
- Another interesting facet of haemophilia which has attracted investigators and researchers is the prevalence of this disorder in the blood of royal families in Great Britain and some European countries.
Pathogenesis:
Haemophilia A is caused by a quantitative reduction of factor VIII in 90% of cases, while 10% of cases have normal or increased levels of factor VIII with reduced activity.
- Factor VIII is synthesised in hepatic parenchymal cells and regulates the activation of factor X in the intrinsic coagulation pathway.
- Factor VIII circulates in blood complexed to another larger protein, von Willebrand’s factor (vWF), which comprises 99% of the factor VIII-vWF complex.
- The genetic coding, synthesis and functions of vWF are different from those of factor VIII and are considered separately below under von Willebrand’s disease. Normal haemostasis requires 25% factor VIII activity.
- Though occasional patients with 25% factor VIII level may develop bleeding, most symptomatic haemophilic patients have factor VIII levels below 5%.
Clinical Features:
Patients of haemophilia suffer from bleeding for hours or days after the injury. The clinical severity of the disease correlates well with the plasma level of factor VIII activity.
- Haemophilic bleeding can involve any organ but occurs most commonly as recurrent painful haemarthroses and muscle haematomas, and sometimes as haematuria.
- Spontaneous intracranial haemorrhage and oropharyngeal bleeding are rare, but when they occur they are the most feared complications.
Laboratory Findings:
The following tests are abnormal:
- Whole blood coagulation time is prolonged in severe cases only.
- Prothrombin time is usually normal.
- Activated partial thromboplastin time (APTT or PTTK) is typically prolonged.
- Specific assay for factor VIII shows lowered activity. The diagnosis of female carriers is made by the findings of about half the activity of factor VIII, while the manifest disease is associated with factor VIII activity below 25%.
Principles Of Treatment:
- Symptomatic patients with bleeding episodes are treated with factor VIII replacement therapy, consisting of factor VIII concentrates or plasma cryoprecipitates.
- With the availability of this treatment, the life expectancy of even severe haemophilic patients was approaching normal but the occurrence of AIDS in multitransfused haemophilic patients has adversely affected the life expectancy.
2. Christmas Disease (Haemophilia B):
Inherited deficiency of factor IX (Christmas factor or plasma thromboplastin component) produces Christmas disease or haemophilia Haemophilia B is rarer than haemophilia A constituting about 20% of cases; its estimated incidence is 1 in 100,000 male births.
- The inheritance pattern and clinical features of factor IX deficiency are indistinguishable from those of classic haemophilia but accurate laboratory diagnosis is critical since haemophilia B requires treatment with different plasma fractions.
- The usual screening tests for coagulation are similar to those in classic haemophilia but a bioassay of factor IX reveals lowered activity.
Principles Of Treatment:
- Therapy in symptomatic haemophilia B consists of an infusion of either fresh frozen plasma or plasma enriched with factor IX.
- Besides the expected possibilities of complications of hepatitis, chronic liver disease and AIDS, the replacement therapy for factor IX deficiency may activate the coagulation system and cause thrombosis and embolism.
3. Von Willebrand’s Disease:
Definition And Pathogenesis:
- von Willebrand’s disease (vWD) is the most common hereditary coagulation disorder occurring due to qualitative or quantitative defects in von Willebrand’s factor (vWF). Its incidence is estimated to be 1 in 1,000 individuals of either sex.
- The vWF comprises the larger fraction of the factor VIII-vWF complex which circulates in the blood.
- The two components of the factor VIII-vWF complex circulate together as a unit and perform the important function of clotting and facilitate platelet adhesion to subendothelial collagen.
vWF differs from factor VIII in the following respects:
- The gene for vWF is located at chromosome 12, while that of factor VIII is in X chromosome. Thus, vWD is inherited as an autosomal dominant trait which may occur in either sex, while factor VIII deficiency (haemophilia A) is a sex (X-)-linked recessive disorder.
- The vWF is synthesised in the endothelial cells, megakaryocytes and platelets but not in the liver cells, while the principal site of synthesis of factor VIII is the liver.
- The main function of vWF is to facilitate the adhesion of platelets to subendothelial collagen, while factor VIII is involved in the activation of factor X in the intrinsic coagulation pathway.
Clinical Features:
Clinically, the patients of vWD are characterised by spontaneous bleeding from mucous membranes and excessive bleeding from wounds.
There are 3 major types of vWD:
- Type I disease is the most common and is characterised by a mild to moderate decrease in plasma vWF (50% activity). The synthesis of vWF is normal but the release of its multimers is inhibited.
- Type II disease is much less common and is characterised by normal or near-normal levels of vWF which is functionally defective.
- Type III disease is extremely rare and is the most severe form of the disease. These patients have no detectable vWF activity and may have sufficiently low factor VIII levels.
- Bleeding episodes in vWD are treated with cryoprecipitates or factor VIII concentrates.
LABORATORY FINDINGS These are as under:
- Prolonged bleeding time
- Normal platelet count
- Reduced plasma vWF concentration
- Defective platelet aggregation with ristocetin, an antibiotic
- Reduced factor VIII activity
4. Vitamin K Deficiency:
Vitamin K is a fat-soluble vitamin which plays an important role in haemostasis since it serves as a cofactor in the formation of 6 prothrombin complex proteins (vitamin K-dependent coagulation factors) synthesised in the liver: factor II, VII, IX, X, protein C and protein S. Vitamin K is obtained from green vegetables, absorbed in the small intestine and stored in the liver.
Some quantity of vitamin K is endogenously synthesised by the bacteria in the colon. Vitamin K deficiency may present in the newborn or in subsequent childhood or adult life:
- Neonatal vitamin K deficiency Deficiency of vitamin K in the newborn causes haemorrhagic disease of the newborn.
- Liver cell immaturity, lack of gut bacterial synthesis of the vitamin and low quantities in breast milk, all contribute to vitamin K deficiency in the newborn and may cause haemorrhage on the 2nd to 4th day of life.
- Routine administration of vitamin K to all newborn infants has led to the disappearance of neonatal vitamin K deficiency.
- Vitamin K deficiency in children and adults There are 3 major causes of vitamin K deficiency in childhood or adult life:
- Inadequate dietary intake
- Intestinal malabsorption
- Loss of storage site due to hepatocellular disease
With the onset of vitamin K deficiency, the plasma levels of all the 6 vitamin K-dependent factors (prothrombin complex proteins) fall. This, in turn, results in prolonged PT and PTTK. Parenteral administration of vitamin K rapidly restores vitamin K levels in the liver.
5. Coagulation Disorders In Liver Disease:
Since the liver is the major site for the synthesis and metabolism of coagulation factors, liver disease often leads to multiple haemostatic abnormalities.
- The liver also produces inhibitors of coagulation such as antithrombin III and proteins C and S and plays a role in the clearance of activated factors and fibrinolytic enzymes.
- Thus, patients with liver disease may develop hypercoagulability and are predisposed to develop DIC and systemic fibrinolysis.
The major causes of bleeding in liver diseases are as under:
1. Morphologic lesions:
- Portal hypertension for example., varices, splenomegaly with secondary thrombocytopenia
- Peptic ulceration
- Gastritis
2. Hepatic dysfunctions:
- Impaired hepatic synthesis of coagulation factors
- Impaired hepatic synthesis of coagulation inhibitors: protein C, protein S and antithrombin III
- Impaired absorption and metabolism of vitamin K
- Failure to clear activated coagulation factors causing DIC and systemic fibrinolysis
3. Complications of therapy:
- Following massive transfusion leads to dilution of platelets and coagulation factors.
- Infusion of activated coagulation proteins.
- Following heparin therapy
Many times, the haemostatic abnormality in liver disease is complex but most patients have prolonged PT and PTTK, mild thrombocytopenia, normal fibrinogen level and decreased hepatic stores of vitamin K.
Bleeding Due To Fibrinolytic Defects:
Normally, fibrinolysis consisting of plasminogen-plasmin and fibrin degradation products (FDPs) is an essential protective physiologic mechanism to limit blood coagulation in the body. However, unchecked and excessive fibrinolysis may sometimes be the cause of bleeding.
The causes of primary pathologic fibrinolysis leading to haemorrhagic defects are as under:
Deficiency of α2-plasmin inhibitor following trauma or surgery.
- Impaired clearance of tissue plasminogen activator such as in cirrhosis of the liver.
- At times, it may be difficult to distinguish primary pathologic fibrinolysis from secondary fibrinolysis accompanying DIC.
Bleeding due to Coagulation Disorders:
- Disorders of coagulation factors may be hereditary or acquired.
- Common hereditary coagulation disorders are haemophilia A (due to deficiency or reduced activity of factor VIII), haemophilia B (due to deficiency of factor IX), and von Willebrand disease (due to qualitative or quantitative defect in von Willebrand’s factor (vWF)
- Common acquired causes are vitamin K deficiency (in neonates, children and adults) and liver diseases causing reduced synthesis of coagulation factors (e.g. morphologic liver lesions, hepatic dysfunction or as a complication of therapy).
- Uncontrolled and excessive fibrinolysis may sometimes be the cause of bleeding.
Disseminated Intravascular Coagulation (Dic):
Disseminated intravascular coagulation (DIC), also termed defibrination syndrome or consumption coagulopathy, is a complex thrombo-haemorrhagic disorder (intravascular coagulation and haemorrhage) occurring as a secondary complication in some systemic diseases.
Aetiology:
Although there are numerous conditions associated with DIC, the most frequent causes are listed below:
Massive tissue injury In obstetrical syndromes (e.g. abruptio placentae, amniotic fluid embolism, retained dead foetus), massive trauma, metastatic malignancies, surgery.
- Infections Especiallyendotoxaemia, gram-negative and meningococcal septicaemia, certain viral infections, malaria, and aspergillosis.
- Widespread endothelial damage In aortic aneurysm, haemolytic-uraemic syndrome, severe burns, acute glomerulonephritis.
- Miscellaneous: Snakebite, shock, acute intravascular haemolysis, heat stroke.

Pathogenesis:
Although in each case, a distinct triggering mechanism has been identified, the sequence of events, in general, can be summarised as under:
1. Activation of coagulation The etiologic factors listed above initiate widespread activation of the coagulation pathway by the release of tissue factors.
2. Thrombotic phase Endothelial damage from the various thrombogenic stimuli causes generalised platelet aggregation and adhesion with resultant deposition of small thrombi and emboli throughout the microvasculature.
3. Consumption phase The early thrombotic phase is followed by a phase of consumption of coagulation factors and platelets.
4. Secondary fibrinolysis As a protective mechanism, the fibrinolytic system is secondarily activated at the site of intravascular coagulation. Secondary fibrinolysis causes a breakdown of fibrin resulting in the formation of FDPs in the circulation. The pathophysiology of DIC is summed up schematically.
Clinical Features:
There are 2 main features of DIC—bleeding as the most common manifestation, and organ damage due to ischaemia caused by the effect of widespread intravascular thrombosis such as in the kidney and brain.
Less common manifestations include:
Microangiopathic haemolytic anaemia and thrombosis in larger arteries and veins.

Laboratory Findings:
The laboratory investigations include the following:
- The platelet count is low.
- Blood film shows the features of microangiopathic haemolytic anaemia. There is the presence of schistocytes and fragmented red cells due to damage caused by trapping and passage through the fibrin thrombi.
- Prothrombin time, thrombin time and activated partial thromboplastin time, are all prolonged.
- Plasma fibrinogen levels are reduced due to consumption in microvascular coagulation.
- Fibrin degradation products (FDPs) are raised due to secondary fibrinolysis.

A summary of important laboratory findings in common causes of haemostatic abnormalities is summed up in Table.
Disseminated intravascular Coagulat (DIC):
- Disseminated intravascular coagulation (DIC) is a complex of intravascular coagulation and bleeding. It may occur from various causes e.g. massive tissue injury, fulminant infections, profuse endothelial damage etc.
- DIC passes through phases of activation of coagulation, thrombotic phase, consumptive phase and secondary fibrinolysis.
- Major findings in DIC are thrombocytopenia, prolonged prothrombin time, reduced fibrinogen, and the presence of FDPs
Transfusion Biology
Karl Landsteiner described the existence of major human blood groups in 1900 and was awarded the Nobel Prize in 1930.
- The term blood group is applied to any well-defined system of red blood cell antigens which are inherited characteristics.
- Over 20 blood group systems having approximately 400 blood group antigens are currently recognised. The ABO and Rhesus (Rh) blood group systems are of major clinical significance.
Other minor and clinically less important blood group systems are:
- Lewis system, P system, I system, MNS system, Kell and Duffy system, and Luthern system.
- Besides transfusion and its complications, a brief account of haemolytic disease of newborns which is closely related to blood group systems between mother and developing foetus, is also given here.
Blood Group Antigens And Antibodies:
- Individuals who lack the corresponding antigen and have not been previously transfused have naturally-occurring antibodies in their serum. The most important are anti-A and anti-B antibodies, usually of the IgM class.
- Immune antibodies, on the other hand, are acquired in response to transfusion and by transplacental passage during pregnancy. These are warm antibodies, usually of IgG class.
Abo System:
This system consists of 3 major allelic genes: A, B and O, located on the long arm of chromosome 9. These genes control the synthesis of blood group antigens A and B.
- The serum of an individual contains naturally-occurring antibodies to A and/or B antigen, whichever antigen is lacking in the person’s red cells.
- Two subgroups of A—A1 and A2, and thus of AB also, A1B and A2B, are recognised but are of minor clinical significance. In routine practice, the ABO type is determined by testing the red blood cells with anti-A and anti-B and by testing the serum against A, B and O red blood cells.
- Red blood cells of type O and A2 have large amounts of another antigen called H substance which is genetically different from ABO but is a precursor of A and B antigens.
- An O group individual who inherits A or B genes but fails to inherit the H gene from either parent is called Oh phenotype or Bombay blood group.
- In such rare individuals, despite the presence of all three antibodies in serum (anti-A, anti-B and anti-H), the red cells are not agglutinated by the antisera.
Rhesus System:
The Rhesus (Rh) blood group system was first discovered on human red cells by the use of antisera prepared by immunising rabbits with red cells from a Rhesus monkey. The Rh allelic genes are C or c, D or d and E or e, located on chromosome 1.
- One set of 3 genes is inherited from each parent giving rise to various complex combinations. The corresponding antigens are similarly named Cc, Ee and only D since no d antigen exists.
- However, out of all these, D antigen is most strongly immunogenic and, therefore, clinically most important. In practice, Rh grouping is performed with anti-D antiserum. Individuals who are D-positive are referred to as Rh-positive and those who lack D antigen are termed Rh-negative. Practically, there are no naturally occurring Rh antibodies.
- All Rh antibodies in Rh-negative individuals are acquired from immunisation such as by transfusion and during pregnancy, resulting in fatal haemolytic transfusion reaction and haemolytic disease of the newborn (described later).

*Persons with blood group AB Rh-positive are called universal recipients.
**Individuals with blood group O Rh-negative are called universal donors.
Transfusion Process:
Haemostatic System, Bleeding Disorders and Transfusion Biology the ABO blood groups
- Healthy blood donor selection with haemoglobin of more than 12.5 g/dl and free of infectious diseases.
- Blood collection by phlebotomy in a sterile plastic blood bag containing anticoagulant, commonly acid citrate, mixing blood and coagulant gently while blood is flowing into the bag.
- Storage of blood bag in refrigerator at 2° to 6° C.
- Donor testing for ABO-Rh grouping, and infectious organisms (HBV, HBsAg, HCV, syphilis and malaria).
- Blood grouping (ABO-Rh) of the recipient.
- Pre-transfusion compatibility testing of donor and recipient is as under:
- Antibody screening of the patient’s serum to detect the presence of clinically significant antibodies.
- Cross-matching the patient’s serum against donor red cells to confirm donor-recipient compatibility.
- Supervised blood transfusion to observe reactions if any.
- Anticoagulants used for the collection of whole blood are CPD or CPDA (C=Citrate, prevents clotting; P=Phosphate, source of 2,3 DPG and maintains pH; D=Dextrose, generates ATP by glycolysis; A=Adenine, a substrate for ATP synthesis):
- CPD: preserves unit of blood for 21 days at 1-6°C
- CPDA: preserves unit of whole blood for 35 days at 1-6°C
- Indications for blood transfusion are acute blood loss and various hematologic disorders discussed already.
Blood Components:
Blood from donors is collected as whole blood in a suitable anticoagulant. Nowadays it is a common practice to divide whole blood into components which include: packed RBCs, platelets, fresh-frozen plasma (FFP) and cryoprecipitate.
- The procedure consists of initial centrifugation at low speed to separate whole blood into two parts: packed RBCs and platelet-rich plasma (PRP).
- Subsequently, PRP is centrifuged at high speed to yield two parts: random donor platelets and FFP.
- Cryoprecipitates are obtained by thawing of FFP followed by centrifugation. Apheresis is the technique of direct collection of a large excess of platelets from a single donor.
Applications of these blood components in clinical use are as under:
1. Packed RBCs These are used to raise the oxygen-carrying capacity of blood and are used in normovolaemic patients of anaemia without cardiac disease. One unit of packed RBCs may raise haemoglobin by 1 g/dl.
2. Platelets Transfusion of platelets is done in patients of thrombocytopenia who have haemorrhage. Optimally, platelet transfusions can be given to a patient with a platelet count below 10,000/µl. Each unit of platelets can raise platelet count by 5,000 to 10,000/µl.
3. Fresh frozen plasma FFP contains plasma proteins and coagulation factors that include albumin, protein C and S and antithrombin. FFP transfusion in indicated in patients of coagulation failure and TTP. Each unit of FFP raises coagulation factors by about 2%.
4. Cryoprecipitate Cryoprecipitate is a source of insoluble plasma proteins, fibrinogen, factor VIII and vWF. Indications for transfusion of cryoprecipitate are for patients requiring fibrinogen, factor VIII and vWF. Transfusion of a single unit of cryoprecipitate yields about 80 IU of factor VIII.
Complications Of Blood Transfusion:
A carefully prepared and supervised blood transfusion is quite safe. However, in 5-6% of transfusions, untoward complications occur, some of which are minor while others are more serious and at times fatal.
Transfusion reactions are generally classified into 2 types: immune and non-immune.
- Immunologic transfusion reactions may be against red blood cells (haemolytic reactions), leucocytes, platelets or immunoglobulins.
- Non-immune transfusion reactions include circulatory overload, massive transfusion, or transmission of an infectious agent.
These transfusion reactions are considered below:
1. Immunologic Transfusion Reactions:
These are as under:
1. Haemolytic transfusion reactions Haemolytic transfusion reactions may be immediate or delayed, intravascular or extravascular.
- Very rapid cell destruction associated with intravascular haemolysis is usually due to ABO incompatibility since both naturally-occurring antibodies, anti-A and anti-B, are capable of fixing complement. The symptoms include restlessness, anxiety, flushing, chest or lumbar pain, tachypnoea, tachycardia and nausea, followed by shock and renal failure.
- Extravascular: Haemolysis is more often due to immune antibodies of the Rh system. The clinical manifestations are relatively less severe and usually consist of malaise and fever but shock and renal failure may rarely occur.
- Some patients develop delayed reactions in which the patient develops anaemia due to the destruction of red cells in the RE system about a week after transfusion. Such delayed reactions are generally the result of a previous transfusion or pregnancy (anamnestic reaction).
2. Transfusion-related acute lung injury (TRALI):
- This is an uncommon reaction resulting from transfusion of donor plasma containing high levels of anti-HLA antibodies which bind to the leucocytes of the recipient.
- These leucocytes then aggregate in pulmonary microcirculation and release mediators of increased vascular permeability resulting in acute pulmonary oedema and signs and symptoms of respiratory failure.
3. Other allergic reactions:
Besides haemolytic transfusion reaction, other reactions are as
follows:
- The febrile reaction which is usually attributed to an immunologic reaction against white blood cells, platelets, or IgA-class immunoglobulins.
- Patients with antibodies against IgA molecules sometimes develop anaphylactic shock on transfusion of blood from other human subjects.
- Allergic reactions such as urticaria may occur.
- Transfusion-related graft-versus-host disease mediated by donor T lymphocytes may occur.
Nonimmune Transfusion Reactions:
This category includes the following adverse effects:
1. Circulatory overload resulting in pulmonary congestion and acute heart failure is the most important and most common complication that may result in death following transfusion. The risk of circulatory overload is particularly high in patients with chronic anaemia and in infants and the elderly. The onset may be immediate or may be delayed up to 24 hours.
2. Massive transfusion When the volume of stored blood transfused to bleeding patients exceeds their normal blood volume, it results in dilutional thrombocytopenia and dilution of coagulation factors.
3. Transmission of infection Some infectious diseases can be transmitted by transfusion of contaminated blood. These include hepatitis (HBV, HCV), CMV infection, syphilis, malaria, toxoplasmosis, infectious mononucleosis, AIDS (HIV infection), brucellosis and babesiosis.
The incidence increases in patients who receive multiple transfusions such as cases of haemophilia, thalassaemia major, acute leukaemias, acute severe haemorrhage etc. It has, therefore, been mandatory that before any human transfusion, every unit of blood must be screened for the serologic testing of HIV, HBV, HCV and syphilis and for the presence of malarial parasites.
4. Air embolism Air embolism is unlikely to occur if the blood transfusion is carried out with plastic bags with negative pressure as is the usual practice nowadays. A debilitated person may develop symptomatic air embolism even if a small volume (10-40 ml) makes its way into the circulation, whereas a healthy individual is at lesser risk.
5. Thrombophlebitis: The complication of thrombophlebitis is more commonly associated with venesection for blood transfusion, especially if it is done in the saphenous vein of the ankle rather than the veins of the arm. The risk of developing thrombophlebitis is further enhanced if the transfusion is continued longer than 12 hours at a single site.
6. Transfusion haemosiderosis Post-transfusion iron overload with deposition of iron in the tissues of the body occurs after repeated transfusions in the absence of any blood loss for example., in thalassaemia major and severe chronic refractory anaemias.
- The body has no other means of getting rid of extra iron except iron excretion at the rate of 1 mg per day.
- A unit of whole blood (400 ml) contains about 250 mg of iron. After approximately 100 units, the liver, myocardium and endocrine glands are all damaged.
Blood Transfusion:
- The ABO system consists of A, B and O allelic genes, which control the synthesis of blood group antigens A and B. The serum of an individual contains naturally-occurring antibodies to A and or B antigen.
- D antigen is strongly immunogenic and individuals may be D-positive (i.e. Rh-positive) or D-negative (i.e. Rh-negative) but there are no naturally-occurring Rh antibodies.
- Complications of blood transfusion may be immunologic (e.g. haemolytic reactions, acute lung injury) or non-immunologic reactions (e.g. circulatory overload, infections, air embolism, thrombophlebitis etc).
- Nowadays it is a common practice to divide whole units of blood into components for optimum utilisation such as packed RBCs, platelets, fresh-frozen plasma (FFP) and cryoprecipitate
Haemolytic Disease Of Newborn:
Haemolytic disease of the newborn (HDN) results from the passage of IgG antibodies from the maternal circulation across the placenta into the circulation of the foetal red cells.
- Besides pregnancy, sensitisation of the mother may result from previous abortions and previous blood transfusions.
- HDN can occur from incompatibility of the ABO or Rh blood group system. ABO incompatibility is much more common but the HDN in such cases is usually mild, while Rh-D incompatibility results in a more severe form of the HDN.
Pathogenesis:
The pathogenesis of the two main forms of HDN is different.
HDN due to Rh-D incompatibility Rh incompatibility occurs when a Rh-negative mother is sensitised to Rh-positive blood.
- This results most often from an Rh-positive foetus by the passage of Rh-positive red cells across the placenta into the circulation of an Rh-negative mother.
- Normally, during pregnancy, very few foetal red cells cross the placenta but haemorrhage during parturition causes significant sensitisation of the mother.
- Sensitisation is more likely if the mother and foetus are ABO compatible rather than ABO incompatible.
- Though approximately 95% of cases of Rh-HDN are due to anti-D, some cases are due to a combination of anti-D with other immune antibodies of the Rh system such as anti-C and anti-E and rarely anti-C alone.
- It must be emphasised here that the risk of sensitisation of a Rh-negative woman married to a Rh-positive man is small in the first pregnancy but increases during successive pregnancies if prophylactic anti-D immunoglobulin is not given within 72 hours after the first delivery.
- If both the parents are Rh-D positive (homozygous), all the newborns will be Rh-D positive, while if the father is Rh-D positive (heterozygous), there is a 50% chance of producing a Rh-D negative child.
HDN due to ABO incompatibility About 20% of pregnancies with ABO incompatibility between the mother and the foetus develop the HDN.
- Naturally occurring anti-A and anti-B antibodies’ which are usually of IgM class do not cross the placenta, while immune anti-A and anti-B antibodies which are usually of IgG class may cross the placenta into foetal circulation and damage the foetal red cells.
- ABO-HDN occurs most frequently in infants born to group O mothers who possess anti-A and/or anti-B IgG antibodies.
- ABO-HDN differs from Rh(D)-HDN, in that it occurs in a first pregnancy, Coombs’ (antiglobulin) test is generally negative and is less severe than the latter.
Clinical Features:
- The HDN due to Rh-D incompatibility in its severest form may result in intrauterine death from hydrops foetalis.
- Moderate disease produces a baby born with severe anaemia and jaundice due to unconjugated hyperbilirubinaemia.
- When the level of unconjugated bilirubin exceeds 20 mg/dl, it may result in the deposition of bile pigment in the basal ganglia of the CNS called kernicterus and result in permanent brain damage.
- Mild disease, however, causes only severe anaemia with or without jaundice.
Laboratory Findings:
The haematologic findings in cord blood and mother’s blood are as under:
Cord blood shows variable degrees of anaemia, reticulocytosis, elevated serum bilirubin and a positive direct Coombs’ test if the cord blood is Rh-D positive. Mother’s blood is Rh-D negative with a high plasma titre of anti-D.
Course And Prognosis:
The course in HDN may range from minimal haemolysis to mental retardation, or death.
- The practice of administration of anti-Rh immunoglobulin to the mother before or after delivery has reduced the incidence of HDN as well as protects the mother before the baby’s RBCs sensitise the mother’s blood.
- Exchange transfusion of the baby is done to remove the antibodies, remove red cells susceptible to haemolysis and also to lower the bilirubin level.
Haemolytic Disease of Newborn:
- Haemolytic disease of newborn results from the passage of IgG antibodies from the maternal circulation across the placenta into the circulation of the foetal red cells.
- It could be due to incompatibility of Rh or ABO groups.
- Cord blood shows anaemia and is Rh-D positive while mother’s blood is Rh-D negative
Leave a Reply