Introduction To Haematopoietic System And Disorders Of Erythroid Series
Bone Marrow And Haematopoiesis
Haematopoiesis is the production of formed elements of the blood. Normally, it takes place in the bone marrow. Circulating blood normally contains 3 main types of mature blood cells—the red cells (erythrocytes), the white cells (leucocytes) and the platelets (thrombocytes).
Table of Contents
These blood cells perform their respective major physiologic functions:
Erythrocytes are largely concerned with oxygen transport, leucocytes play various roles in body defense against infection and tissue injury, while thrombocytes are primarily involved in maintaining the integrity of blood vessels and in preventing blood loss. The lifespan of these cells in circulating blood is variable—neutrophils have a short lifespan (<24 hours), followed by platelets (8-10 days), while the RBCs have the longest lifespan (90-120 days).
Read And Learn More: General Pathology Notes
The rates of production of these blood cells are normally regulated in healthy individuals in such a way so as to match the rate at which they are lost from circulation. Their concentration is normally maintained within well-defined limits unless the balance is disturbed due to some pathologic processes.
Haematopoietic Organs:
- In the human embryo, the yolk sac is the main site of haematopoiesis in the first few weeks of gestation.
- By about 3rd month, however, the liver and spleen are the main sites of blood cell formation and continue to do so until about 2 weeks after birth.
- Haematopoiesis commences in the bone marrow by 4th and 5th month and becomes fully active by 7th and 8th month so that at birth practically all the bones contain active marrow.
During normal childhood and adult life, therefore, the marrow is the only source of new blood cells. However, during childhood, there is progressive fatty replacement throughout the long bones so that by adult life the haematopoietic marrow is confined to the central skeleton (vertebrae, sternum, ribs, skull, sacrum and pelvis) and proximal ends of femur, tibia and humerus.
Even in these haematopoietic areas, about 50% of the marrow consists of fat. Non-haematopoietic marrow in the adult is, however, capable of reverting to active haematopoiesis in certain pathologic conditions. The spleen and liver can also resume their foetal haematopoietic role in certain pathologic conditions and is called extramedullary haematopoiesis.
In the bone marrow, developing blood cells are situated outside the marrow sinuses, from where after maturation they enter the marrow sinuses, the marrow microcirculation and then released into circulation.
Haematopoietic Stem Cells:
Haematopoiesis involves two stages: mitotic division or proliferation, and differentiation or maturation. It is known for a few decades that blood cells develop from a small population of common multipotent haematopoietic stem cells (HSC).
Haematopoietic stem cells have the appearance of small or intermediate-sized lymphocytes and their presence in the marrow can be demonstrated by cell culture techniques by the growth of colony-forming units (CFU) of different cell lines. The bone marrow provides a suitable microenvironment for growth and development of HSC.
For instance, if HSC are infused intravenously into a suitably-prepared recipient, they seed the marrow successfully but do not thrive at other sites. This principle forms the basis of bone marrow (or HSC) transplantation performed for various haematologic diseases.
HSC have following essential features:


- They have capacity to differentiate into a variety of mature cell types.
- They have capacity for self-renewal.
- They express a variety of cell surface molecules: CD34, CD90 (Thy-1), CD117 (c-kit receptor), CD133, CD164 and CD110 (thrombopoietin factor).
- HSC possess certain adhesion molecules which assigns them the feature of mobility. These are integrins, VCAM-1 and chemokine CXCR4 receptor. When HSC are infused during transplant, they ‘home’ to the bone marrow due to chemokine receptor CXCR4.
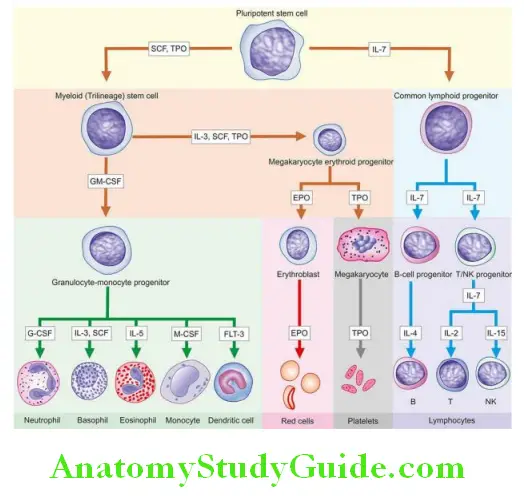
After a series of divisions, HSC differentiate into two types of progenitors—lymphoid (immune system) stem cells, and non-lymphoid or myeloid (trilineage) stem cells. The former develop into T, B and NK cells while the latter differentiate into 3 types of cell lines—granulocyte-monocyte progenitors (producing neutrophils, eosinophils, basophils and monocytes), erythroid progenitors (producing red cells), and megakaryocytes (as the source of platelets).
Monocytes on entering the tissues form a variety of phagocytic macrophages, both of which together constitute mononuclear-phagocyte system. Lymphopoietic cells in the marrow undergo differentiation to form B, T and natural killer (NK) cells of the immune system.
The development of mature cells (i.e. poiesis)—red cells (erythropoiesis), granulocytes (granulopoiesis), monocytes, lymphocytes (lymphopoiesis) and platelets (thrombopoiesis) are considered in detail later under relevant headings. Haematopoiesis or myelopoiesis is regulated by certain endogenous glycoproteins called haematopoietic growth factors, cytokines and hormones.
For example:
- Erythropoietin: for red cell formation
- Granulocyte colony-stimulating factor (G-CSF): for production of granulocytes
- Granulocyte-macrophage colony-stimulating factor (GM-CSF): for production of granulocytes and monocyte-macrophages
- Thrombopoietin; for production of platelets
Each of these growth factors acts on their specific receptors to initiate further cell events as discussed under respective topics later.
Bone Marrow Examination:
Examination of the bone marrow provides an invaluable diagnostic help in some cases, while in others it is of value in confirming a diagnosis suspected on clinical examination or on the blood film.
A peripheral blood smear examination, however, must always precede a bone marrow examination. Bone marrow examination may be performed by two methods—aspiration and trephine biopsy. A comparison of the two methods is summarised in Table.
Bone Marrow Aspiration:
The method involves the suction of marrow via a strong, wide-bore, short-bevelled needle fitted with a stylet and an adjustable guard to prevent excessive penetration; for instance Salah bone marrow aspiration needle.
Smears are prepared immediately from the bone marrow aspirate and are fixed in 95% methanol after air-drying. The usual Romanowsky technique is employed for staining and a stain for iron is performed routinely to assess the reticuloendothelial stores of iron.

The marrow film provides assessment of cellularity, details of developing blood cells (i.e. normoblastic or megaloblastic, myeloid, lymphoid, macrophages and megakaryocytic), ratio between myeloid and erythroid (M: E ratio) cells, storage diseases, and for the presence of cells foreign to the marrow such as secondary carcinoma, granulomatous conditions, fungi (e.g. histoplasmosis) and parasites (e.g. malaria, leishmaniasis, trypanosomiasis).
Estimation of the proportion of cellular components in the marrow, however, can be provided by doing a differential count of at least 500 cells (myelogram) In some conditions, the marrow cells can be used for more detailed special tests such as cytogenetics, microbiological culture, biochemical analysis, and immunological and cytological markers.
Trephine Biopsy:
Trephine biopsy is performed by a simple Jamshidi trephine needle by which a core of tissue from the periosteum to bone marrow cavity is obtained. The tissue is then fixed, soft decalcified and processed for histological sections and stained with haematoxylin and eosin and for reticulin. Trephine biopsy is useful over aspiration since it provides an excellent view of the overall marrow architecture, cellularity, and presence or absence of infiltrates, but is less valuable than aspiration as far as individual cell morphology is concerned.

Bone Marrow and Haematopoiesis:
- Haematopoiesis commences in the bone marrow by 4th and 5th month and becomes fully active by 7th and 8th month. During normal childhood and adult life, the marrow is the only source of new blood cells.
- Haematopoietic stem cells (HSC) in the bone marrow give rise to two types of multipotent stem cells: non-lymphoid which differentiate into committed trilineage series (granulocyte-monocyte, erythroid, megakaryocytic precursors), and lymphoid stem cells which differentiate in the bone marrow and then migrate to the lymphoid tissues (T, B and NK cells).
- Haematopoiesis is regulated by a few factors such as erythropoietin, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor and thrombopoietin.
- Examination of the bone marrow provides invaluable diagnostic help and confirms a suspected diagnosis. Bone marrow examination may be performed by two methods—aspiration and trephine biopsy.
- Major indications of bone marrow aspiration are typing of anaemias, leukaemias, neutropenia and marrow infiltrations, while trephine has additional advantages in dry aspiration, myelofibrosis and aplastic anaemia.
Fat/cell ratio: 50:50
Myeloid/erythroid (M/E) ratio: 2-4:1 (mean 3:1)
Myeloid series: 30-45% (37.5%)
- Myeloblasts: 0.1-3.5%
- Promyelocytes: 0.5-5%
Erythroid series: 10-15% (mean 12.5%)
Megakaryocytes: 0.5%
Lymphocytes: 5-20%
Plasma cells: < 3%
Reticulum cells: 0.1-2%
Erythropoiesis
Erythropoiesis is the production of mature erythrocytes of the peripheral blood which takes place in the bone marrow from morphologically unrecognisable HSC. Red cell production is influenced by growth factors and hormones, notably erythropoietin.
Erythropoietin:
Erythropoietic activity in the body is regulated by erythropoietin, which is produced in response to anoxia. The principal site of erythropoietin production is the kidney though there is evidence of its extra-renal production in certain unusual circumstances. Its levels are, therefore, lowered in chronic renal diseases, while a case of renal cell carcinoma may be associated with its increased production and erythrocytosis.
Erythropoietin acts on the marrow at the various stages of morphologically unidentifiable as well as identifiable erythroid precursors. Immunoassay of erythropoietin in plasma or serum can be done by sensitive techniques (ELISA and radioimmunoassay) due to its quite low values; normal values are 10-25 U/L.
Significance:
- There is an increased production of erythropoietin in most types of anaemias. However, in anaemia of chronic diseases (e.g. in infections and neoplastic conditions) there is no such enhancement of erythropoietin.
- In polycythemia rubra vera, there is erythrocytosis but a depressed production of erythropoietin. This is because of an abnormality of HSC class which is not under erythropoietin control.
Besides erythropoietin, androgens and thyroxine also appear to be involved in red cell production.
Erythroid Series:
Erythroid series is a well-defined and readily recognisable lineage of nucleated red cells normally confined to the marrow.
These are as under:

1. Proerythroblast:
The earliest recognisable cell in the marrow is a proerythroblast or pronormoblast. It is a large cell, 15-20 µm in diameter having deeply basophilic cytoplasm and a large central nucleus containing nucleoli. The deep blue colour of the cytoplasm is due to the high content of RNA which is associated with active protein synthesis.
As the cells mature, the nuclei lose their nucleoli and become smaller and denser, while the cytoplasm on maturation leads to the replacement of dense blue colour progressively by pink-staining haemoglobin. Each proerythroblast undergoes 4-5 replications and forms 16-32 mature RBCs.
2. Basophilic (Early) Erythroblast:
It is a round cell having a diameter of 12-16 µm with a large nucleus which is slightly more condensed than the proerythroblast and contains basophilic cytoplasm. Basophilic erythroblast undergoes rapid proliferation.
3. Polychromatic (Intermediate) Erythroblast:
The next maturation stage has a diameter of 12-14 µm. The nucleus at this stage is coarse and deeply basophilic. The cytoplasm is characteristically polychromatic i.e. contains an admixture of basophilic RNA and acidophilic haemoglobin. The cell at this stage ceases to undergo proliferative activity.
4. Orthochromatic (Late) Erythroblast:
The final stage in the maturation of nucleated red cells is the orthochromatic or late erythroblast. The cell at this stage is smaller, 8-12 µm in diameter, containing a small and pyknotic nucleus with dark nuclear chromatin. The cytoplasm is characteristically acidophilic with diffuse basophilic hue due to the presence of large amounts of haemoglobin.
5. Reticulocyte:
The nucleus is finally extruded from the late erythroblast within the marrow and a reticulocyte results. The reticulocytes are juvenile red cells devoid of nuclei but contain ribosomal RNA so that they are still able to synthesise haemoglobin. A reticulocyte spends 1-2 days in the marrow and circulates for 1-2 days in the peripheral blood before maturing in the spleen, to become a biconcave red cell.
The reticulocytes in the peripheral blood are distinguished from mature red cells by a slightly basophilic hue in the cytoplasm similar to that of an orthochromatic erythroblast. Reticulocytes can be counted in the laboratory by vital staining with dyes such as new methylene blue or brilliant cresyl blue. The reticulocytes by either of these staining methods contain deep blue reticulofilamentous material.
While erythroblasts are not normally present in human peripheral blood, reticulocytes are found normally in the peripheral blood. The normal range of reticulocyte count in health is 0.5-2.5% in adults and 2-6% in infants. Their percentage in the peripheral blood is a fairly accurate reflection of erythropoietic activity. Their proportion is increased in conditions of rapid red cell regeneration e.g. after haemorrhage, haemolysis and haematopoietic response of anaemia to treatment.
The Red Cell:
The mature erythrocytes of the human peripheral blood are non-nucleated cells and lack the usual cell organelles. The normal human erythrocyte is a biconcave disc, 7.2 µm in diameter, and has a thickness of 2.4 µm at the periphery and 1 µm in the centre. The biconcave shape renders the red cells quite flexible so that they can pass through capillaries whose minimum diameter is 3.5 µm. More than 90% of the weight of erythrocytes consists of haemoglobin. The lifespan of red cells is 120 ± 30 days.
Red Cell Membrane:
The red cell membrane is a trilaminar structure having a bimolecular lipid layer interposed between two layers of proteins.
- Important proteins in the red cell membrane are band 3 protein (named based on the order in which it migrates during electrophoresis), glycophorin and spectrin.
- Important lipids are glycolipids, phospholipids and cholesterol.
- Carbohydrates form the skeleton of erythrocytes having a lattice-like network which is attached to the internal surface of the membrane and is responsible for a biconcave form of the erythrocytes.
A number of inherited disorders of the red cell membrane and cytoskeletal components produce abnormalities of the shape such as: spherocytosis (spherical shape from loss of part of the membrane), ovalocytosis (oval shape from loss of elasticity of cytoskeleton), pinocytosis (spiny processes from external surface due to metabolic abnormalities of red cells), and stomatocytosis (bowl-shaped red cells from expansion of inner membrane on one side).

Nutritional Requirements For Erythropoiesis:
New red cells are being produced each day for which the marrow requires certain essential substances. These substances are as under:
1. Metals: Iron is essential for red cell production because it forms part of the haem molecule in haemoglobin. Its deficiency leads to iron deficiency anaemia. Cobalt and manganese are certain other metals required for red cell production.
2. Vitamins: Vitamin B12 and folate are essential for the biosynthesis of nucleic acids. Deficiency of B12 or folate causes megaloblastic anaemia. Vitamin C (ascorbic acid) plays an indirect role by facilitating iron turnover in the body. Vitamin B6 (pyridoxine), vitamin E (tocopherol) and riboflavin are the other essential vitamins required in the synthesis of red cells.
3. Amino acids: Amino acids comprise the globin component of haemoglobin. Severe amino acid deficiency due to protein deprivation causes depressed red cell production.
Haemoglobin:
Haemoglobin consists of a basic protein, globin, and the iron-porphyrin complex, haem. The molecular weight of haemoglobin is 68,000. Normal adult haemoglobin (HbA) constitutes 96-98% of the total haemoglobin content and consists of four polypeptide chains, α2β2.
Small quantities of 2 other haemoglobins present in adults are:
HbF contains α2γ2 globin chains comprising 0.5-0.8% of total haemoglobin, and HbA2 has α2δ2 chains constituting 1.5-3.5% of total haemoglobin. Most of the haemoglobin (65%) is synthesised by the nucleated red cell precursors in the marrow, while the remainder (35%) is synthesised at the reticulocyte stage.
Synthesis of haem occurs largely in the mitochondria by a series of biochemical reactions summarised. Iron in ferrous form may be stored as ferritin or carried in circulation by two vehicle proteins, ferroportin and hephaestin, for transport to transferrin. Coenzyme, pyridoxal-6-phosphate, derived from pyridoxine (vitamin B6) is essential for the synthesis of amino levulinic acid (ALA) which is the first step in the biosynthesis of protoporphyrin.
The reaction is stimulated by erythropoietin and inhibited by haem. Ultimately, protoporphyrin combines with iron supplied from circulating transferrin to form haem. Each molecule of haem combines with a globin chain synthesised by polyribosomes. A tetramer of 4 globin chains, each having its own haem group, constitutes the haemoglobin molecule.

Red Cell Functions:
An essential function of the red cells is to carry oxygen from the lungs to the tissue and to transport carbon dioxide to the lungs. In order to perform these functions, the red cells can generate energy as ATP by an anaerobic glycolytic pathway (EmbdenMeyerhof pathway). This pathway also generates reducing power as NADH and NADPH by the hexose monophosphate (HMP) shunt.
1. Oxygen carrying:
The normal adult haemoglobin, HbA, is an extremely efficient oxygen carrier. The four units of a tetramer of haemoglobin molecule take up oxygen in succession, which, in turn, results in a stepwise rise in affinity of haemoglobin for oxygen. This is responsible for the sigmoid shape of the oxygen dissociation curve.
The oxygen affinity of haemoglobin is expressed in terms of P50 value which is the oxygen tension (pO2 ) at which 50% of the haemoglobin is saturated with oxygen. Pulmonary capillaries have high pO2 and, thus, there is virtual saturation of available oxygen-combining sites of haemoglobin.
The tissue capillaries, however, have relatively low pO2 and, thus, part of haemoglobin is in a deoxy state. The extent to which oxygen is released from haemoglobin at pO2, in tissue capillaries depends upon 3 factors—the nature of globin chains, the pH, and the concentration of 2,3-biphosphoglycerate (2,3-BPG) as follows:
Normal adult haemoglobin (HbA) has lower affinity for oxygen than foetal haemoglobin and, therefore, releases greater amount of bound oxygen at pO2 of tissue capillaries. A fall in the pH (acidic pH) lowers affinity of oxyhaemoglobin for oxygen, so-called the Bohr effect, thereby causing enhanced release of oxygen from erythrocytes at the lower pH in tissue capillaries.
A rise in red cell concentration of 2,3-BPG, an intermediate product of Embden-Meyerhof pathway, as occurs in anaemia and hypoxia, causes decreased affinity of HbA for oxygen. This, in turn, results in enhanced supply of oxygen to the tissue.
2. CO2 transport:
Another important function of the red cells is the CO2 transport. In the tissue capillaries, the pCO2 is high so that CO2 enters the erythrocytes where much of it is converted into bicarbonate ions which diffuse back into the plasma. In the pulmonary capillaries, the process is reversed and bicarbonate ions are converted back into CO2 . Some of the CO2 produced by tissues is bound to deoxyhaemoglobin forming carbamino-haemoglobin. This compound dissociates in the pulmonary capillaries to release CO2 .
Haemoglobin Molecule:

Oxygen Dissociation Curve:

Red Cell Destruction:
Red cells have a mean lifespan of 120 days, after which red cell metabolism gradually deteriorates as the enzymes are not replaced. The destroyed red cells are removed mainly by the macrophages of the reticuloendothelial (RE) system of the marrow, and to some extent by the macrophages in the liver and spleen.
The breakdown of red cells liberates iron for recirculation via plasma transferrin to marrow erythroblasts, and protoporphyrin which is broken down to bilirubin. Bilirubin circulates to the liver where it is conjugated to its diglucuronide which is excreted in the gut via bile and converted to stercobilinogen and stercobilin excreted in the faeces.
Part of stercobilinogen and stercobilin is reabsorbed and excreted in the urine as urobilinogen and urobilin. A small fragment of protoporphyrin is converted to carbon monoxide and excreted in exhaled air from the lungs. Globin chains are broken down to amino acids and reused for protein synthesis in the body.

Normal Values And Red Cell Indices:
Range of normal red cell count in health is 5.5 ± 1.0 × 1012/L in men and 4.8 ± 1.0 × 1012/L in women. The packed cell volume (PCV) or haematocrit is the volume of erythrocytes per litre of whole blood indicating the proportion of plasma and red cells and ranges 0.47 ± 0.07 L/L (40-54%) in men and 0.42 ± 0.05 L/L (37-47%) in women. The haemoglobin content in health is 15.5 ± 2.5 g/dl (13-18 g/dl) in men and 14.0 ± 2.5 g/dl (11.5-16.5 g/dl) in women. Based on these normal values, a series of absolute values or red cell indices can be derived which have diagnostic importance.
These are as under:
1. Mean corpuscular volume (MCV):

Normal value = 85 ± 8 fl (77-93 fl)*.
2. Mean corpuscular haemoglobin (MCH):

Normal range = 29.5 ± 2.5 pg (27-32 pg)*.
3. Mean corpuscular haemoglobin concentration (MCHC):

The normal value is 32.5 ± 2.5 g/dl (30-35 g/dl).
Since MCHC is independent of red cell count and size, it is considered to be of greater clinical significance as compared to other absolute values. It is low in iron deficiency anaemia but is usually normal in macrocytic anaemia.
Red cell distribution width (RDW) RDW is an assessment of the varying volume of red cells based on the size of red cells. For example, fragmented red cells have a tiny size while the macrocytes and reticulocytes have a large size.
Erythropoiesis:
- Erythropoietic activity in the body is regulated by erythropoietin, which is produced mainly from kidneys, in response to anoxia. Its levels are increased in most forms of anaemias.
- Erythroid precursors are a series of recognisable nucleated red cells normally seen in the marrow and include proerythroblast, polychromatic erythroblast, orthochromatic erythroblast, and finally reticulocytes.
- Mature erythrocytes of the human peripheral blood are non-nucleated cells. A red cell membrane is a trilaminar structure having a bimolecular lipid layer interposed between two layers of proteins.
- Erythropoiesis requires various nutrients, namely iron, vitamins and amino acids.
- Haemoglobin consists of a basic protein, globin, and the iron-porphyrin complex, haem.
- Essential functions performed by red cells are oxygen-carrying and carbon dioxide transport.
- At the end of lifespan, red cells are destroyed and phagocytosed by macrophages
Anaemia—General Considerations
Anaemia is defined as reduced haemoglobin concentration in blood below the lower limit of the normal range for the age and sex of the individual; range of haemoglobin value according to gender and age is as follows:
At birth: 17 (±1) g/dl
Children: 11.5 (±0.5) g/dl
Adult men: 16 (±2) g/dl
Adult women (menstruating): 13 (±2) g/dl
Adult women (post-menopausal): 14 (±2) g/dl
Women during pregnancy: 12 (±2) g/dl
An alternate means of determining whether or not anaemia is present and its severity, is by red cell counts, haematocrit (PCV) and absolute values (MCV, MCH and MCHC).
Pathophysiology:
Based on haemoglobin value, the severity of anaemia can be graded as mild, moderate and severe. Subnormal level of haemoglobin causes lowered oxygen-carrying capacity of the blood. This, in turn, initiates compensatory physiologic adaptations such as follows:

- Increased release of oxygen from haemoglobin
- Increased blood flow to the tissues
- Maintenance of the blood volume
- Redistribution of blood flow to maintain the cerebral blood supply.
Eventually, however, tissue hypoxia develops causing impaired functions of the affected tissues. The degree of functional impairment of individual tissues is variable depending upon their oxygen requirements. Tissues with high oxygen requirements such as the heart, CNS and the skeletal muscle during exercise, bear the brunt of clinical effects of anaemia.
General Clinical Features:
The haemoglobin level at which symptoms and signs of anaemia develop depends upon 4 main factors:
- The speed of onset of anaemia Rapidly progressive anaemia causes more symptoms than anaemia of slow-onset as there is less time for physiologic adaptation.
- The severity of anaemia Mild anaemia produces no symptoms or signs but a rapidly developing severe anaemia (haemoglobin below 6.0 g/dl) may produce significant clinical features.
- The age of the patient The young patients due to good cardiovascular compensation tolerate anaemia quite well as compared to the elderly. The elderly patients develop cardiac and cerebral symptoms more prominently due to associated cardiovascular disease.
- The haemoglobin dissociation curve In anaemia, the affinity of haemoglobin for oxygen is depressed as 2,3-BPG in the red cells increases. As a result, oxyhaemoglobin is dissociated more readily to release free oxygen for cellular use, causing a shift of the oxyhaemoglobin dissociation curve to the right.
Symptoms:
In symptomatic cases of anaemia, the presenting features are: tiredness, easy fatiguability, generalised muscular weakness, lethargy and headache. In older patients, there may be symptoms of cardiac failure, angina pectoris, intermittent claudication, confusion and visual disturbances.
Signs:
A few general signs common to all types of anaemias are as under:
- Pallor: Pallor is the most common and characteristic sign which may be seen in the mucous membranes, conjunctivae and skin.
- Cardiovascular system: A hyperdynamic circulation may be present with tachycardia, collapsing pulse, cardiomegaly, midsystolic flow murmur, dyspnoea on exertion, and in the case of elderly, congestive heart failure.
- Central nervous system: The older patients may develop symptoms referable to the CNS such as attacks of faintness, giddiness, headache, tinnitus, drowsiness, numbness and tingling sensations of the hands and feet.
- Ocular manifestations: Retinal haemorrhages may occur if there is associated vascular disease or bleeding diathesis.
- Reproductive system: Menstrual disturbances such as amenorrhoea menorrhagia and loss of libido are some of the manifestations involving the reproductive system in anaemic subjects.
- Renal system: Mild proteinuria and impaired concentrating capacity of the kidney may occur in severe anaemia.
- Gastrointestinal system: Anorexia, flatulence, nausea, constipation and weight loss may occur.
In addition to the general features, specific signs may be associated with particular types of anaemia which are described later together with discussion of specific types of anaemias.
General Scheme Of Investigations Of Anaemia:
After obtaining the full medical history about different general and specific signs and symptoms, the patient is examined for evidence of anaemia. Special emphasis is placed on the colour of the skin, conjunctivae, sclerae and nails.
Changes in the retina, atrophy of the papillae of the tongue, rectal examination for evidence of bleeding, and presence of hepatomegaly, splenomegaly, lymphadenopathy and bony tenderness are looked for.
In order to confirm or deny the presence of anaemia, its type and its cause, the following plan of investigation is generally followed, of which complete blood counts (CBC) with reticulocyte count is the basic test.
1. Haemoglobin Estimation:
The first and foremost investigation in any suspected case of anaemia is to carry out a haemoglobin estimation. Several methods are available but most reliable and accurate is the cyanmethaemoglobin (HiCN) method employing Drabkin’s solution and a spectrophotometer.
If the haemoglobin value is below the lower limit of the normal range for particular age and sex, the patient is said to be anaemic. In pregnancy, there is haemodilution and, therefore, the lower limit in normal pregnant women is less (10.5 g/dl) than in the nonpregnant state.
2. Peripheral Blood Smear Examination:
The haemoglobin estimation is invariably followed by an examination of a peripheral blood film for morphologic features after staining it with the Romanowsky dyes (e.g. Leishman’s stain, May-Grünwald-Giemsa’s stain, Jenner-Giemsa’s stain, Wright’s stain etc).
The blood smear is evaluated in an area where there is neither Rouleaux formation nor so thin as to cause red cell distortion. Such an area can usually be found at junction of the body with the tail of the film, but not actually at the tail.
The following abnormalities in the erythroid series of cells are particularly looked for in a blood smear:
1. Variation in size (Anisocytosis):
Normally, there is a slight variation in the diameter of the red cells from 6.7-7.7 µm (mean value 7.2 µm). Increased variation in size of the red cell is termed anisocytosis. Anisocytosis may be due to the presence of cells larger than normal (macrocytosis) or cells smaller than normal (microcytosis). Sometimes both microcytosis and macrocytosis are present (dimorphic).
- Macrocytes are classically found in megaloblastic anaemia; other causes are aplastic anaemia, other dyserythropoietic anaemias, chronic liver disease and in conditions with increased erythropoiesis.
- Microcytes are present in iron deficiency anaemia, thalassaemia and spherocytosis. They may also result from fragmentation of erythrocytes such as in haemolytic anaemia.
2. Variation in shape (Poikilocytosis):
Increased variation in shape of the red cells is termed poikilocytosis. The nature of the abnormal shape determines the cause of anaemia. Poikilocytes are produced in various types of abnormal erythropoiesis
Example: In megaloblastic anaemia, iron deficiency anaemia, thalassaemia, myelosclerosis and microangiopathic haemolytic anaemia.
3. Inadequate haemoglobin formation (Hypochromasia):
Normally, the intensity of pink staining of haemoglobin in a Romanowsky-stained blood smear gradually decreases from the periphery to the centre of the cell. Increased central pallor is referred to as hypochromasia. It may develop either from lowered haemoglobin content (e.g. in iron deficiency anaemia, chronic infections), or due to thinness of the red cells (e.g. in thalassaemia, sideroblastic anaemia). Unusually deep pink staining of the red cells due to increased haemoglobin concentration is termed hyperchromasia and may be found in megaloblastic anaemia, spherocytosis and in neonatal blood.
4. Compensatory erythropoiesis:
A number of changes are associated with compensatory increases in erythropoietic activity. These are as under:
- Polychromasia is defined as the red cells having more than one type of colour. Polychromatic red cells are slightly larger, generally stained bluish-grey and represent reticulocytes and, thus, correlate well with reticulocyte count.
- Erythroblastaemia is the presence of nucleated red cells in the peripheral blood film. A small number of erythroblasts (or normoblasts) may be normally found in cord blood at birth. They are found in large numbers in haemolytic disease of the newborn, other haemolytic disorders and in extramedullary erythropoiesis. They may also appear in the blood in various types of severe anaemias except in aplastic anaemia. Erythroblastaemia may also occur after splenectomy.
- Punctate basophilia or basophilic stippling is diffuse and uniform basophilic granularity in the cell which does not stain positively with Perls’ reaction (in contrast to Pappenheimer bodies which stain positively). Classical punctate basophilia is seen in aplastic anaemia, thalassaemia, myelodysplasia, infections and lead poisoning.
- Howell-Jolly bodies are purple nuclear remnants, usually found in basophilic stippling. They are present in megaloblastic anaemia and after splenectomy.
5. Red cell morphologic abnormalities:
In addition to the features of red cells described above, several morphologic abnormalities of red cells may be found in different haematological disorders.
Some of these are as follows:
- Spherocytosis is characterised by the presence of spheroidal rather than biconcave disc-shaped red cells. Spherocytes are seen in hereditary spherocytosis, autoimmune haemolytic anaemia and in ABO haemolytic disease of the newborn.
- Schistocytosis is identified by the fragmentation of erythrocytes. Schistocytes are found in thalassaemia, hereditary elliptocytosis, megaloblastic anaemia, iron deficiency anaemia, microangiopathic haemolytic anaemia and in severe burns.
- Irregularly contracted red cells are found in drug and chemical-induced haemolytic anaemia and in unstable haemoglobinopathies.
- Leptocytosis is the presence of unusually thin red cells. Leptocytes are seen in severe iron deficiency and thalassaemia. The target cell is a form of leptocyte in which there is a central round stained area and a peripheral rim of haemoglobin. Target cells are found in thalassaemia, chronic liver disease, and after splenectomy.
- Sickle cells or drepanocytes are sickle-shaped red cells found in sickle cell disease.
- Created red cells are the erythrocytes which develop numerous projections from the surface. They are present in blood films due to alkaline pH, the presence of traces of fatty substances on the slides and in cases where the film is made from blood that has been allowed to stand overnight.
- Acanthocytosis is the presence of coarsely crenated red cells. Acanthocytes are found in large numbers in blood film made from splenectomised subjects, and in chronic liver disease.
- Burr cells are cell fragments having one or more spines. They are particularly found in uraemia.
- Stomatocytosis is the presence of stomatocytes which have central area having slit-like or mouth-like appearance. They are found in hereditary stomatocytosis or may be seen in chronic alcoholism.
- Ovalocytosis or elliptocytosis is the oval or elliptical shape of red cells. Their highest proportion (79%) is seen in hereditary ovalocytosis and elliptocytosis; other conditions showing such abnormal shapes of red cells are megaloblastic anaemia and hypochromic anaemia.
3. Red Cell Indices:
An alternative method to diagnose and detect the severity of anaemia is by measuring the red cell indices:
- In iron deficiency and thalassaemia, MCV, MCH and MCHC are reduced. In early stage of iron deficiency, RDW is increased while in the thalassaemia trait RDW is normal (with low MCV) and can be distinguished from iron deficiency.
- In anaemia due to acute blood loss and haemolytic anaemias, MCV, MCH and MCHC are all within normal limits.
- In megaloblastic anaemias, MCV is raised above the normal range.
4. Leucocyte And Platelet Count:
Measurement of leucocyte and platelet count helps to distinguish pure anaemia from pancytopenia in which red cells, granulocytes and platelets are all reduced. In anaemias due to haemolysis or haemorrhage, the neutrophil count and platelet counts are often elevated. In infections and leukaemias, the leucocyte counts are high and immature leucocytes appear in the blood.
5. Reticulocyte Count:
Reticulocyte count (normal 0.5-2.5%) is done in each case of anaemia to assess the marrow erythropoietic activity. In acute haemorrhage and in haemolysis, raised reticulocyte count is indicative of hyperfunctioning marrow.
6. Erythrocyte Sedimentation Rate:
The ESR is a non-specific test used as a screening test for anaemia. It usually gives a clue to the underlying organic disease but anaemia itself may also cause rise in the ESR.
7. Bone Marrow Examination:
Bone marrow aspiration is done in cases where the cause for anaemia is not obvious. The procedures involved for marrow aspiration and trephine biopsy and their relative advantages and disadvantages. In addition to these general tests, certain specific tests are done in different types of anaemias which are described later under the discussion of specific anaemias.

Classification Of Anaemias:
Several schemes of classifications of anaemias have been proposed. Two of the widely accepted classifications are based on two criteria:
- Pathophysiology of anaemia
- Morphologic features in blood smear
1. Pathophysiologic classification:
Depending upon the pathophysiologic mechanism, anaemias are classified into 3 groups:
- Anaemia due to blood loss
- Anaemia due to impaired red cell formation
- Anaemia due to increased red cell destruction (haemolytic anaemias)
The term hypoproliferative anaemias is also used to denote impaired marrow proliferative activity and includes 2 main groups:
- Hypoproliferation due to iron deficiency
- Due to other hypoproliferative disorders that includes anaemia of chronic inflammation/infection, renal disease, hypometabolic states, and causes of bone marrow failure.
2. Morphologic classification:
Based on the red cell size, haemoglobin content and red cell indices, anaemias are classified into 3 types:
- Microcytic, hypochromic MCV, MCH, and MCHC are all reduced e.g. in iron deficiency anaemia and in certain non-iron deficient anaemias (sideroblastic anaemia, thalassaemia, anaemia of chronic disorders).
- Normocytic, normochromic MCV, MCH, and MCHC are all normal e.g. after acute blood loss, haemolytic anaemias, bone marrow failure, anaemia of chronic disorders.
- Macrocytic MCV is raised
- Example: In megaloblastic anaemia due to a deficiency of vitamin B12 or folic acid.
1. Pathophysiologic
- Anaemia due to increased blood loss
1. Acute post-haemorrhagic anaemia
2. Chronic blood loss
2. Anaemias due to impaired red cell production
- Cytoplasmic maturation defect
1. Deficient haem synthesis: Iron deficiency anaemia
2. Deficient globin synthesis: Thalassaemic syndromes - Nuclear maturation defects Vitamin B12 and/or folic acid deficiency: Megaloblastic anaemia
- Defects in stem cell proliferation and differentiation
1. Aplastic anaemia
2. Pure red cell aplasia - Anaemia of chronic disorders
- Bone marrow infiltration
- Congenital anaemia
3. Anaemias due to increased red cell destruction (Haemo-lytic anaemias)
- Extrinsic (extracorpuscular) red cell abnormalities
- Intrinsic (intracorpuscular) red cell abnormalities
2. Morphologic
- Microcytic, hypochromic
- Normocytic, normochromic
- Macrocytic, normochromic
Most common form of anaemia, however, in the world, is due to nutritional deficiency of iron and vitamin B12/folate, causing iron deficiency anaemia and megaloblastic anaemia respectively, together termed as nutritional anaemias. It is not unusual for a patient of nutritional anaemia to suffer from combined deficiency as well.
After these general comments on anaemias, a discussion of the specific types of anaemias is given in the following pages.
Anaemias: General Considerations:
- Anaemia is reduced haemoglobin concentration in blood below the lower limit of the normal for the age and sex of the individual.
- Anaemia causes lowered oxygen-carrying capacity and eventually tissue hypoxia andimpaired function.
- Major symptoms of anaemia are tiredness, weakness and lethargy, and main signs are pallor, hyperdynamic circulation, and impaired functions of CNS and kidneys.
- A suspected case of anaemia is investigated by haemoglobin estimation, peripheral blood smear examination, and complete blood counts including reticulocyte count and ESR.
- Bone marrow aspiration and trephine biopsy are done to confirm and type the anaemia.
- Anaemias are classified based on pathophysiology (into anaemia due to blood loss, impaired red cell production, increased red cell destruction) or on morphology (into microcytic hypochromic, macrocytic, normocytic normochromic)
Hypochromic Anaemias
Hypochromic anaemia due to iron deficiency is the most common cause of anaemia the world over. It is estimated that about 20% of women in child-bearing age group are iron deficient, while the overall prevalence in adult males is about 2%.
It is the most important, though not the sole, cause of microcytic hypochromic anaemia in which all the three red cell indices (MCV, MCH and MCHC) are reduced and occurs due to defective haemoglobin synthesis.
Hypochromic anaemias, therefore, are classified into 2 groups:
- Hypochromic anaemia due to iron deficiency
- Hypochromic anaemias other than iron deficiency
The latter category includes 3 groups of disorders—sideroblastic anaemia, thalassaemia and anaemia of chronic disorders. These anaemias are discussed below except thalassaemia which is discussed along with other haemolytic anaemias later.
Iron Deficiency Anaemia:
The most common nutritional deficiency disorder present throughout the world is iron deficiency but its prevalence is higher in developing countries. The factors responsible for iron deficiency in different populations are variable and are best understood in the context of normal iron metabolism.
Iron Metabolism:
The amount of iron obtained from the diet should replace the losses from the skin, bowel and genitourinary tract. These losses together are about 1 mg daily in an adult male or in a nonmenstruating female, while in a menstruating woman there is an additional iron loss of 0.5-1 mg daily.
The iron required for haemoglobin synthesis is derived from 2 primary sources—ingestion of foods containing iron (e.g. leafy vegetables, beans, meats, liver etc) and recycling of iron from senescent red cells.

Absorption:
The average Western diet contains 10-15 mg of iron, out of which only 5-10% is normally absorbed. In pregnancy and in iron deficiency, the proportion of absorption is raised to 20-30%. Iron is absorbed mainly in the duodenum and proximal jejunum.
The absorption is regulated by the mucosal block mechanism—when iron stores are low (e.g. during pregnancy, menstruation, periods of growth and various diseases) absorption is enhanced, and when iron stores are increased (e.g. in haemosiderosis) little iron is absorbed orm transported.
Iron from a diet containing haem is better absorbed than non-haem iron:
- Absorption of non-haem iron is enhanced by factors such as ascorbic acid (vitamin C), citric acid, amino acids, sugars, gastric secretions and hydrochloric acid of the stomach. Iron absorption is impaired by factors like medicinal antacids, milk, pancreatic secretions, phytates, phosphates, ethylene diamine tetra-acetic acid (EDTA) and tannates contained in tea. Non-haem iron is released as either ferrous or ferric form but it is absorbed almost exclusively as ferrous form. Reduction of ferric to ferrous form, when required, takes place at the brush border of the proximal intestine by ferric reductase enzyme called duodenal cytochrome b (dcytb) reductase. Transport across the enterocyte membrane is accomplished by divalent metal transporter 1 (DMT)
- Once inside the gut cells, ferrous iron may be either stored as ferritin or further transported to transferrin by two vehicle proteins—ferroportin and hephaestin. The function of ferroportin is inversely regulated by hepcidin released from the liver and is the main iron-regulating hormone.
- The mechanism of dietary haem iron absorption is not clearly understood yet but it is through a transporter other than DMT 1.
After absorption of both non-haem and haem forms of iron, it comes into the mucosal pool.
Transport:
Iron is transported in plasma bound to a β-globulin, transferrin, synthesised in the liver. Transferrin-bound iron is made available to the marrow where the developing erythroid cells having transferrin receptors utilise iron for haemoglobin synthesis.
It may be mentioned here that transferrin receptors are present on cells of many tissues of the body but their number is greatest in the developing erythroblasts. Transferrin is reutilised after iron is released from it.
A small amount of transferrin iron is delivered to other sites such as parenchymal cells of the liver. Normally, transferrin is about one-third saturated. But in conditions where transferrin-iron saturation is increased, parenchymal iron uptake is increased.
Virtually, no iron is deposited in the mononuclear-phagocyte cells (RE cells) from the plasma transferrin-iron but instead these cells derive most of their iron from phagocytosis of senescent red cells.
Excess of iron beyond haemoglobin synthesis binds to a storage protein, apoferritin, forming ferritin in RE cells. Ferritin can be readily mobilised in response to increased demands for erythropoiesis. However, conditions such as malignancy, infection and inflammation interfere with the release of iron from iron stores causing ineffective erythropoiesis.

Excretion:
The body is unable to regulate its iron content by excretion alone. The amount of iron lost per day is 0.5-1 mg which is independent of iron intake. This loss is nearly twice more (i.e. 1-2 mg/day) in menstruating women. Iron is lost from the body in both sexes as a result of the desquamation of epithelial cells from the gastrointestinal tract, from excretion in the urine and sweat, and loss via hair and nails. Iron excreted in the faeces mainly consists of unabsorbed iron and desquamated mucosal cells.
Distribution:
In an adult, iron is distributed in the body as under:
- Haemoglobin—present in the red cells, contains most of the body iron (65%).
- Myoglobin—comprises a small amount of iron in the muscles (3.5%).
- Haem and non-haem enzymes—e.g. cytochrome, catalase, peroxidases, succinic dehydrogenase and flavoproteins constitute a fraction of total body iron (0.5%).
- Transferrin-bound iron—circulates in the plasma and constitutes another fraction of total body iron (0.5%).
All these forms of iron are in functional form. - Ferritin and haemosiderin—are the storage forms of excess iron (30%). They are stored in the mononuclear-phagocyte cells of the spleen, liver and bone marrow and in the parenchymal cells of the liver.
Pathogenesis:
Iron deficiency anaemia develops when the supply of iron is inadequate for the requirement of haemoglobin synthesis. Initially, negative iron balance is covered by mobilisation from the tissue stores to maintain haemoglobin synthesis.
It is only after the tissue stores of iron are exhausted that the supply of iron to the marrow becomes insufficient for haemoglobin formation and thus a state of iron deficiency anaemia develops. The development of iron deficiency depends upon one or more of the following factors:
- Increased blood loss
- Increased requirements
- Inadequate dietary intake
- Decreased intestinal absorption.
The relative significance of these factors varies with the age and sex of the patient. Accordingly, certain groups of individuals at increased risk of developing iron deficiency can be identified (see below).
In general, in developed countries, the mechanism of iron deficiency is usually due to chronic occult blood loss, while in developing countries poor intake of iron or defective absorption are responsible for iron deficiency anaemia.
Aetiology:
Iron deficiency anaemia is always secondary to an underlying disorder. Correction of the underlying cause, therefore, is an essential part of its treatment. Based on the above-mentioned pathogenetic mechanisms, the following etiologic factors are involved in the development of iron deficiency anaemia at different ages and sex.
1. Increased Blood Loss:
- Uterine e.g. excessive menstruation in reproductive years, repeated miscarriages, at onset of menarche, post-menopausal uterine bleeding
- Gastrointestinal e.g. peptic ulcer, haemorrhoids hookworm infestation, cancer of stomach and large bowel, oesophageal varices, hiatus hernia, chronic aspirin ingestion, ulcerative colitis, diverticulosis
- Renal tract e.g. haematuria, haemoglobinuria
- Nose e.g. repeated epistaxis
- Lungs e.g. haemoptysis
2. Increased Requirements:
- Spurts of growth in infancy, childhood and adolescence
- Prematurity
- Pregnancy and lactation
3. Inadequate Dietary Intake:
- Poor economic status
- Anorexia e.g. in pregnancy
- Elderly individuals due to poor dentition, apathy and financial constraints
4. Decreased absorption:
- Partial or total gastrectomy
- Achlorhydria
- Intestinal malabsorption such as in coeliac disease
1. Women In Reproductive Years Of Life:
The highest incidence of iron deficiency anaemia is in women during their reproductive years of life.
It may be from one or more of the following causes:
- Blood loss This is the most important cause of anaemia in women during child-bearing age group. Commonly, it is due to persistent and heavy menstrual blood loss such as occurs in various pathological states and due to the insertion of IUCDs. Young girls at the onset of menstruation may develop mild anaemia due to blood loss. Significant blood loss may occur as a result of repeated miscarriages.
- Inadequate intake Inadequate intake of iron is prevalent in women of lower economic status. Besides diet deficient in iron, other factors such as anorexia, impaired absorption and diminished bioavailability may act as contributory factors.
- Increased requirements During pregnancy and adolescence, the demand of the body for iron is increased. During a normal pregnancy, about 750 mg of iron may be siphoned off from the mother—about 400 mg to the foetus, 150 mg to the placenta, and 200 mg is lost at parturition and lactation. If several pregnancies occur at short intervals, iron deficiency anaemia certainly follows.
2. Post-Menopausal Women:
Though the physiological demand for iron decreases after cessation of menstruation, iron deficiency anaemia may develop in post-menopausal women due to chronic blood loss. Following are among the important causes during these years:
Post-menopausal uterine bleeding due to carcinoma of the uterus. Bleeding from the alimentary tract such as due to carcinoma of the stomach and large bowel and hiatus hernia.
3. Adult Males:
It is uncommon for adult males to develop iron deficiency anaemia in the presence of normal dietary iron content and iron absorption. The vast majority of cases of iron deficiency anaemia in adult males are due to chronic blood loss.
The cause for chronic haemorrhage may lie at one of the following sites:
- The gastrointestinal tract is the usual source of bleeding which may be due to peptic ulcer, haemorrhoids, hookworm infestation, carcinoma of stomach and large bowel, oesophageal varices, hiatus hernia, chronic aspirin ingestion and ulcerative colitis. Other causes in GIT are malabsorption and following gastrointestinal surgery.
- Urinary tract
- Example: Due to haematuria and haemoglobinuria.
- Nose
- Example: In repeated epistaxis.
- Lungs
- Example: In haemoptysis from various causes.
4. Infants And Children:
Iron deficiency anaemia is fairly common during infancy and childhood with a peak incidence at 1-2 years of age. The principal cause for anaemia at this age is increased demand of iron which is not met by the inadequate intake of iron in the diet. Normal full-term infant has sufficient iron stores for the first 4-6 months of life, while premature infants have inadequate reserves because iron stores from the mother are mainly laid down during the last trimester of pregnancy. Therefore, unless the infant is given supplemental feeding of iron or iron-containing foods, iron deficiency anaemia develops.
Clinical Features:
As already mentioned, iron deficiency anaemia is much more common in women between the age of 20 and 45 years than in men; at periods of active growth in infancy, childhood and adolescence; and is also more frequent in premature infants. Initially, there are usually no clinical abnormalities. But subsequently, in addition to features of the underlying disorder causing the anaemia, the clinical consequences of iron deficiency manifest in 2 ways—anaemia itself and epithelial tissue changes.
1. Anaemia:
The onset of iron deficiency anaemia is generally slow. The usual symptoms are weakness, fatigue, dyspnoea on exertion, palpitations and pallor of the skin, mucous membranes and sclerae. Older patients may develop angina and congestive cardiac failure. Patients may have unusual dietary cravings such as pica. Menorrhagia is a common symptom in iron-deficient women.
2. Epithelial Tissue Changes:
Long-standing chronic iron deficiency anaemia causes epithelial tissue changes in some patients. The changes occur in the nails (koilonychia or spoon-shaped nails), tongue (atrophic glossitis), mouth (angular stomatitis), and oesophagus causing dysphagia from the development of thin, membranous webs at the postcricoid area (Plummer-Vinson syndrome).
Laboratory Findings:
The development of anaemia progresses in 3 stages:
- Firstly, storage iron depletion occurs during which iron reserves are lost without compromise of the iron supply for erythropoiesis.
- The next stage is iron deficient erythropoiesis during which the erythroid iron supply is reduced without the development of anaemia.
- The final stage is the development of frank iron deficiency anaemia when the red cells become microcytic and hypochromic.
The following laboratory tests can be used to assess the varying degree of iron deficiency:
Blood smear and red cell indices:
The degree of anaemia varies. It is usually mild to moderate but occasionally it may be marked (haemoglobin less than 6.5 g/dl) due to persistent and severe blood loss. The salient haematological findings in these cases are as under.
1. Haemoglobin:
The essential feature is a fall in haemoglobin concentration up to a variable degree.
2. Red cells:
The red cells in the blood film are hypochromic and microcytic, and there is anisocytosis and poikilocytosis. Hypochromia generally precedes microcytosis. Hypochromia is due to poor filling of the red cells with haemoglobin so that there is increased central pallor. In severe cases, there may be only a thin rim of pink staining at the periphery. Target cells, elliptical forms and polychromatic cells are often present. Normoblasts are uncommon. RBC count is below normal but is generally not proportionate to the fall in haemoglobin value. When iron deficiency is associated with severe folate or vitamin B12 deficiency, a dimorphic blood picture occurs with a dual population of red cells—macrocytic as well as microcytic hypochromic.
3. Reticulocyte count:
The reticulocyte count is normal or reduced but may be slightly raised (2-5%) in cases after haemorrhage.
4. Absolute values:
The red cell indices reveal a diminished MCV (below 50 fl), diminished MCH (below 15 pg), and diminished MCHC (below 20 g/dl).
5. Leucocytes:
The total and differential white cell counts are usually normal.
6. Platelets:
Platelet count is usually normal but there may be elevated mildly to moderately in patients who had recent bleeding.
2. Bone Marrow Findings:
Bone marrow examination is not essential in such cases routinely but is done in complicated cases so as to distinguish from other hypochromic anaemias. The usual findings are as follows:
1. Marrow cellularity: The marrow cellularity is increased due to erythroid hyperplasia (myeloid-erythroid ratio decreased).
2. Erythropoiesis: There is normoblastic erythropoiesis with predominance of small polychromatic erythroblasts (or micronormoblasts). These erythroblasts have a thin rim of cytoplasm around the nucleus and a ragged and irregular cell border. The cytoplasmic maturation lags so that the late erythroblasts have a pyknotic nucleus but persisting polychromatic cytoplasm (compared to megaloblastic anaemia in which the nuclear maturation lags behind)
3. Other cells: Myeloid, lymphoid and megakaryocytic cells are normal in number and morphology.
4. Marrow iron: Iron staining (Prussian blue reaction) on bone marrow aspirate smear shows deficient reticuloendothelial iron stores and the absence of siderotic iron granules from developing normoblasts.
3. Biochemical Findings:
In addition to blood and bone marrow examination, the following biochemical tests are of value:
- The serum iron level is low (normal 40-140 µg/dl); it is often under 50 µg/dl. When serum iron falls below 15 µg/dl, marrow iron stores are absent.
- Total iron binding capacity (TIBC) is high (normal 250-450 µg/dl) and rises to give less than 10% saturation (normal 33%). In anaemia of chronic disorders, however, serum iron as well as TIBC are reduced.
- Serum ferritin is very low (normal 30-250 ng/ml) indicating poor tissue iron stores. The serum ferritin is raised in iron overload and is normal in anaemia of chronic disorders.
- Red cell protoporphyrin is very low (normal 20-40 µg/dl) as a result of insufficient iron supply to form haem.
- Serum transferrin receptor protein which is normally present on developing erythroid cells and reflects total red cell mass, is raised in iron deficiency due to its release in circulation (normal level 4-9 µg/L as determined by immunoassay).


Principles Of Treatment:
The management of iron deficiency anaemia consists of 2 essential principles:
correction of disorder causing the anaemia, and correction of iron deficiency.
Correction Of The Disorder:
The underlying cause of iron deficiency is established after thorough check-up and investigations. Appropriate surgical, medical or preventive measures are instituted to correct the cause of blood loss.
Correction Of Iron Deficiency:
The lack of iron is corrected with iron therapy as under:
1. Oral therapy: Iron deficiency responds very effectively to the administration of oral iron salts such as ferrous sulfate, ferrous fumarate, ferrous gluconate and polysaccharide iron. These preparations have varying amounts of elemental iron in each tablet ranging from 39 mg to 150 mg.
Optimal absorption is obtained by giving iron fasting, but if side-effects occur (e.g. nausea, abdominal discomfort, diarrhoea) iron can be given with food or by using a preparation of lower iron content (e.g. ferrous gluconate containing 39 mg elemental iron).
Oral iron therapy is continued long enough, both to correct the anaemia and to replenish the body’s iron stores. The response to oral iron therapy is observed by reticulocytosis which begins to appear in 3-4 days with a peak in about 10 days.
Poor response to iron replacement may occur from various causes such as:
incorrect diagnosis, non-compliance, continuing blood loss, bone marrow suppression by tumour or chronic inflammation, and malabsorption.
2. Parenteral therapy Parenteral iron therapy is indicated in following types of cases:
- Intolerance to oral iron therapy
- In GIT disorders such as malabsorption
- Post-operative cases
- Cases requiring a rapid replenishment of iron stores
- Example: In women with severe anaemia a few weeks before the expected date of delivery.
Parenteral iron therapy is hazardous and expensive when compared with oral administration. The haematological response to parenteral iron therapy is no faster than the administration of an adequate dose of oral iron but the stores are replenished much faster.
Before giving the parenteral iron, the total dose is calculated by a simple formula by multiplying the grams of haemoglobin below normal with 250 (250 mg of elemental iron is required for each gram of deficit haemoglobin), plus an additional 500 mg is added for building up iron stores.
A common preparation is iron dextran which may be given as a single intramuscular injection, or as an intravenous infusion after dilution with dextrose or saline. The adverse effects of iron dextran include hypersensitivity or anaphylactoid reactions, haemolysis, hypotension, circulatory collapse, vomiting and muscle pain. Newer iron complexes such as sodium ferric gluconate and iron sucrose can be administered as repeated intravenous injections with much lesser side effects.
Sideroblastic Anaemia:
Sideroblastic anaemias comprise a group of disorders of diverse etiology in which the nucleated erythroid precursors in the bone marrow, show characteristic ‘ringed sideroblasts.’
Siderocytes And Sideroblasts:
Siderocytes and sideroblasts are erythrocytes and normoblasts respectively which contain cytoplasmic granules of iron.
Siderocytes:
These are red cells containing granules of non-haem iron. These granules stain positively with Prussian blue reaction as well as stain with Romanowsky dyes when they are referred to as Pappenheimer bodies. Siderocytes are normally not present in the human peripheral blood but a small number may appear following splenectomy.
This is because the reticulocytes on release from the marrow are finally sequestered in the spleen to become mature red cells. In the absence of spleen, the final maturation step takes place in the peripheral blood and hence siderocytes make their appearance in the blood after splenectomy.
Sideroblasts:
These are nucleated red cells (normoblasts) containing siderotic granules which stain positively with Prussian blue reaction.
Depending upon the number, size and distribution of siderotic granules, sideroblasts may be normal or abnormal:

Normal sideroblasts: Contain a few fine, scattered cytoplasmic granules representing iron which has not been utilised for haemoglobin synthesis. These cells comprise 30-50% of normoblasts in the normal marrow but are reduced or absent in iron deficiency.
Abnormal sideroblasts are further of 2 types:
- One type is a sideroblast containing numerous, diffusely scattered, coarse cytoplasmic granules and are seen in conditions such as dyserythropoiesis and haemolysis. In this type, there is no defect of haem or globin synthesis but the percentage saturation of transferrin is increased.
- The other type is ringed sideroblast in which haem synthesis is disturbed as occurs in sideroblastic anaemias. Ringed sideroblasts contain numerous large granules, often forming a complete or partial ring around the nucleus. The ringed arrangement of these granules is due to the presence of iron-laden mitochondria around the nucleus.
Types Of Sideroblastic Anaemias:
Based on etiology, sideroblastic anaemias are classified into hereditary and acquired types.
The acquired type is further divided into primary and secondary forms:

1. Hereditary Sideroblastic Anaemia:
This is a rare X-linked disorder associated with defective enzyme activity of aminolevulinic acid (ALA) synthetase required for haem synthesis. The affected males have moderate to marked anaemia while the females are carriers of the disorder and do not develop anaemia. The condition manifests in childhood or in early adult life.
2. Acquired Sideroblastic Anaemia:
The acquired sideroblastic anaemias are classified into primary and secondary types.
1. Primary acquired sideroblastic anaemia: Primary, idiopathic, or refractory acquired sideroblastic anaemia occurs spontaneously in middle-aged and older individuals of both sexes. The disorder has its pathogenesis in disturbed growth and maturation of erythroid precursors at the level of haematopoietic stem cell, possibly due to reduced activity of the enzyme, ALA synthetase.
The anaemia is of moderate to severe degree and appears insidiously. The bone marrow cells commonly show chromosomal abnormalities, neutropenia and thrombocytopenia with associated bleeding diathesis.
The spleen and liver may be either normal or mildly enlarged, while the lymph nodes are not enlarged. Unlike other types of sideroblastic anaemia, this type is regarded as a myelodysplastic disorder in the FAB (French-American-British) classification and thus, can be a pre-leukaemic disorder (page 379). About 10% of individuals with refractory acquired sideroblastic anaemia develop acute myelogenous leukaemia.
2. Secondary acquired sideroblastic anaemia: Acquired sideroblastic anaemia may develop secondary to a variety of drugs, chemicals, toxins, haematological and various other diseases.
- Drugs, chemicals and toxins Isoniazid, an anti-tuberculous drug and a pyridoxine antagonist, is most commonly associated with development of sideroblastic anaemia by producing abnormalities in pyridoxine metabolism.
Other drugs occasionally causing acquired sideroblastic anaemia are: Cycloserine, chloramphenicol and alkylating agents (e.g. cyclophosphamide). Alcohol and lead also cause sideroblastic anaemia. All these agents cause reversible sideroblastic anaemia which usually resolves following removal of the offending agent. - Haematological disorders These include myelofibrosis, polycythaemia vera, acute leukaemia, myeloma, lymphoma and haemolytic anaemia.
- Miscellaneous Occasionally, secondary sideroblastic anaemia may occur in association with a variety of inflammatory, neoplastic and autoimmune diseases such as carcinoma, myxoedema, rheumatoid arthritis and SLE.
Laboratory Findings:
Sideroblastic anaemias usually show the following haematological features:
- There is generally a moderate to severe degree of anaemia.
- The blood smear shows hypochromic anaemia which may be microcytic, or there may be some normocytic red cells as well (dimorphic).
- Absolute values (MCV, MCH and MCHC) are reduced in the hereditary type but MCV is often raised in the acquired type.
- Bone marrow examination shows erythroid hyperplasia with usually macronormoblastic erythropoiesis. Marrow iron stores are raised and pathognomonic ring sideroblasts are present.
- Serum ferritin levels are raised.
- Serum iron is usually raised with almost complete saturation of TIBC.
- There is increased iron deposition in the tissue.

Principles Of Treatment:
The treatment of secondary sideroblastic anaemia is primarily focussed on removal of the offending agent. No definite treatment is available for hereditary and idiopathic types of sideroblastic anaemias. However, pyridoxine is administered routinely to all cases of sideroblastic anaemia (200 mg per day for 2-3 months). Blood transfusions and other supportive therapy are indicated in all patients. Differential diagnosis of various types of hypochromic anaemias by laboratory tests is summarised in Table.
Anaemia Of Chronic Disorders:
One of the commonly encountered anaemia is in patients of a variety of chronic systemic diseases in which anaemia develops secondary to a disease process but there is no actual invasion of the bone marrow. A list of such chronic systemic diseases is given in Table.
Generally, anaemia in chronic disorders is usually normocytic normochromic but can have a mild degree of microcytosis and hypochromia unrelated to iron deficiency. The severity of anaemia is usually directly related to the primary disease process. The anaemia is corrected only if the primary disease is alleviated.
Anaemias secondary to chronic systemic disorders:
1. Anaemia In Chronic Infections/Inflammation
- Infections e.g. tuberculosis, lung abscess, pneumonia, osteomyelitis, subacute bacterial endocarditis, pyelonephritis.
- Non-infectious inflammations e.g. rheumatoid arthritis, SLE, vasculitis, dermatomyositis, scleroderma, sarcoidosis, Crohn’s disease.
- Disseminated malignancies e.g. Hodgkin’s disease, disseminated carcinomas and sarcomas.
2. Anaemia Of Renal Disease
e.g. uraemia, renal failure
3. Anemia of hypometabolic state
e.g. endocrinopathies (myxoedema, Addison’s disease, hyperthyroidism, hypopituitarism, Addison’s disease), protein malnutrition, scurvy and pregnancy, liver disease.
Pathogenesis:
Several factors may contribute to the development of anaemia in chronic systemic disorders, and in many conditions, the anaemia is complicated by other causes such as iron, B12 and folate deficiency, hypersplenism, renal failure with consequent reduced erythropoietic activity, endocrine abnormalities etc. However, in general, 2 factors appear to play a significant role in the pathogenesis of anaemia in chronic disorders.
These are: Defective red cell production and reduced red cell lifespan.
1. Defective red cell production: Though there is abundance of storage iron in these conditions but the amount of iron available to developing erythroid cells in the marrow is subnormal. The mononuclear phagocyte system is hyperplastic which traps all the available free iron due to the activity of iron binding protein, lactoferrin. A defect in the transfer of iron from macrophages to the developing erythroid cells in the marrow leads to reduced availability of iron for haem synthesis despite adequate iron stores, elevating serum ferritin levels.
The defect lies in suppression of erythropoietin by inflammatory cytokines at some stage in erythropoiesis, and hepcidin which is the key iron regulatory hormone. These inflammatory cytokines include TNF and IFN-β released in bacterial infections and tumours, and IL-1 and IFN-γ released in patients of rheumatoid arthritis and autoimmune vasculitis.

Reduced red cell lifespan Decreased survival of circulating red cells in chronic renal disease is attributed to hyperplastic mononuclear phagocyte system.
Laboratory Findings:
The characteristic features of anaemia in these patients uncomplicated by other deficiencies are as under:
- Haemoglobin: Anaemia is generally mild to moderate. A haemoglobin value of less than 8 g/dl suggests the presence of additional contributory factors.
- Blood smear The type of anaemia in these cases is generally normocytic normochromic but may have slight microcytosis and hypochromia.
- Absolute values Red cell indices indicate that in spite of normocytic normochromic anaemia, MCHC is slightly low.
- Reticulocyte count The reticulocyte count is generally low.
- Red cell survival Measurement of erythrocyte survival generally reveals mild to moderate shortening of their lifespan.
- Bone marrow Examination of the marrow generally reveals normal erythroid maturation. However, the red cell precursors have reduced stainable iron than normal, while macrophages in the marrow usually contain increased amount of iron. Cases of chronic infection often have myeloid hyperplasia and increase in plasma cells.
- Serum iron and TIBC Serum iron is characteristically reduced in this group of anaemias while TIBC is low-to-normal (in contrast to iron deficiency where there is reduction in serum iron but high TIBC.
- Serum ferritin Serum ferritin levels are increased in these patients and is the most distinguishing feature between true iron-deficiency anaemia and iron-deficient erythropoiesis in anaemia of chronic diseases.
- Other plasma proteins In addition, certain other plasma proteins called ‘phase reactants’ are raised in patients with chronic inflammation, probably under the stimulus of interleukin-1 released by activated macrophages. These proteins include γ-globulin, C3, haptoglobin, α1-antitrypsin and fibrinogen. Elevation of these proteins is responsible for raised ESR commonly present in these patients
Hypochromic Anaemias:
- Hypochromic anaemias are classified into 2 groups: due to iron deficiency, and from causes other than iron deficiency (i.e. sideroblastic anaemia, thalassaemia and anaemia of chronic disorders).
- The commonest nutritional deficiency throughout the world is iron deficiency but its prevalence is higher in the developing countries
- The development of iron deficiency may be due to increased blood loss, increased requirement, inadequate dietary intake or decreased intestinal absorption. Accordingly, the causes depend upon age and sex.
- Clinical consequences of iron deficiency manifest as anaemia itself and from epithelial changes.
- Laboratory diagnosis of iron deficiency is made by CBC and blood picture showing microcytosis and hypochromia, reduced red cell indices, serum iron and ferritin and bone marrow examination showing micronormoblastic picture with depleted iron stores.
- Sideroblastic anaemia is classified into hereditary and acquired. Marrow iron stores are raised and pathognomonic ring sideroblasts are present. Serum ferritin and serum iron are usually raised.
- Anaemia of chronic disorders is due to chronic systemic diseases, such as inflammatory or infectious and chronic renal diseases. Serum ferritin is increased and is the most distinguishing feature from true iron-deficiency anaemia.
Megaloblastic Anaemias
Megaloblastic anaemias are associated with macrocytic blood picture and megaloblastic marrow erythropoiesis.
This group is due to deficiency of vitamin B12 and/or folate and includes megaloblastic picture from the following two types of etiologies:
- Nutritional deficiency of vitamin B12 or folate, or combined deficiency, most common in developing countries.
- Deficiency of intrinsic factor, causing impaired absorption of vitamin B12 called pernicious anaemia, is rare in India but more prevalent in individuals of European and Caucasian descent.
Megaloblastic Anaemia Due To Nutritional Vitamin B12 And Folate Deficiency:
The megaloblastic anaemias are disorders caused by impaired DNA synthesis and are characterised by a distinctive abnormality in the haematopoietic precursors in the bone marrow in which the maturation of the nucleus is delayed relative to that of the cytoplasm.
Since cell division is slow but cytoplasmic development progresses normally, the nucleated red cell precursors tend to be larger which Ehrlich in 1880 termed megaloblasts. Megaloblasts are both morphologically and functionally abnormal with the result that the mature red cells formed from them and released into the peripheral blood are also abnormal in shape and size, the most prominent abnormality being macrocytosis.
The underlying defect for the asynchronous maturation of the nucleus is defective DNA synthesis due to deficiency of vitamin B12 (cobalamin) and/or folic acid (folate). Less common causes are interference with DNA synthesis by congenital or acquired abnormalities of vitamin B12 or folic acid metabolism.
Before considering the megaloblastic anaemia, an outline of vitamin B12 and folic acid metabolism is given for a better understanding of the subject. The salient nutritional aspects and metabolic functions of vitamin B12 and folic acid are summarised in Table.
Vitamin B12 Metabolism:
Biochemistry:
Vitamin B12 or cobalamin is a complex organometallic compound having a cobalt atom situated within a corrin ring, similar to the structure of porphyrin from which haem is formed. In humans, there are 2 metabolically active forms of cobalamin—methyl-cobalamin and adenosyl-cobalamin, which act as coenzymes. The therapeutic vitamin B12 preparation is called cyanocobalamin.

Sources: The only dietary sources of vitamin B12 are foods of animal protein origin such as kidney, liver, heart, muscle meats, fish, eggs, and in vegetarian meal as cheese and milk. In contrast to folate, fruits and vegetables contain practically no vitamin B12 unless contaminated with bacteria.
Cooking has little effect on its activity. Vitamin B12 is synthesised in the human large bowel by microorganisms but is not absorbed from this site and, thus, the humans are entirely dependent upon dietary sources. The average daily requirement for vitamin B12 is 2-4 µg.
Absorption:
After ingestion, vitamin B12 in food is released and forms a stable complex with gastric R-binder. R-binder is a form of glycoprotein found in various secretions (e.g. saliva, milk, gastric juice, bile), phagocytes and plasma. On entering the duodenum, the vitamin B12-R-binder complex is digested releasing vitamin B12 which then binds to intrinsic factor (IF).
The IF is a secretion roughly parallels that of hydrochloric acid. The vitamin B12-IF complex, on reaching the distal ileum, binds to the specific receptors on the mucosal brush border, thereby enabling the vitamin to be absorbed. The IF, therefore, acts as cell-directed carrier protein similar to transferrin.
The receptor-bound vitamin B12-IF complex is taken into the ileal mucosal cells where after several hours the IF is destroyed, vitamin B12 released and is transferred to another transport protein, transcobalamin (TC) II. The vitamin B12-TC II complex is finally secreted into the portal circulation from where it is taken by the liver, bone marrow and other cells.
There are 2 major vitamin Bv binding proteins—TC I and TC II, and a minor protein TC III. TC I is not essential for vitamin B12 transport but functions primarily as a storage protein while TC III is similar to TC II and binds a small amount of vitamin B12.
Tissue Stores:
Normally, the liver is the principal storage site of vitamin B12and stores about 2 mg of the vitamin, while other tissues like the kidney, heart and brain together store about 2 mg. The body stores of vitamin B12 are adequate for 2-4 years. Major source of loss is via bile and shedding of intestinal epithelial cells. A major part of the excreted vitamin B12 is reabsorbed in the ileum by the IF resulting in enterohepatic circulation.
Functions:
Vitamin B12plays an important role in general cell metabolism, particularly essential for normal haematopoiesis and for maintenance of integrity of the nervous system. Vitamin B12 acts as a co-enzyme for 2 main biochemical reactions in the body:
- Firstly, as methyl cobalamin (methyl B12) in the methylation of homocysteine to methionine by methyl tetrahydrofolate (THF).The homocysteine-methionine reaction is closely linked to folate metabolism:
 When this reaction is impaired, folate metabolism is deranged and results in defective DNA synthesis responsible for megaloblastic maturation.
When this reaction is impaired, folate metabolism is deranged and results in defective DNA synthesis responsible for megaloblastic maturation. - Secondly, as adenosyl cobalamin (adenosyl B12) in propionate metabolism for the conversion of methyl malonyl co-enzyme A to succinyl co-enzyme A:
 Lack of adenosyl B12 leads to large increase in the level of methyl malonyl CoA and its precursor, propionyl CoA. This results in synthesis of certain fatty acids which are incorporated into the neuronal lipids. This biochemical abnormality may contribute to the neurologic complications of vitamin B12 deficiency.
Lack of adenosyl B12 leads to large increase in the level of methyl malonyl CoA and its precursor, propionyl CoA. This results in synthesis of certain fatty acids which are incorporated into the neuronal lipids. This biochemical abnormality may contribute to the neurologic complications of vitamin B12 deficiency.
Folate Metabolism:
Biochemistry:
Folate or folic acid, a yellow compound, is a member of water-soluble B complex vitamins with the chemical name of pteroyl glutamic acid (PGA). Folic acid does not exist as such in nature but exists as folates in polyglutamate form (conjugated folates). For its metabolic action as co-enzyme, polyglutamates must be reduced to dihydro- and tetrahydrofolate forms.
Sources:
Folate exists in different plants, bacteria and animal tissues. Its main dietary sources are fresh green leafy vegetables, fruits, liver, kidney, and to a lesser extent, muscle meats, cereals and milk. Folate is labile and is largely destroyed by cooking and canning.
Some amount of folate synthesised by bacteria in the human large bowel is not available to the body because its absorption takes place in the small intestine. Thus, humans are mainly dependent upon diet for its supply. The average daily requirement is 100-200 µg.
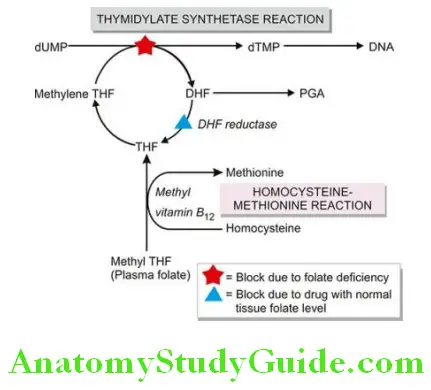
Absorption And Transport: Folate is normally absorbed from the duodenum and upper jejunum and to a lesser extent, from the lower jejunum and ileum. However, absorption depends upon the form of folate in the diet. Polyglutamate form in the foodstuffs is first cleaved by the enzyme, folate conjugase, in the mucosal cells to mono- and diglutamates which are readily assimilated.
Synthetic folic acid preparations in polyglutamate form are also absorbed as rapidly as mono- and diglutamate form because of the absence of natural inhibitors. Mono- and diglutamates undergo further reduction in the mucosal cells to form tetrahydrofolate (THF), a monoglutamate. THF circulates in the plasma as methylated compound, methyl THF, bound to a protein. Once methyl THF is transported into the cell by a carrier protein, it is reconverted to polyglutamate.
Tissue Stores:
The liver and red cells are the main storage sites of folate, largely as methyl THF polyglutamate form. The total body stores of folate are about 10-12 mg enough for about 4 months. Normally, folate is lost from the sweat, saliva, urine and faeces.
Functions:
Folate plays an essential role in cellular metabolism. It acts as a co-enzyme for 2 important biochemical reactions involving transfer of 1-carbon units (viz. methyl and formyl groups) to various other compounds.
These reactions are as under:
Thymidylate synthetase reaction Formation of deoxy thymidylate monophosphate (dTMP) from its precursor form, deoxy uridylate monophosphate (dUMP). Methylation of homocysteine to methionine This reaction is linked to vitamin B12 metabolism. These biochemical reactions are considered in detail below together with biochemical basis of the megaloblastic anaemia.
Biochemical Basis Of Megaloblastic Anaemia:
The basic biochemical abnormality common to both vitamin B12 and folate deficiency is a block in the pathway of DNA synthesis and that there is an inter-relationship between vitamin B12 and folate metabolism in the methylation reaction of homocysteine to methionine.
As stated above, folate as co-enzyme methylene THF, is required for transfer of 1-carbon moieties (e.g. methyl and formyl) to form building blocks in DNA synthesis. These 1-carbon moieties are derived from serine or formiminoglutamic acid (FIGLU). Two of the important folate-dependent (1-carbon transfer) reactions for formation of building blocks in DNA synthesis are as under:
1. Thymidylate synthetase reaction: This reaction involves synthesis of deoxy thymidylate monophosphate (dTMP) from deoxy uridylate monophosphate (dUMP). The methyl group of dUMP → dTMP reaction is supplied by the co-enzyme, methylene-THF.
After the transfer of 1- carbon from methylene-THF, dihydrofolate (DHF) is produced which must be reduced to active THF by the enzyme DHF-reductase before it can participate in further 1-carbon transfer reaction. Drugs like methotrexate (anti-cancer) and pyrimethamine (antimalarial) are inhibitory to the enzyme, DHF-reductase, thereby inhibiting the DNA synthesis.
2. Homocysteine-methionine reaction: Homocysteine is converted into methionine by transfer of a methyl group from methylene-THF. After transfer of 1-carbon from methylene-THF, THF is produced. This reaction requires the presence of vitamin B12 (methyl-B12).
Deficiency of folate from any cause results in reduced supply of the coenzyme, methyleneTHF, and thus interferes with the synthesis of DNA. Deficiency of vitamin B12 traps folate as its transport form, methyl-THF, thereby resulting in reduced formation of the active form, methylene-THF, needed for DNA synthesis. This is referred to as methyl-folate trap hypothesis.
An alternative hypothesis of inter-relationship of B12 and folate is the formate-saturation hypothesis. According to this hypothesis, the active substrate is formyl-THF. Vitamin B12 deficiency results in reduced supply of formate to THF causing reduced generation of the active compound, formyl THF.
Etiology And Classification Of Megaloblastic Anaemia:
The etiology of megaloblastic anaemia varies in different parts of the world. As outlined megaloblastic anaemia is classified into 3 broad groups: vitamin B12 deficiency, folate deficiency, and deficiency from other causes.
Vitamin B12 Deficiency:
In Western countries, deficiency of vitamin B12 is more commonly due to pernicious (Addisonian) anaemia. True vegetarians like traditional Indian Hindus and breast-fed infants have dietary lack of vitamin B12.
Gastrectomy by lack of intrinsic factor, and small intestinal lesions involving distal ileum where absorption of vitamin B12 occurs, may cause deficiency of the vitamin. Deficiency of vitamin B12 takes at least 2 years to develop when the body stores are totally depleted.
Vitamin B12 Deficiency:
- Inadequate dietary intake e.g. strict vegetarians, breastfed infants.
- Malabsorption
- Gastric causes: pernicious anaemia, gastrectomy, congenital lack of intrinsic factor.
- Intestinal causes: tropical sprue, ileal resection, Crohn’s disease, intestinal blind loop syndrome, fishtapeworm infestation.
2. Folate Deficiency:
- Inadequate dietary intake e.g. in alcoholics, teenagers, infants, old age, poverty.
- Malabsorption e.g. in tropical sprue, coeliac disease, partial gastrectomy, jejunal resection, Crohn’s disease.
- Excess demand
- Physiological: pregnancy, lactation, infancy.
- Pathological: malignancy, increased haematopoiesis, chronic exfoliative skin disorders, tuberculosis, rheumatoid arthritis.
- Excess urinary folate loss e.g. in active liver disease, congestive heart failure.
3. Other causes:
Impaired metabolism e.g. inhibitors of dihydrofolate (DHF) reductase such as methotrexate and pyrimethamine; alcohol, congenital enzyme deficiencies. Unknown etiology e.g. in Di Guglielmo’s syndrome, congenital dyserythropoietic anaemia, refractory megaloblastic anaemia.
2. Folate Deficiency:
Folate deficiency is more often due to poor dietary intake. Other causes include malabsorption, excess folate utilisation such as in pregnancy and in various disease states, chronic alcoholism, and excess urinary folate loss.
Folate deficiency arises more rapidly than vitamin B12 deficiency since the body’s stores of folate are relatively low which can last for up to 4 months only. Patients with tropical sprue are often deficient in both vitamin B12 and folate. Combined deficiency of vitamin B12 and folate may occur from severe deficiency of vitamin B12because of the biochemical interrelationship with folate metabolism.
3. Other Causes:
In addition to deficiency of vitamin B12 and folate, megaloblastic anaemias may occasionally be induced by other factors unrelated to vitamin deficiency. These include many drugs which interfere with DNA synthesis, acquired defects of haematopoietic stem cells, and rarely, congenital enzyme deficiencies.
Clinical Features:
Deficiency of vitamin B12 and folate may cause following clinical manifestations which may be present singly or in combination and in varying severity:
- Anaemia Macrocytic megaloblastic anaemia is the cardinal feature of deficiency of vitamin B12 and/or folate. The onset of anaemia is usually insidious and gradually progressive.
- Glossitis Typically, the patient has a smooth, beefy, red tongue.
- Neurologic manifestations Vitamin B12 deficiency, particularly in patients of pernicious anaemia, is associated with significant neurological manifestations in the form of subacute combined, degeneration of the spinal cord and peripheral neuropathy deficiency may occasionally develop neuropathy only. The underlying pathologic process consists of demyelination of the peripheral nerves, the spinal cord and the cerebrum. Signs and symptoms include numbness, paraesthesia, weakness, ataxia, poor finger coordination and diminished reflexes.
- Others In addition to the cardinal features mentioned above, patients may have various other symptoms. These include: mild jaundice, angular stomatitis, purpura, melanin pigmentation, symptoms of malabsorption, weight loss and anorexia.
Laboratory Findings:
The investigations of a suspected case of megaloblastic anaemia are aimed at 2 aspects:
General laboratory investigations of anaemia which include blood picture, red cell indices, bone marrow findings, and biochemical tests. Special tests to establish the cause of megaloblastic anaemia as to know whether it is due to deficiency of vitamin B12 or folate.
Based on these principles, the following scheme of investigations is followed:
General Laboratory Findings:
Blood picture and red cell indices: Estimation of haemoglobin, examination of a blood film and evaluation of absolute values are essential preliminary investigations:
- Haemoglobin Haemoglobin estimation reveals values below the normal range. The fall in haemoglobin concentration may be of a variable degree.
- Red cells Red blood cell morphology in a blood film shows the characteristic macrocytosis. However, macrocytosis can also be seen in several other disorders such as: haemolysis, liver disease, chronic alcoholism, hypothyroidism, aplastic anaemia, myeloproliferative disorders and reticulocytosis. In addition, the blood smear demonstrates marked anisocytosis, poikilocytosis and presence of macroovalocytes. Basophilic stippling and occasional erythroblast may also be seen.
- Reticulocyte count The reticulocyte count is generally low to normal in untreated cases.
- Absolute values The red cell indices reveal an elevated MCV (above 120 fl) proportionate to the severity of macrocytosis, elevated MCH (above 50 pg) and normal or reduced MCHC.
- Leucocytes The total white blood cell count may be reduced. Presence of characteristic hypersegmented neutrophils (having more than 5 nuclear lobes) in the blood film should raise the suspicion of megaloblastic anaemia. An occasional myelocyte may also be seen.
- Platelets Platelet count may be moderately reduced in severely anaemic patients. Bizarre forms of platelets may be seen.
2. Bone Marrow Findings:
The bone marrow examination is very helpful in the diagnosis of megaloblastic anaemia.
Significant findings of marrow examination are as under:
- Marrow cellularity The marrow is hypercellular with a decreased myeloid-erythroid ratio.
- Erythropoiesis: There is erythroid hyperplasia due to characteristic megaloblastic erythropoiesis. Megaloblasts are abnormal, large, nucleated erythroid precursors, having nuclear-cytoplasmic asynchrony i.e. the nuclei are less mature than the development of cytoplasm.The nuclei are large, having fine, sieve-like and open chromatin that stains lightly, while the haemoglobinisation of the cytoplasm proceeds normally or at a faster rate i.e. nuclear maturation lags behind that of cytoplasm (compared from iron deficiency anaemia in which cytoplasmic maturation lags behind).Megaloblasts with abnormal mitoses may be seen. Features distinguishing megaloblast from erythroblast are summed up in Table. Ineffective erythropoiesis such as presence of degenerated erythroid precursors may be present.
- Other cells: Granulocyte precursors are also affected to some extent. Giant forms of metamyelocytes and band cells may be present in the marrow. Megakaryocytes are usually present in normal number but may occasionally be decreased and show abnormal morphology such as hypersegmented nuclei and agranular cytoplasm.
- Marrow iron: Prussian blue staining for iron in the marrow shows an increase in the number and size of iron granules in the erythroid precursors. Ring sideroblasts are, however, rare. Iron in the reticulum cells is increased.
- Chromosomes: Marrow cells may show variety of random chromosomal abnormalities such as chromosome breaks, centromere spreading etc.
Biochemical Findings:
In addition to the general blood and marrow investigations and specific tests to determine the cause of deficiency (described below), the following biochemical abnormalities are observed in cases of megaloblastic anaemia:
- There is rise in serum unconjugated bilirubin and LDH as a result of ineffective erythropoiesis causing marrow cell breakdown.
- The serum iron and ferritin may be normal or elevated.
2. Special Tests for Cause of Specific Deficiency: In evaluating a patient of megaloblastic anaemia, it is important to determine the specific vitamin deficiency by assay of vitamin B12 and folate. In sophisticated clinical laboratories, currently automated multiparametric, random access analysers are employed based on separation techniques by chemiluminescence and enzyme-linked fluorescence detection systems which have largely replaced the traditional microbiologic assays for vitamin B12 and folate. Traditional tests are briefly described here.
Tests For Vitamin B12 Deficiency: The normal range of vitamin B12 in serum is 280-1000 pg/ml. Values less than 100 pg/ml indicate clinically deficient stage. Traditional tests employed to establish vitamin B12 deficiency are serum vitamin B12 assay, Schilling (24- hour urinary excretion) test and serum enzyme levels.
Serum Vitamin B12 Assay: Assay of vitamin B12 in blood can be done by 2 methods—microbiological assay and radioassay.
- Microbiological assay In this test, the serum sample to be assayed is added to a medium containing all other essential growth factors required for a vitamin B12-dependent microorganism.The medium along with microorganism is incubated and the amount of vitamin B12 is determine turbimetrically which is then compared with the growth produced by a known amount of vitamin B12. Several organisms have been used for this test such as Euglena gracilis, Lactobacillus leichmannii, Escherichia coli and Ochromonas malhamensis but E. gracilis is considered more sensitive and accurate. The addition of antibiotics to the test interferes with the growth and yields false low result.
- Radioassay Assays of serum B12 by radioisotope dilution (RID) and radioimmunoassay (RIA) have been developed. These tests are more sensitive and have the advantage over microbiologic assays in that they are simpler and more rapid, and the results are unaffected by antibiotics and other drugs which may affect the living organisms.
2. Schilling Test (24-Hour Urinary Excretion Test): Schilling test is done to detect vitamin B12 deficiency as well as to distinguish and detect lack of IF and malabsorption syndrome. The results of test also depend upon good renal function and proper urinary collection. Radioisotope used for labelling B12 is either 58Co or 57Co.
The test is performed in two stages as under:
Stage I: Without IF The patient after an overnight fasting is administered oral dose of 1 mg of radioactively labelled vitamin B12 (‘hot’ B12) in water. At the same time, 1 mg of unlabelled vitamin B12 (‘cold’ B12) is given by intramuscular route; this ‘cold’ B12 will saturate the serum as well as the tissue binding sites to create excess of circulating vitamin B12.
If parenteral administration is not done, whole of orally administered radiolabelled vitamin B12 will be taken up by the tissues in the case of dietary vitamin B12 deficiency and little will be excreted in the urine.
- Urine is collected over the next 24 hours and excretion of radioactive-labelled vitamin B12 is estimated:
- Urinary excretion >10% of the oral dose of ‘hot’ B12 indicates normal absorption of this vitamin and is diagnostic of dietary B12 deficiency. Low urinary excretion of ‘hot’ B12 rules out dietary deficiency and is due to poor absorption of this vitamin, which could be from IF deficiency (stage II) or from malabsorption and therefore, proceed to stage II.
Stage II: With IF If the 24-hour urinary excretion of ‘hot’ B12 low, the test is repeated using the same procedure as in stage I but concomitantly high oral dose of IF is administered with ‘hot’ B12.
- If the 24-hour urinary excretion of ‘hot’ BB12 is now normal or high, it is diagnostic of IF deficiency (i.e. pernicious anaemia). Patients with pernicious anaemia have abnormal test even after treatment with vitamin B12 due to IF deficiency.
- However, if 24-hour urinary excretion of ‘hot’ B12 remains low, then vitamin B12 deficiency is due to other causes in intestinal malabsorption (e.g. pancreatic deficiency, ileal disease, intestinal bacterial overgrowth etc).
Serum Enzyme Levels:
Besides Schilling test, another way of distinguishing whether megaloblastic anaemia is due to cobalamine or folate is by serum determination of methylmalonic acid and homocysteine by sophisticated enzymatic assays. Both are elevated in cobalamine deficiency, while in folate deficiency there is only elevation of homocysteine and not of methylmalonic acid.
Tests For Folate Deficiency:
The normal range of serum folate is 6-18 ng/ml. Values of 4 ng/ml or less are generally considered to be diagnostic of folate deficiency. Measurement of formiminoglutamic acid (FIGLU) urinary excretion after histidine load was used formerly for assessing folate status but it is less specific and less sensitive than the serum assays. Currently, there are 3 tests used to detect folate deficiency—urinary excretion of Figlu, serum and red cell folate assay.
1. Urinary Excretion Of Figlu:
Folic acid is required for conversion of formiminoglutamic acid (FIGLU) to glutamic acid in the catabolism of histidine. Thus, on oral administration of histidine, urinary excretion of FIGLU is increased if folate deficiency is present.
2. Serum Folate Assay:
Folate in serum can be estimated by 2 methods—microbiological assay and radioassay.
- Microbiological assay: This test is based on the principle that theserum folate acid activity is mainly due to the presence of a folic acid co-enzyme, 5-methyl THF, and that this compound is required for growth of the microorganism, Lactobacillus casei. The growth of L. casei is inhibited by addition of antibiotics.
- Radioassay: The principle and method of radioassay by radioisotope dilution (RID) test are similar to that for serum B12 assay. The test employs labelled pteroylglutamic acid or methylTHF. Commercial kits are available which permit simultaneous assay of both vitamin B12 and folate.
3. Red Cell Folate Assay:
Red cells contain 20-50 times more folate than the serum; thus red cell folate assay is more reliable indicator of tissue stores of folate than serum folate assay. Microbiological radioassay and protein-binding assay methods can be used for estimation of red cell folate. Red cell folate values are decreased in patients with megaloblastic anaemia as well as in patients with pernicious anaemia.

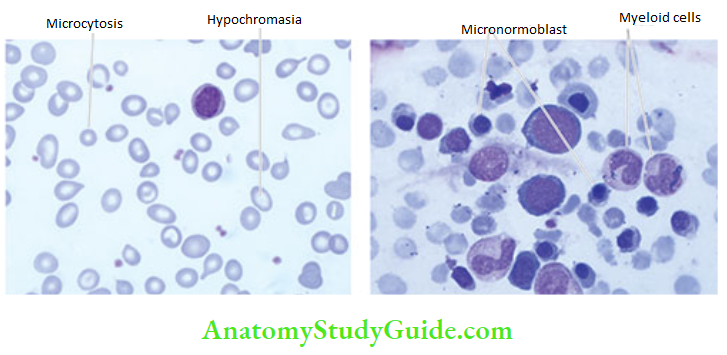

Principles Of Treatment:
Most cases of megaloblastic anaemia need therapy with appropriate vitamin. This includes:
Hydroxycobalamin as intramuscular injection 1000 µg for 3 weeks and oral folic acid 5 mg tablets daily for 4 months. Severely-anaemic patients in whom a definite deficiency of either vitamin cannot be established with certainty are treated with both vitamins concurrently.
Blood transfusion should be avoided since it may cause circulatory overload. Packed cells may, however, be infused slowly. Treatment of megaloblastic anaemia is quite gratifying. The marrow begins to revert back to normal morphology within a few hours of initiating treatment and becomes normoblastic within 48 hours of start of treatment.
Reticulocytosis appears within 4-5 days after therapy is started and peaks at day 7. Haemoglobin should rise by 2-3 g/dl each fortnight. Peripheral neuropathy may show some improvement but subacute combined degeneration of the spinal cord is irreversible.
Pernicious Anaemia:
Pernicious anaemia (PA) was first described by Addison in 1855 as a chronic disorder of middleaged and elderly individual of either sex in which intrinsic factor (IF) secretion ceases owing to atrophy of the gastric mucosa.
The condition is, therefore, also termed Addisonian megaloblastic anaemia. The average age at presentation is 60 years but rarely it can be seen in children under 10 years of age (juvenile pernicious anaemia).
PA is seen most frequently in individuals of northern European descent and African Americans and is uncommon in South Europeans and Orientals. Besides, PA occurs in close relatives of persons with organ-specific autoimmune diseases.
Pathogenesis:
There is evidence to suggest that the atrophy of gastric mucosa in PA resulting in absence or low level of IF is caused by an autoimmune reaction against gastric parietal cells. The evidences in support of immune basis are by disease association with immune abnormalities and detection of antibodies in serum:
1. Disease associations:
These evidences are as under:
- The incidence of PA is high in patients with other autoimmune diseases such as Graves’ disease, myxoedema, thyroiditis, vitiligo, diabetes and idiopathic adrenocortical insufficiency.
- Relatives of patients with PA have an increased incidence of the disease or increased presence of autoantibodies.
- Corticosteroids have been reported to be beneficial in curing the disease both pathologically
and clinically. - PA is more common in patients with agammaglobulinaemia supporting the role of cellular immune system in destruction of parietal cells.
- HLA-3 and HLA-B8 types have higher association with PA II.
2. Serum antibodiea in PA:
Definite evidence of autoimmune phenomena in PA is supported by detection of serum autoantibodies against IF and against parietal cells:
- IF antibodies Patients of PA have two types of IF immunoglobulin G antibodies in their sera:
- Type I or blocking antibody seen in sera of about 55% of patients of PA. It prevent combination of IF with cobalamin. Type I antibody is also seen sometimes in other autoimmune diseases and in sera of relatives of PA patients. Besides sera, type I antibody may also be detected in gastric juice.
- Type II or binding antibody is seen in approximately 35% cases of PA. It prevents attachment of IF with ileal mucosa.
- Parietal cell antibody is present in sera of ~90% cases of PA. This antibody acts against α and β subunits of gastric proton pump i.e. against H+, K+ and ATPase.
Morphologic Features:
The most characteristic pathologic finding in PA is gastric atrophy affecting the acid- and pepsin-secreting portion of the stomach and sparing the antrum (page 573). Gastric epithelium may show cellular atypia. About 2-3% cases of PA develop carcinoma of the stomach. Other pathologic changes are secondary to vitamin B12 deficiency and include megaloblastoid alterations in the gastric and intestinal epithelium and neurologic abnormalities such as peripheral neuropathy and spinal cord damage.
Clinical Features:
The disease has insidious onset and progresses slowly. The clinical manifestations are mainly due to vitamin B12 deficiency. These include: anaemia, glossitis, neurological abnormalities (neuropathy, subacute combined degeneration of the spinal cord, retrobulbar neuritis), gastrointestinal manifestations (diarrhoea, anorexia, weight loss, dyspepsia), hepatosplenomegaly, congestive heart failure and haemorrhagic manifestations. Other autoimmune diseases such as autoimmune thyroiditis may be associated.
Principles Of Treatment:
Patients of PA are treated with vitamin B12 in the following way:
- Parenteral vitamin B12 replacement therapy.
- Symptomatic and supportive therapy such as physiotherapy for neurologic deficits and occasionally blood transfusion.
- Follow-up for early detection of cancer of the stomach. Most of the abnormalities due to vitamin B12 deficiency can be corrected except the irreversible damage to the spinal cord. Corticosteroid therapy can improve the gastric lesion with a return of acid secretion but the higher incidence of gastric polyps and cancer of the stomach in these patients can only be detected by frequent follow-up.
Megaloblastic Anaemias:
- Megaloblastic anaemias are associated with macrocytic blood picture and megaloblastic marrow erythropoiesis. This group is due to either due to nutritional deficiency of vitamin B12 and/or folate or due to pernicious anaemia from deficiency of IF.
- The only dietary sources of vitamin B12 are foods of animal protein origin while folate exists in different plants, bacteria and animal tissues.
- Basic biochemical abnormality in both vitamin B12 and folate deficiency is a block in the pathway of DNA synthesis.
- Major clinical features of megaloblastic anaemia are anaemia, glossitis, and neurological manifestations.
- Laboratory diagnosis is made by macrocytic blood picture, raised MCV and MCH, and megaloblastic erythropoiesis in the bone marrow with raised iron stores. Specific deficiency of vitamin B12 and folate can be made by serum assay and Schilling test.
- Pernicious anaemia is megaloblastic erythropoiesis from impaired absorption of vitamin B12 which is due to deficiency of intrinsic factor from atrophic gastritis. Pernicious anaemia has autoimmune basis as seen by disease associations and by detection of antibodies against IF and parietal cells of the stomach
Haemolytic Anaemias And Anaemia Due To Blood Loss
Definition And Classification:
Haemolytic anaemias are defined as anaemias resulting from an increase in the rate of red cell destruction. Normally, effete red cells undergo lysis at the end of their lifespan of 120 ± 30 days within the cells of reticuloendothelial (RE) system in the spleen and elsewhere (extravascular haemolysis), and haemoglobin is not liberated into the plasma in appreciable amounts.
The red cell lifespan is shortened in haemolytic anaemia i.e. there is accelerated haemolysis. However, shortening of red cell lifespan does not necessarily result in anaemia. In fact, compensatory bone marrow hyperplasia may cause 6 to 8-fold increase in red cell production without causing anaemia to the patient, so-called compensated haemolytic disease.
The premature destruction of red cells in haemolytic anaemia may occur at either of the following 2 sites:
- Firstly, the red cells undergo lysis in the circulation and release their contents into plasma (intravascular haemolysis). In these cases the plasma haemoglobin rises substantially and part of it may be excreted in the urine (haemoglobinuria).
- Secondly, the red cells are taken up by cells of the RE system where they are destroyed and digested (extravascular haemolysis). In extravascular haemolysis, plasma haemoglobin level is, therefore, barely raised.
Extravascular haemolysis is more common than the former. One or more factors may be involved in the pathogenesis of various haemolytic anaemias. Clinically, haemolytic anaemias may be acute or chronic, mild to severe, hereditary or acquired.
Haemolytic anaemias are broadly classified into 2 main categories:
- Acquired haemolytic anaemias caused by a variety of extrinsic environmental factors (i.e. extracorpuscular). However, PNH is an acquired HA with intracorpuscular defect.
- Hereditary haemolytic anaemias are usually the result of intrinsic red cell defects (i.e. intracorpuscular). However, familial haemolytic-uraemic syndrome is hereditary HA with extracorpuscular defect. A simplified classification based on these mechanisms is given in Table and diagrammatically represented.
General Considerations:
A number of clinical and laboratory features are shared by various types of haemolytic anaemias.
These are briefly described below:
General Clinical Features:
Some of the general clinical features common to most congenital and acquired haemolytic anaemias are as under:
- Presence of pallor of mucous membranes.
- Positive family history with life-long anaemia in patients with congenital haemolytic anaemia.
- Mild fluctuating jaundice due to unconjugated hyperbilirubinaemia.
- Urine turns dark on standing due to excess of urobilinogen in urine.
- Splenomegaly is found in most chronic haemolytic anaemias, both congenital and acquired.
- Pigment gallstones are found in some cases.
Laboratory Evaluation Of Haemolysis:
Pathways by which haemoglobin derived from effete red cells is metabolised.
The investigations of a patient suspected to have haemolytic anaemia should provide answers to 3 vital questions:
- Is there evidence of haemolysis?
- What is the type of haemolytic mechanism?
- What is the precise diagnosis?
In order to find answer to these questions, laboratory work-up is divided into the following 4 groups:
1. Tests of Increased Red Cell Breakdown
- Serum bilirubin: unconjugated (indirect) bilirubin raised
- Urine urobilinogen: raised
- `Bilirubinuria: Absent
- Faecal stercobilinogen: raised
- Serum haptoglobin (α-globulin binding protein): reduced or absent due to its binding to liberated haemoglobin
- Plasma lactic dehydrogenase: raised
- Haemoglobinaemia, haemoglobinuria, methaemoglobinaemia and haemosiderinuria: present as evidence of intravascular haemolysis
2. Tests of Increased Red Cell Production
- Reticulocyte count: raised (reticulocytosis); generally appears early and is hence most useful initial test of marrow erythroid hyperplasia
- Routine blood smear: macrocytosis, polychromasia and presence of erythroblasts
- Bone marrow: erythroid hyperplasia with usually raised iron stores
- X-ray of bones: expanded marrow space, especially in tubular bones and skull
3. Tests of Damage to Red Cells
- Routine blood smear shows a variety of abnormal morphological appearances of red cells described on page 307 and illustrated in earlier. A summary of contributory features of morphology of RBCs in arriving at the diagnosis of haemolytic anaemia and its cause is given in Table.
- Osmotic fragility is increased or decreased
- Autohaemolysis test with or without addition of glucose
- Coombs’ antiglobulin test
- Electrophoresis for abnormal haemoglobins
- Estimation of HbA2
- Estimation of HbF
- Tests for sickling
- Screening test for G6PD deficiency and other enzymes (e.g. Heinz bodies test)
4. Tests for Shortened Red Cell Lifespan A shortened red cell survival is best tested by 51Cr labelling method. Normal RBC lifespan of 120 days is shortened to 20-40 days in moderate haemolysis and to 5-20 days in severe haemolysis.
Contrasting features of intravascular and extravascular haemolysis are given in Table. After these general comments on clinical and laboratory featureof haemolysis, we now turn to discuss various types of haemolytic anaemia as given in the classification in Table.

1. Acquired:
- Antibody: Immunohaemolytic anaemias
- Autoimmune haemolytic anaemia (AIHA)
- Warm antibody AIHA
- Cold antibody AIHA
- Drug-induced immune haemolytic anaemia
- Isoimmune haemolytic anaemia
- Autoimmune haemolytic anaemia (AIHA)
- Mechanical trauma: Microangiopathic haemolytic anaemia
- Direct toxic effects: Malaria, bacterial, infection and other agents
- Acquired red cell membrane abnormalities: Paroxysmal nocturnal haemoglobinuria (PNH)
- Splenomegaly
2. Hereditary:
- Abnormalities of red cell membrane
- Hereditary spherocytosis
- Hereditary elliptocytosis (hereditary ovalocytosis)
- Hereditary stomatocytosis
3. Disorders of red cell interior:
- Red cell enzyme defects (Enzymopathies)
- Defects in the hexose monophosphate shunt: G6PD deficiency
- Defects in the Embden-Meyerhof (or glycolytic) pathway: pyruvate kinase deficiency
- Disorders of haemoglobin (Haemoglobinopathies)
- Structurally abnormal haemoglobins: sickle syndromes, other haemoglobinopathies
- Reduced globin chain synthesis: thalassaemias (G6PD, glucose-6-phosphate dehydrogenase).
Haemolytic Anaemias: General Considerations:
- Haemolytic anaemias (HA) result from an increased rate of red cell destruction and premature destruction of red cells either at intravascular (peripheral blood) or in extravascular sites (reticuloendothelial organs); the latter is more common.
- HA are divided into hereditary and acquired, the former are more common.
- Clinically, HA cases have pallor, mild jaundice, and often splenomegaly.
- Laboratory work-up of HA includes 4 groups of investigations:
-
- Tests for increased red cell destruction (e.g. raised bilirubin, raised urobilinogen, reduced serum haptoglobin etc),
- Tests for increased red cell production (reticulocytosis, blood smear and bone marrow examination),
- Tests for damage to red cells (osmotic fragility, Coombs’ test, electrophoresis etc), and
- Tests for red cell lifespan (by radioisotope labelling)
Acquired Haemolytic Anaemias:
Acquired haemolytic anaemias are caused by a variety of extrinsic factors, namely: antibody (immunohaemolytic anaemia), mechanical factors (microangiopathic haemolytic anaemia), direct toxic effect (in malaria, clostridial infection etc), splenomegaly, and certain acquired membrane abnormalities (paroxysmal nocturnal haemoglobinuria). These are discussed below.
1. Immunohaemolytic Anaemias:
Immunohaemolytic anaemias are a group of anaemias occurring due to antibody production by the body against its own red cells. Based on type of antibody inducing immune haemolysis
These are of following three types:
Autoimmune haemolytic anaemia (AIHA) characterised by formation of autoantibodies against patient’s own red cells.
Depending upon the reactivity of autoantibody, AIHA is further divided into 2 types:

- ‘Warm’ antibody AIHA in which the autoantibodies are reactive at body temperature (37°C).
- ‘Cold’ antibody AIHA in which the autoantibodies react better with patient’s own red cells at 4°C.
2. Drug-induced immunohaemolytic anaemia.

3. Isoimmune haemolytic anaemia in which the antibodies are acquired by blood transfusions, pregnancies and haemolytic disease of the newborn. An important diagnostic tool in all cases of immunohaemolytic anaemias is Coombs’ antiglobulin test for detection of incomplete Rh-antibodies in saline directly (direct Coombs’) or after addition of albumin (indirect Coombs’).
Autoimmune Haemolytic Anaemia (Aiha):
‘Warm’ Antibody AIHA:
Pathogenesis:
Warm antibodies reactive at body temperature and coating the red cells are generally IgG class antibodies and occasionally they are IgA.
These red cell antibodies are idiopathic in origin but the mechanism of destruction of red cells coated with IgG is better understood:
- Human red cells coated with IgG antibodies are bound to the surface of RE cells, especially splenic macrophages. The spleen is particularly efficient in trapping red cells coated with IgG antibodies, and is, thus, the major site of red cell destruction in warm antibody AIHA.
- A part of the coated cell membrane is lost resulting in spherical transformation of the red cells (acquired spherocytosis).
- Red cells coated with IgG along with C3 on the surface further promote this red cellleucocyte interaction, accounting for more severe haemolysis.
Clinical Features:
Warm antibody AIHA may occur at any age and in either sex. The disease may occur without any apparent cause (idiopathic) but about a quarter of patients develop this disorder as a complication of an underlying disease affecting the immune system such as SLE, chronic lymphocytic leukaemia, lymphomas and certain drugs such as methyl DOPA, penicillin etc.
The disease tends to have remissions and relapses. The usual clinical features are as follows:
- Chronic anaemia of varying severity with remissions and relapses
- Splenomegaly
- Occasionally hyperbilirubinaemia
Laboratory Findings:
The haematological and biochemical findings in such cases are as under:
- Mild to moderate chronic anaemia
- Reticulocytosis
- Prominent spherocytosis in the peripheral blood film
- Positive direct Coombs’ (antiglobulin) test for presence of warm antibodies on the red cell best detected at 37°C
- A positive indirect Coombs’ (antiglobulin) test at 37°C may indicate presence of large quantities of warm antibodies in the serum.
- Unconjugated (indirect) hyperbilirubinaemia
- Co-existent immune thrombocytopenia along with occasional venous thrombosis may be present (termed Evans’ syndrome)
- In more severe cases, haemoglobinaemia and haemoglobinuria may be present.
- Warm antibody AIHA
- Idiopathic (primary)
- Lymphomas-leukaemias e.g. non-Hodgkin’s lymphoma, CLL, Hodgkin’s disease.
- Collagen vascular diseases e.g SLE
- Drugs e.g. methyldopa, penicillin, quinidine group
- Post-viral
- Cold antibody AIHA
- Cold agglutinin disease
- Acute: Mycoplasma infection, infectious mononucleosis
- Chronic: Idiopathic, lymphomas
- PCH (Mycoplasma infection, tertiary syphilis) (AIHA, autoimmune haemolytic anaemia; CLL, chronic lymphocytic leukaemia; SLE, systemic lupus erythematosus; PCH, paroxysmal cold haemoglobinuria).
- Cold agglutinin disease
Treatment of these cases consists of removal of the cause whenever present, corticosteroid therapy, and in severe cases blood transfusions. Splenectomy is the second line of therapy in this disorder.
‘Cold’ Antibody AIHA:
Pathogenesis:
Antibodies which are reactive in the cold (4°C) may induce haemolysis under 2 conditions: cold agglutinin disease and paroxysmal cold haemoglobinuria.
1. Cold agglutinin disease: In cold agglutinin disease, the antibodies are IgM type which bind to the red cells best at 4°C. These cold antibodies are usually directed against the I antigen on the red cell surface. Agglutination of red blood cells by IgM cold agglutinins is most profound at very low temperature but upon warming to 37°C or above, disagglutination occurs quickly. Haemolytic effect is mediated through fixation of C3 to the red blood cell surface and not by agglutination alone. Most cold agglutinins affect juvenile red blood cells Cold antibody disease is seen in the course of certain viral infections (e.g. EBV, CMV, infectious mononucleosis) and secondary to lymphomas.
2. Paroxysmal cold haemoglobinuria (PCH): In PCH, cold antibody is an IgG antibody (Donath-Landsteiner antibody) which is directed against P blood group antigen and causes complement-mediated haemolysis. Attacks of PCH are precipitated by exposure to cold. PCH is uncommon and may be seen in Mycoplasma pneumonia and as a complication of tertiary syphilis.
Clinical Features:
The clinical manifestations are due to haemolysis and not due to agglutination. These include the following:
- Chronic anaemia which is worsened by exposure to cold
- Raynaud’s phenomenon
- Cyanosis affecting the cold exposed regions such as tips of nose, ears, fingers and toes
- Haemoglobinaemia and haemoglobinuria occur on exposure to cold
Laboratory Findings:
The haematologic and biochemical findings are somewhat similar to those found in warm antibody AIHA except the thermal amplitude.
These findings are as follows:
- Chronic anaemia
- Low reticulocyte count since young red cells are affected more
- Spherocytosis is less marked
- Positive direct Coombs’ test for detection of C3 on the red cell surface but IgM responsible for C3 coating on red cells is not found.
- The cold antibody titre is very high at 4°C and very low at 37°C (Donath-Landsteiner test). IgM class cold antibody has specificity for I antigen, while the rare IgG class antibody of PCH has P blood group antigen specificity.
Treatment consists of keeping the patient warm and treating the underlying cause.
Drug-Induced Immunohaemolytic Anaemia:
Drugs may cause immunohaemolytic anaemia by 3 different mechanisms:
1. α-Methyl Dopa Type Antibodies:
A small proportion of patients receiving α-methyl dopa develop immunohaemolytic anaemia which is identical in every respect to warm antibody AIHA described above.
2. Penicillin-Induced Immunohaemolysis:
Patients receiving large doses of penicillin or penicillin-type antibiotics develop antibodies against the red blood cell-drug complex which induces haemolysis.
3. Innocent Bystander Immunohaemolysis:
Drugs such as quinidine form a complex with plasma proteins to which an antibody forms. This drug-plasma protein-antibody complex may induce lysis of bystanding red blood cells or platelets. In each type of drug-induced immunohaemolytic anaemia, discontinuation of the drug results in gradual disappearance of haemolysis.
Isoimmune Haemolytic Anaemia:
Isoimmune haemolytic anaemias are caused by acquiring isoantibodies or alloantibodies by blood transfusions, pregnancies and in haemolytic disease of the newborn. These antibodies produced by one individual are directed against red blood cells of the other. These conditions are considered.
2. Microangiopathic Haemolytic Anaemia:
Microangiopathic haemolytic anaemia is caused by abnormalities in the microvasculature. It is generally due to mechanical trauma to the red cells in circulation and is characterised by red cell fragmentation (schistocytosis). There are 3 different ways by which microangiopathic haemolytic anaemia results:
1. External Impact:
Direct external trauma to red blood cells when they pass through microcirculation, especially over the bony prominences, may cause haemolysis during various activities e.g. in prolonged marchers, joggers, karate players etc. These patients develop haemoglobinaemia, haemoglobinuria (march haemoglobinuria), and sometimes myoglobinuria as a result of damage to muscles.
2. Cardiac Haemolysis:
A small proportion of patients who receive prosthetic cardiac valves or artificial grafts develop haemolysis. This has been attributed to direct mechanical trauma to the red cells or shear stress from turbulent blood flow.
3. Fibrin Deposit In Microvasculature:
Deposition of fibrin in the microvasculature exposes the red cells to physical obstruction and eventual fragmentation of red cells and trapping of the platelets.
Fibrin deposits in the small vessels may occur in the following conditions:
- Abnormalities of the vessel wall e.g. in hypertension, eclampsia, disseminated cancers, transplant rejection, haemangioma etc.
- Thrombotic thrombocytopenic purpura
- Haemolytic-uraemic syndrome
- Disseminated intravascular coagulation (DIC)
- Vasculitis in collagen diseases
All these conditions are described in relevant sections separately.
3. Haemolytic Anaemia From Direct Toxic Effects:
Haemolysis may result from direct toxic effects of certain agents.
These include the following examples:
- Malaria by direct parasitisation of red cells (black-water fever).
- Bartonellosis by direct infection of red cells by the microorganisms.
- Septicaemia with Clostridium welchii by damaging the red cells.
- Other microorganisms such as pneumococci, staphylococci and Escherichia coli.
- Copper by direct haemolytic effect on red cells in Wilson’s disease and patients on haemodialysis.
- Lead poisoning shows basophilic stippling of red blood cells.
- Snake and spider bites cause haemolysis by their venoms.
- Extensive burns.

4. Paroxysmal Nocturnal Haemoglobinuria (PNH):
PNH is a rare acquired disorder of red cell membrane in which red cells are unduly sensitive to destruction by activated complement due to intrinsic defect in red cell membrane. The disorder generally presents in adult life.
Pathogenesis:
In PNH, there is chronic intravascular haemolysis (as compared to extravascular haemolysis in other conditions of acquired HA) by the following mechanism:
- PNH is a clonal disease of the cell membrane while normal clone also continues to proliferate. Being clonal in origin, the defect affects all the cells of myeloid progenitor lineage (RBCs, WBCs, platelets), resulting in deficient haematopoiesis.
- Due to intrinsic red cell membrane defect, there is deficient synthesis of a membrane glycolipid called glycosyl phosphatidyl inositol (GPI), essential for anchoring of the cell.
- The shortage in synthesis of GPI is due to a mutation in an X-linked gene called PIG-A (phosphatidyl inositol glycan). The mutation in PIG-A is not inherited but instead it affects haematopoietic stem cells as somatic mutations. Thus, the patient’s marrow continues to have dual population of cells: mutant and nonmutant cells, and accordingly blood too contains PNH cells and non-PNH (normal) cells.
- Normally, red cells are protected from complement activation and subsequent lysis due to presence of two GPI-linked anchor proteins, decay accelerating factor (DAF, CD55) and membrane inhibitor of reactive lysis (MIRL, CD59).
- As a result of PIG-A mutation, there is partial or complete deficiency of CD55 and CD59. This mutated state renders the red cells unduly sensitive to the lytic effect of activated complement, mainly by membrane attack complex (MAC).
- Haemolysis in PNH is exacerbated during the course of a viral or bacterial infection due to activation of complement by any pathway.

Clinical And Laboratory Findings:
Clinical and laboratory findings are as under:
- Haemolytic anaemia
- Pancytopenia (mild granulocytopenia and thrombocytopenia frequent)
- Intermittent clinical haemoglobinuria; acute haemolytic episodes occur at night identified by passage of brown urine in the morning.
- Haemosiderinuria very common.
- Venous thrombosis as a common complication due to inappropriate activation of PNH platelets.
The presence of inordinate sensitivity of red blood cells, leucocytes and platelets to complement in PNH can be demonstrated in vitro by Ham’s test using red cell lysis at acidic pH or by sucrose haemolysis test.
Since PNH is a stem cell disorder, about 20% cases of PNH may develop myeloproliferative or myelodysplastic syndrome, and some may even develop acute myeloid leukaemia and aplastic anaemia.
5. Haemolytic Anaemia In Splenomegaly:
Haemolytic anaemia is common in splenic enlargement from any cause. Normally, the spleen acts as a filter and traps the damaged red blood cells, destroys them and the splenic macrophages phagocytose the damaged red cells.
A normal spleen poses no risk to normal red blood cells. But splenomegaly exaggerates the damaging effect to which the red cells are exposed. Besides haemolytic anaemia, splenomegaly is usually associated with pancytopenia. Splenectomy or reduction in size of spleen by appropriate therapy relieves the anaemia as well as improves the leucocyte and platelet counts.
Acquired Haemolytic Anaemias:
- Acquired HA may be due to immune causes, mechanical factors, direct toxic effects, splenomegaly, and PNH.
- Immune HA are due to antibody production in the body against its own red cells. These may be autoimmune (warm and cold antibody), drug-induced or is immune antibodies
- Microangiopathic HA are caused due to abnormalities in the microvasculature causing trauma to red cells from external impact, cardiac disease and fibrin deposits in the microvasculature.
- Direct toxic effects of malaria and some bacterial infections can cause HA. PNH is a chronic acquired HA due to defect in the stem cells (deficiency of CD55 and CD59) and hence affects all cells in myeloid progenitor lineage.
- Splenomegaly is associated with HA due to damage induced during passage through splenic macrophages in enlarged spleen.
2. Hereditary Haemolytic Anaemias:
Hereditary haemolytic anaemias are usually the result of intracorpuscular defects (except familial haemolytic-uraemic syndrome). Accordingly, they are broadly classified into 2 groups:
- Hereditary abnormalities of red cell membrane
- Hereditary disorders of the interior of the red cells
1. Hereditary Abnormalities Of Red Cell Membrane:
The abnormalities of red cell membrane are readily identified on blood smear examination. Inherited red cell membrane defects are: hereditary spherocytosis (most common), hereditary elliptocytosis (or hereditary ovalocytosis) and hereditary stomatocytosis.
Hereditary Spherocytosis:
Hereditary spherocytosis is a common type of hereditary haemolytic anaemia of autosomal inheritance, mostly dominant but sometimes recessive too, in which the red cell membrane is abnormal.
Pathogenesis:
The molecular abnormality in hereditary spherocytosis is a mutation in one of the proteins which anchor the lipid bilayer to the underlying cytoskeleton.
These protein abnormalities are as under and are schematically illustrated:
1. Spectrin deficiency:
Almost all cases have a deficiency in the structural protein of the red cell membrane, spectrin. Spectrin deficiency correlates with the severity of anaemia. Mutation in spectrin by recessive inheritance called α-spectrin causes a more severe form of anaemia, while mutation by dominant inheritance forming β-spectrin results in mild form of the disease.
2. Ankyrin abnormality:
About half the cases of hereditary spherocytosis have defects in ankyrin, a protein that binds protein 3 and spectrin. Homozygous state with a recessive inheritance pattern has severe anaemia while heterozygotes with more common dominant inheritance pattern have milder anaemia.
Inherited mutation in spectrin or ankyrin causes defect in the anchoring of lipid bilayer cell membrane. When red cells with such an unstable membrane but having normal volume, are released in circulation, they lose their membrane further, till they can accommodate the given volume.
This results in the formation of spheroidal contour and smaller size of red blood cells, termed microspherocytes. These deformed red cells are not flexible, unlike normal biconcave red cells. membrane further. This produces a subpopulation of hyperspheroidal red cells in the peripheral blood which are subsequently destroyed in the spleen.
Clinical Features:
The disorder may be clinically apparent at any age from infancy to old age and has equal sex incidence.
The family history may be present. The major clinical features are as under:
- Anaemia is usually mild to moderate.
- Splenomegaly is a constant feature.
- Jaundice occurs due to increased concentration of unconjugated (indirect) bilirubin in the plasma (also termed congenital haemolytic jaundice).
- Pigment gallstones are frequent due to increased bile pigment production. Splenectomy offers the only reliable mode of treatment.
Laboratory Findings:
The usual haematological and biochemical findings are as under:
- Anaemia of mild to moderate degree.
- Reticulocytosis, usually 5-20%.
- Blood film shows the characteristic abnormality of erythrocytes in the form of microspherocytes.
- MCV is usually normal or slightly decreased but MCHC is increased.
- The osmotic fragility test helps test the spheroidal nature of red cells which lyse more readily in solutions of low salt concentration i.e. osmotic fragility is increased.
- The autohaemolysis test is similar to the osmotic fragility test after incubation and shows increased spontaneous autohaemolysis (10-15% red cells) as compared to normal red cells (less than 4%). Autohaemolysis is correctable by the addition of glucose.
- Direct Coombs’ (antiglobulin) test is negative to distinguish this condition from acquired spherocytosis of AIHA in which case it is positive.
Spherocytes may also be seen in blood film in acquired immune haemolytic anaemia and following red cell transfusion.
Hereditary Elliptocytosis (Hereditary Ovalocytosis):
Hereditary elliptocytosis or hereditary ovalocytosis is another autosomal dominant disorder involving red cell membrane protein spectrin. Some patients have an inherited defect in erythrocyte membrane protein 4.1 that interconnects spectrin with actin in the cytoskeleton.
The disorder is similar in all respects to hereditary spherocytosis except that the blood smear shows oval or elliptical red cells and is clinically a milder disorder than hereditary spherocytosis. Acquired causes of elliptocytosis include iron deficiency and myeloproliferative disorders.
Hereditary Stomatocytosis:
Stomatocytes are cup-shaped RBCs having one surface concave and the other side convex. This causes a central slit-like or mouth-like appearance of red cells. The underlying defect is in membrane protein, stomatin, having an autosomal dominant pattern of inheritance. The stomatocytes are swollen red cells (overhydrated red cells) due to increased permeability to sodium and potassium. The affected patients have mild anaemia and splenomegaly.

Hereditary Disorders Of Red Cell Interior:
Inherited disorders involving the interior of the red blood cells are classified into 2 groups:
1. Red cell enzyme defects (Enzymopathies): These cause defective red cell metabolism involving 2 pathways
- Defects in the hexose monophosphate (HMP) shunt: e.g. glucose-6-phosphate dehydrogenase (G6PD) deficiency.
- Defects in the Embden-Meyerhof (glycolytic) pathway: e.g. pyruvate kinase (PK) deficiency.
2. Disorders of haemoglobin (haemoglobinopathies) These are divided into 2 subgroups:
- Structurally abnormal haemoglobin (Qualitative disorders): e.g. sickle syndromes and other haemoglobinopathies.
- Reduced globin chain synthesis (Quantitative disorders): e.g. various types of thalassaemias.
These disorders are discussed below:

Red Cell Enzyme Defects (Enzymopathies):
G6pd Deficiency:
Among the defects in HMP shunt, the most common is G6PD deficiency. It affects millions of people throughout the World but is more prevalent in Mediterranean countries, the Middle East and India. The G6PD gene is located on the X chromosome and its deficiency is, therefore, a sex (X)-linked trait affecting males, while the females are carriers and are asymptomatic. Several variants of G6PD have been described.
The normal G6PD variant is designated as type B but blacks have normal A+ (positive) type G6PD variant. The most common and significant clinical variant is A–(negative) type found in black males. Like the HbS gene, the A–type G6PD variant confers protection against malaria.
Individuals with A–-type G6PD variant have shortened red cell lifespan but without anaemia. However, these individuals develop haemolytic episodes on exposure to oxidant stress such as viral and bacterial infections, certain drugs (antimalarials, sulfonamides, nitrofurantoin, aspirin, vitamin K), metabolic acidosis and on ingestion of fava beans (favism).

Pathogenesis:
Normally, red blood cells are well protected against oxidant stress because of adequate generation of reduced glutathione via the HMP shunt. Individuals with inherited deficiency of G6PD, an enzyme required for HMP shunt for glucose metabolism, fail to develop adequate levels of reduced glutathione in their red cells.
This results in oxidation and precipitation of haemoglobin within the red cells forming Heinz bodies. Besides G6PD deficiency, deficiency of various other enzymes involved in the HMP shunt may also infrequently cause clinical problems.
Clinical Features:
The clinical manifestations are as under:
1. Acute haemolytic anaemia: This develops in males, being X-linked disorder, within hours of exposure to oxidant stress such as drugs like primaquin, infection, favism and metabolic acidosis. The haemolysis is, however, self-limiting even if the exposure to the oxidant is continued since it affects the older red cells only.
Haemoglobin level may return to normal when the older population of red cells has been destroyed and only younger cells remain. Some patients may have only darkening of the urine from haemoglobinuria but more severely affected ones develop constitutional symptoms including jaundice.
2. Chronic haemolytic anaemia: Cases having severe enzyme deficiency have chronic persistent haemolysis throughout life.
3. Neonatal jaundice: Infants born with G6PD deficiency may continue to have unconjugated hyperbilirubinaemia and may even develop kernicterus.
Laboratory Findings:
These are as under:
1. During the period of acute haemolysis, there is a rapid fall in haematocrit by 25-30%, features of intravascular haemolysis such as rise in plasma haemoglobin, haemoglobinuria, rise in unconjugated bilirubin and fall in plasma haptoglobin.
Formation of Heinz bodies is visualised using supravital stains such as crystal violet, also called Heinz body haemolytic anaemia. However, Heinz bodies are not seen after the first one or two days since they are removed by the spleen, leading to the formation of ‘bite cells’ and fragmented red cells.
2. Between the crises, the affected patient generally has no anaemia. The red cell survival is, however, shortened. The diagnosis of G6PD enzyme deficiency is made by one of the screening tests (e.g. methaemoglobin reduction test or MRT, fluorescent screening test, ascorbate cyanide screening test), or by direct enzyme assay on red cells. Treatment is directed towards the prevention of haemolytic episodes such as the stoppage of the offending drug. Blood transfusions are rarely indicated.
PK Deficiency:
Pyruvate kinase (PK) deficiency is the only significant enzymopathy of the Embden-Meyerhof glycolytic pathway. The disorder is inherited as an autosomal recessive pattern. The Heterozygote state is entirely asymptomatic, while the homozygous individual presents during early childhood with anaemia, jaundice and splenomegaly.
Laboratory Findings:
These are as under:
- Normocytic and normochromic anaemia.
- Reticulocytosis.
- Blood film shows bizarre red cells.
- Osmotic fragility is usually normal but after incubation, it is increased.
- Autohaemolysis is increased, but unlike hereditary spherocytosis, is not corrected by the addition of glucose.
- Direct specific enzyme assay on red cells is the only method of establishing the diagnosis.
Haemoglobinopathies:
Haemoglobin in RBCs may be abnormally synthesised due to inherited defects.
These disorders may be of two types:
- Qualitative disorders in which there is a structural abnormality in the synthesis of haemoglobin e.g. sickle cell syndrome, other haemoglobinopathies.
- Quantitative disorders in which there quantitatively decreased globin chain synthesis of haemoglobin e.g. thalassaemias.
Both these groups of disorders may occur as the homozygous state in which both genes coding for that character are abnormal, or heterozygous when one gene is abnormal and the other gene is normal.
However, there are examples of combined disorders too in which there are two different mutations in loci of two corresponding genes (i.e. double heterozygous) e.g. HbS gene from one parent and the β-thal gene from the other parent resulting in bSbthal. These disorders may vary in presentation from asymptomatic laboratory abnormalities to intrauterine death.
There are geographic variations in the distribution of various haemoglobinopathies the world over. Major examples of these disorders are described below.
1. Structurally Abnormal Haemoglobins (Qualitative Disorders):
Sickle Syndromes:
The most important and widely prevalent type of haemoglobinopathy is due to the presence of sickle haemoglobin (HbS) in the red blood cells. The red cells with HbS develop ‘sickling’ when they are exposed to low oxygen tension. Sickle syndromes have the highest frequency in black race and in Central Africa where falciparum malaria is endemic. Patients with HbS are relatively protected against falciparum malaria.
Sickle syndromes occur in 3 different forms:

- As heterozygous state for HbS: sickle cell trait (AS)
- As homozygous state for HbS: sickle cell anaemia (SS)
- As double heterozygous states e.g. sickle β-thalassaemia, sickle-C disease (SC), sickle-D disease (SD)
Heterozygous State: Sickle Cell Trait:
Sickle cell trait (AS) is a benign heterozygous state of HbS in which only one abnormal gene is inherited. Patients with AS develop no significant clinical problems except when they become severely hypoxic and may develop sickle cell crises.
Laboratory Findings:
These patients have no anaemia and have normal appearance of red cells. But in a hypoxic crisis, sickle cell crises develop.
The diagnosis is made by 2 tests:
Demonstration of sickling done under the condition of reduced oxygen tension by an oxygen-consuming reagent, sodium metabisulfite. Haemoglobin electrophoresis reveals 35-40% of the total haemoglobin as HbS.
Homozygous State: Sickle Cell Anaemia:
Sickle cell anaemia (SS) is a homozygous state of HbS in the red cells in which an abnormal gene is inherited from each parent. SS is a severe disorder associated with protean clinical manifestations and decreased life expectancy.
Pathogenesis:
Following abnormalities are observed:

1. Basic molecular lesion: In HbS, the basic genetic defect is the single point mutation in one amino acid out of 146 in the haemoglobin molecule—there is the substitution of valine for glutamic acid at 6- residue position of the β-globin, producing Hb α2β2s.
2. Mechanism of sickling: During deoxygenation, the red cells containing HbS change from a biconcave disc shape to an elongated crescent-shaped or sickle-shaped cell. This process termed sickling occurs both within the intact red cells and in vitro in free solution.
The mechanism responsible for sickling upon deoxygenation of HbS-containing red cells is the polymerisation of deoxygenated HbS which aggregates to form elongated rod-like polymers. These elongated fibres align and distort the red cell into a classic sickle shape.
3. Reversible-irreversible sickling: The oxygen-dependent sickling process is usually reversible. However, damage to red cell membrane leads to the formation of irreversibly sickled red cells even after they are exposed to normal oxygen tension.
4. Factors determining the rate of sickling: The following factors determine the rate at which the polymerisation of HbS and consequent sickling take place
- Presence of non-HbS haemoglobins: The red cells in patients of SS have a predominance of HbS and a small part consists of non-HbS haemoglobins, chiefly HbF (2-20% of the total haemoglobin). HbF-containing red cells are protected from sickling while HbA-containing red cells participate readily in co-polymerisation with HbS.
- Intracellular concentration of HbS.
- Total haemoglobin concentration.
- Extent of deoxygenation.
- Acidosis and dehydration.
- Increased concentration of 2,3-BPG in the red cells.
Clinical Features:
The clinical manifestations of homozygous sickle cell disease are
widespread. The symptoms begin to appear after 6th month of life when most of the HbF is replaced by HbS. Infection and folic acid deficiency result in more severe clinical manifestations.
These features are as under:
1. Anaemia: There is usually severe chronic haemolytic anaemia (primarily extravascular) with onset of aplastic crisis in between. The symptoms of anaemia are generally mild since HbS gives up oxygen more readily than HbA to the tissues.
2. Vaso-occlusive phenomena: Patients of SS develop recurrent vaso-occlusive episodes throughout their lives due to obstruction to capillary blood flow by sickled red cells upon deoxygenation or dehydration.
Vaso-obstruction affecting different organs and tissues results in infarcts which may be of 2 types:
- Microinfarcts affecting particularly the abdomen, chest, back and joints and are the cause of recurrent painful crises in SS.
- Macroinfarcts involving most commonly the spleen (splenic sequestration, auto splenectomy), bone marrow (pains), bones (aseptic necrosis, osteomyelitis), lungs (pulmonary infections), kidneys (renal cortical necrosis), CNS (stroke), retina (damage) and skin (ulcers), and result in anatomic and functional damage to these organs.
3. Constitutional symptoms In addition to the features of anaemia and infarction, patients with SS have impaired growth and development and increased susceptibility to infection due to markedly impaired splenic function.

Laboratory Findings:
The diagnosis of SS is considered high in blacks with haemolytic anaemia. The laboratory findings in these cases are as under:
- Moderate to severe anaemia (haemoglobin concentration 6-9 g/dl).
- The blood smear shows sickle cells and target cells and features of splenic atrophy such as the presence of Howell-Jolly bodies.
- A positive sickling test with a reducing substance such as sodium metabisulfite.
- Haemoglobin electrophoresis shows no normal HbA but shows a predominance of HbS and 2-20% HbF.
Double Heterozygous States:
Double heterozygous conditions involving a combination of HbS with other haemoglobinopathies may occur. The most common among these are sickle-β-thalassaemia (bSbthal), sickle C disease (SC), and sickle D disease (SD). All these disorders behave like mild forms of sickle cell disease. Their diagnosis is made by haemoglobin electrophoresis and separating the different haemoglobins.
Other Structural Haemoglobinopathies:
Besides sickle haemoglobin, about 400 structurally different abnormal human haemoglobins have been discovered in different parts of the world. Some of them are associated with clinical manifestations, while others are of no consequence.
A few important and common variants are briefly described below:

HbC Haemoglobinopathy:
HbC haemoglobinopathy is prevalent in West Africa and in American blacks. The molecular lesion in HbC is the substitution of lysine for glutamic acid at β-6 globin chain position. The disorder of HbC may occur as benign homozygous HbC disease, or as asymptomatic heterozygous HbC trait, or as double heterozygous combinations such as sickle-HbC disease and HbC-β thalassaemia.
HbD Haemoglobinopathy:
HbD occurs in North-West India, Pakistan and Iran. About 3% of Sikhs living in Punjab are affected with HbD haemoglobinopathy (called HbD Punjab, also known as Hb-Los Angeles). HbD Punjab arises from the substitution of glutamine for glutamic acid at β-121 globin chain position.
HbE Haemoglobinopathy:
HbE is predominantly found in South-East Asia, India, Burma and Sri Lanka. HbE arises from the substitution of lysine for glutamic acid at β-26 globin chain position. Like other abnormal haemoglobins, HbE haemoglobinopathy may also occur as asymptomatic heterozygous HbE trait, compensated haemolytic homozygous HbE disease, or as double heterozygous states in combination with other haemoglobinopathies such as HbE-β thalassaemia and HbE-α thalassaemia.
Haemoglobin O-Arab Disease:
Hb O-Arab disease was first identified in an Arab family but has now been detected in American blacks too. The homozygous form of the disease appears as mild haemolytic anaemia with splenomegaly.
Unstable-Hb Haemoglobinopathy:
The unstable haemoglobins are those haemoglobin variants which undergo denaturation and precipitation within the red cells as Heinz bodies. These give rise to what is known as congenital non-spherocytic haemolytic anaemia or congenital Heinz body haemolytic anaemia.
These disorders have either autosomal dominant inheritance or develop from spontaneous mutations. The unstable haemoglobins arise from either a single amino acid substitution in the globin chain or due to deletion of one or more amino acids within.
β-globin chain so that the firm bonding of the haem group within the molecule is disturbed leading to the formation of methaemoglobin and precipitation of globin chains as Heinz bodies. Over 100 unstable haemoglobins have been described. They are named according to the city where they are encountered.
For instance:
Hb-Koln, Hb-Hammersmith, Hb-Zurich, Hb-Sydney, and so on. The diagnosis of unstable Hb disease is made by test for Heinz bodies and by haemoglobin electrophoresis.
Reduced Globin Chain Synthesis (Quantitative Disorder): Thalassaemias:
Definition:
The thalassaemias are a diverse group of hereditary disorders in which there is reduced synthesis of one or more of the globin polypeptide chains. Thus, thalassaemias, unlike haemoglobinopathies which are qualitative disorders of haemoglobin, are quantitative abnormalities of polypeptide globin chain synthesis.
Thalassaemias were first described in people of Mediterranean countries (North Africa, Southern Europe) from where it derives its name ‘Mediterranean anaemia.’ The Word ‘Thalassa’ in Greek means ‘the sea’ since the condition was found commonly in regions surrounding the Mediterranean Sea.
The condition also occurs in the Middle East, the Indian subcontinent, Southeast Asia and, in general in blacks. In India, thalassaemia is seen throughout the country and an estimated 35 million have β-thalassaemia major. Its incidence is particularly high in some races such as Punjabis who migrated from Pakistan after partition, Bengalis, Gujaratis and Parsis.
Genetics And Classification:
Thalassaemias are genetically transmitted disorders.
- Normally, an individual inherits two β-globin genes located one each on two chromosomes 11, and two α-globin genes one each on two chromosomes 16, from each parent i.e. normal adult haemoglobin (HbA) is α2β2*.
- Depending upon whether the genetic defect or deletion lies in the transmission of α- or β-globin chain genes, thalassaemias are classified into α- and β-thalassaemias. Thus, patients with α- thalassaemia have structurally normal α-globin chains but their production is impaired. Similarly, in β-thalassaemia, β-globin chains are structurally normal but their production is decreased.
- Each of the two main types of thalassaemias may occur as heterozygous (called α- and β- thalassaemia minor or trait), or as homozygous state (termed α- and β-thalassaemia major). The former is generally asymptomatic, while the latter is a severe congenital haemolytic anaemia.
A classification of various types of thalassaemias along with the clinical syndromes produced and salient laboratory findings are given in Table.
Pathophysiology Of Anaemia In Thalassaemia:
A constant feature of all forms of thalassaemia is the presence of anaemia which occurs from following mechanisms:
α-Thalassaemia: In α-thalassaemia major, the obvious cause of anaemia is the inability to synthesise adult haemoglobin, while in α-thalassaemia trait there is reduced production of normal adult haemoglobin.
β-Thalassaemia: In β-thalassaemia major, the most important cause of anaemia is premature red cell destruction brought about by erythrocyte membrane damage caused by the precipitated α-globin chains. Other contributory factors are: shortened red cell lifespan, ineffective erythropoiesis, and haemodilution due to increased plasma volume.
A deficiency of β-globin chains in β-thalassaemia leads to a large excess of α-chains within the developing red cells. Part of these excessive α-chains are removed by pairing with γ-globin chains as HbF, while the remainder unaccompanied α-chains precipitate rapidly within the red cell as Heinz bodies. The precipitated α-chains cause red cell membrane damage. During their passage through the splenic sinusoids, these red cells are further damaged and develop pitting due to the removal of the precipitated aggregates.
Thus, such red cells are irreparably damaged and are phagocytosed by the RE cells of the spleen and the liver causing anaemia, hepatosplenomegaly, and excess of tissue iron stores. Patients with β-thalassaemia minor, on the other hand, have very mild ineffective erythropoiesis, haemolysis and shortening of red cell lifespan.
α-Thalassaemia:
Molecular Pathogenesis:
α-thalassaemias are disorders in which there is defective synthesis of α-globin chains resulting in depressed production of haemoglobins that contain α- chains i.e. HbA, HbA2 and HbF. The α-thalassaemias are most commonly due to the deletion of one or more of the α-chain genes located on short arm of chromosome 16. Since there is a pair of α-chain genes, the clinical manifestations of α-thalassaemia depend upon the number of genes deleted.
Accordingly, α-thalassaemias are classified into 4 types:


- Four α-gene deletions: Hb Bart’s hydrops foetal
- Three α-gene deletions: HbH disease
- Two α-gene deletions: α-thalassaemia trait
- One α-gene deletion: α-thalassaemia trait (carrier)
Hb Bart’s Hydrops Foetalis:
When there is deletion of all four α-chain genes (homozygous state) it results in total suppression of α-globin chain synthesis causing the most severe form of α-thalassaemia called Hb Bart’s hydrops foetalis. Hb Bart’s is a gamma globin chain tetramer (γ4) which has high oxygen affinity leading to severe tissue hypoxia.
Clinical Features:
Hb Bart’s hydrops foetal is incompatible with life due to severe tissue hypoxia. The condition is either fatal in utero or the infant dies shortly after birth. If born alive, features similar to severe Rh haemolytic disease are present (page 357).
Laboratory Findings:
Infants with Hb Bart’s hydrops foetalis born alive may have the
following laboratory findings:
- Severe anaemia (haemoglobin below 6 g/dl).
- Blood smear show marked anisopoikilocytosis, hypochromia, microcytosis, polychromasia, basophilic stippling, numerous erythroblasts and target cells.
- Reticulocyte count is high.
- Serum bilirubin level is elevated.
- Haemoglobin electrophoresis shows 80-90% Hb-Bart’s and a small amount of Hb-H and Hb-Portland but no HbA, HbA2 or HbF.
HbH Disease:
Deletion of three α-chain genes produces HbH which is a β-globin chain tetramer (β4) and markedly impaired α-chain synthesis. HbH is precipitated as Heinz bodies within the affected red cells. An elongated α-chain variant of HbH disease is termed Hb Constant Spring.
Clinical Features:
HbH disease is generally present as a well-compensated haemolytic anaemia. The features are intermediate between that of β-thalassaemia minor and major. The severity of anaemia fluctuates and may fall to very low levels during pregnancy or infections. Majority of patients have splenomegaly and may develop cholelithiasis.
Laboratory Findings:
These are as follows:
- Moderate anaemia (haemoglobin 8-9 g/dl).
- Blood smear shows severe microcytosis, hypochromia, basophilic stippling, target cells and erythroblasts.
- Mild reticulocytosis.
- HbH inclusions as Heinz bodies can be demonstrated in mature red cells with brilliant cresyl blue stain.
Haemoglobin electrophoresis shows 2-4% HbH and the remainder consists of HbA, HbA2 and HbF.
α-Thalassaemia Trait:
α-thalassaemia trait may occur by the following molecular pathogenesis:
By deletion of two of the four α-chain genes in a homozygous form called homozygous α-thalassaemia, or in double heterozygous form termed heterozygous α-thalassaemia. By deletion of a single α-chain gene causes a heterozygous α-thalassaemia trait called heterozygous α-thalassaemia.
Clinical Features:
α-thalassaemia trait due to two α-chain gene deletion is asymptomatic. It is suspected in a patient of refractory microcytic hypochromic anaemia in whom iron deficiency and β-thalassaemia minor have been excluded and the patient belongs to the high-risk ethnic group. One gene deletion α-thalassaemia trait is a silent carrier state.
Laboratory Findings:
The patients of α-thalassaemia trait may have the following haematological findings:
- Haemoglobin level normal or mildly reduced.
- The blood smear shows microcytic and hypochromic red cell morphology but no evidence of haemolysis or anaemia.
- MCV, MCH and MCHC may be slightly reduced.
- Haemoglobin electrophoresis reveals a small amount of Hb-Bart’s in the neonatal period (1-2% in α-thalassaemia 2 and 5-6% in α-thalassaemia 1) which gradually disappears by adult life. HbA2 is either normal or slightly decreased (contrary to the elevated HBA2 levels in β-thalassaemia trait).
β-Thalassaemias:
Molecular Pathogenesis:
β-thalassaemias are caused by a decreased rate of β-chain synthesis resulting in reduced formation of HbA in the red cells. The molecular pathogenesis of the β-thalassaemias is more complex than that of α-thalassaemias.
In contrast to α-thalassaemia, gene deletion rarely ever causes β-thalassaemia and is only seen in an entity called hereditary persistence of foetal haemoglobin (HPFH). Instead, most β-thalassaemias arise from different types of mutations of the β-globin gene resulting from single base changes.
The symbol β° is used to indicate the complete absence of β-globin chain synthesis while β+ denotes partial synthesis of the β-globin chains. More than 100 such mutations have been described affecting the preferred sites in the coding sequences e.g. in the promoter region, termination region, splice junctions, exons, and introns. Some of the important ones having effects on β-globin chain synthesis are as under:
Transcription defect: Mutation affecting transcriptional promoter sequence causing reduced synthesis of β-globin chain. Hence the result is partially preserved synthesis i.e. β+ thalassaemia.
Translation defect: Mutation in the coding sequence causing stop codon (chain termination) interrupting β-globin messenger RNA. This would result in no synthesis of β-globin chain i.e. β° thalassaemia.
mRNA splicing defect: Mutation leads to defective mRNA processing forming abnormal mRNA that is degraded in the nucleus. Depending upon whether part of splice site remains intact or is totally degraded, it may result in β+ thalassaemia or β° thalassaemia.
Depending upon the extent of reduction in β-chain synthesis, there are 3 types of β-thalassaemia:
Homozygous form: β-Thalassaemia major It is the most severe form of congenital haemolytic anaemia.
It is further of 2 types:
β° thalassaemia major is characterised by the complete absence of β-chain synthesis.
β+ thalassaemia major having incomplete suppression of β-chain synthesis.
β-Thalassaemia intermedia It is β-thalassaemia of intermediate degree of severity that does not require regular blood transfusions. These cases are genetically heterozygous (β°/β or β+/β).
Heterozygous form: β-thalassaemia minor (trait) It is a mild asymptomatic condition in which there is moderate suppression of β-chain synthesis.
Besides β-thalassaemia minor, a few uncommon globin chain combinations resulting in the β-thalassaemia trait are as under:

- δβ-thalassaemia minor in which there is the total absence of both β and δ chain synthesis and is characterised by elevated HbF level but unlike β-thalassaemia minor, there is normal or reduced HbA2 level.
- Hb Lepore syndrome is characterised by a non-homologous fusion of β- and δ-genes forming an abnormal haemoglobin called Hb Lepore (named after a family called Lepore). There is the total absence of normal β-chain synthesis.
An individual may inherit one β-chain gene from each parent and produce heterozygous, homozygous, or double heterozygous states. Statistically, 25% of offspring born to two heterozygotes (i.e. β-thalassaemia trait) will have the homozygous state i.e. β-thalassaemia major.
β-Thalassaemia Major:
β-thalassaemia major, also termed Mediterranean or Cooley’s anaemia is the most common form of congenital haemolytic anaemia. β-thalassaemia major is a homozygous state with either a complete absence of β-chain synthesis (β° thalassaemia major) or only small amounts of β-chains are formed (β+ thalassaemia major). These result in excessive formation of alternate haemoglobins, HbF (α2γ2) and HbA2 (α2δ2).
Clinical Features:
Clinical manifestations appear insidiously and are as under:
- Anaemia starts appearing within the first 4-6 months of life when the switch over from γ-chain to β-chain production occurs.
- Marked hepatosplenomegaly occurs due to excessive red cell destruction, extramedullary haematopoiesis and iron overload.
- Expansion of bones occurs due to marked erythroid hyperplasia leading to thalassaemic facies and malocclusion of the jaw.


Iron overload due to repeated blood transfusions causes damage to the endocrine organs resulting in a slow rate of growth and development, delayed puberty, diabetes mellitus and damage to the liver and heart.
Laboratory Findings:
The haematological investigations reveal the following findings:
- Anaemia, is usually severe.
- The blood smear shows severe microcytic hypochromic red cell morphology, marked anisopoikilocytosis, basophilic stippling, presence of many target cells, tear drop cells and normoblasts.
- Serum bilirubin (unconjugated) is generally raised.
- Reticulocytosis is generally present.
- MCV, MCH and MCHC are significantly reduced.
- WBC count is often raised with some shift to left of the neutrophil series, with presence of some myelocytes and metamyelocytes.
- Platelet count is usually normal but may be reduced in patients with massive splenomegaly.
- Osmotic fragility characteristically reveals increased resistance to saline haemolysis i.e. decreased osmotic fragility.
- Haemoglobin electrophoresis shows presence of increased amounts of HbF, increased amount of HbA2, and almost complete absence or presence of variable amounts of HbA. The increased level of HbA2 has not been found in any other haemoglobin abnormality except β-thalassaemia. The increased synthesis of HbA2 is probably due to increased activity at both δ-chain loci.
- Bone marrow aspirate examination shows normoblastic erythroid hyperplasia with predominance of intermediate and late normoblasts which are generally smaller in size than normal. Iron staining demonstrates siderotic granules in the cytoplasm of normoblasts, increased reticuloendothelial iron but ring sideroblasts are only occasionally seen.
Principles Of Treatment:
Treatment of β-thalassaemia major is largely supportive.
- Anaemia is generally severe and patients require regular blood transfusions (4-6 weekly) to maintain haemoglobin above 8 g/dl.
- In order to maintain increased demand of hyperplastic marrow, folic acid supplement is given.
- Splenectomy is beneficial in children over 6 years of age since splenic sequestration contributes to shortened red cell lifespan.
- Prevention and treatment of iron overload is done by chelation therapy (desferrioxamine). Oral chelation with kelfer or deferiprone is also available now.
- Bone marrow transplantation from HLA-matched donor that provides stem cells which can form normal haemoglobin is being done in many centres with fair success rate, especially when done at an early stage before end-organ damage has supervened.
- Some workers have found success with cord blood transfusion.
- Gene therapy of thalassaemia involving genetic manipulation in haematopoietic stem cells may become an option for future.
Since these patients require multiple blood transfusions, they are at increased risk of developing AIDS. In general, patients with β-thalassaemia major have short life expectancy. The biggest problem is iron overload and consequent myocardial siderosis leading to cardiac arrhythmias, congestive heart failure, and ultimately death.

β-Thalassaemia Minor:
The β-thalassaemia minor or β-thalassaemia trait, a heterozygous state, is a common entity characterised by moderate reduction in β-chain synthesis.
Clinical Features:
Clinically, the condition is usually asymptomatic and the diagnosis is generally made when the patient is being investigated for mild chronic anaemia. The spleen may be palpable.
Laboratory Findings:
These are as under:
- Mild anaemia; mean haemoglobin level is about 15% lower than in normal person for the age and sex.
- Blood smear shows mild anisopoikilocytosis, microcytosis and hypochromia, occasional target cells and basophilic stippling.
- Serum bilirubin may be normal or slightly raised.
- Mild reticulocytosis is often present.
- MCV, MCH and MCHC may be slightly reduced.
- Osmotic fragility test shows increased resistance to haemolysis i.e. decreased osmotic fragility.
- Haemoglobin electrophoresis is confirmatory for the diagnosis and shows about two-fold increase in HbA2 and a slight elevation in HbF (2-3%).
Principles Of Treatment:
Patients with β-thalassaemia minor do not require any treatment. But they should be explained about the genetic implications of the disorder, particularly to those of child-bearing age. If the two subjects of β-thalassaemia trait marry, there is a 25% chance of developing thalassaemia major in offsprings. Patients with β-thalassaemia minor have normal life expectancy but those who are treated under the mistaken diagnosis of iron deficiency may develop siderosis and its complications.
Prevention Of Thalassaemia:
Finally, since thalassaemia is an inheritable disease, its prevention is possible by making an antenatal diagnosis. This is done by chorionic villous biopsy or cells obtained by amniocentesis and foetal DNA studied by PCR amplification technique for presence of genetic mutations of thalassaemias.
Anaemia Due To Blood Loss:
Depending upon the rate of blood loss due to haemorrhage, variable effects of post-haemorrhagic anaemia appear.
Acute Blood Loss:
When the loss of blood occurs suddenly, the following events take place:
Immediate threat to life due to hypovolaemia which may result in shock and death. If the patient survives, shifting of interstitial fluid to intravascular compartment with consequent haemodilution with low haematocrit. Hypoxia stimulates production of erythropoietin resulting in increased marrow erythropoiesis.
Laboratory Findings:
- Normocytic and normochromic anaemia
- Low haematocrit
- Increased reticulocyte count in peripheral blood (10-15% after one week) reflecting accelerated marrow erythropoiesis.
Chronic Blood Loss:
When the loss of blood is slow and insidious, the effects of anaemia will become apparent only when the rate of loss is more than rate of production and the iron stores are also depleted. This results in iron deficiency anaemia.
Hereditary Haemolytic Anaemias and Anaemia due to Blood Loss:
- Hereditary HA are due to abnormalities in the red cell membrane, or disorders due to red cell interior (enzymopathies and haemoglobin disorders). Defects in the red cell membrane are: spherocytosis (spectrin deficiency or ankyrin defect) most common, elliptocytosis (spectrin or protein 4.1 defect) and stomatocytosis.
- Inherited defects in red cell enzymes are deficiency of G6PD (due to defect in HMP shunt) and pyruvate kinase (due to defect in Embden-Meyerhof glycolytic pathway).
- Haemoglobinopathies may be qualitative (structural e.g. sickle cell syndrome and other haemoglobin disorders), or quantitative (reduced globin synthesis e.g. thalassaemia) disorders of haemoglobin.
- Sickle cell syndrome is widely prevalent and is due to genetic defect of a single point mutation in an amino acid. Red cells having sickle haemoglobin undergo morphologic change to sickle cells when they are exposed to low oxygen tension. Patient’s symptoms are due to vasoocclusion by sickle cells.
- Thalassaemias are hereditary defects of quantitatively reduced synthesis of either α-(α-thalassaemia) or β-(β-thalassaemia) globin chains which are otherwise structurally normal.
- β-thalassaemia is more common and may occur as a trait (heterozygous) or major (homozygous) form of disease.
- β-thalassaemia major has high foetal haemoglobin, anaemia, hepatosplenomegaly and iron overload.
- β-thalassaemia trait cases have mild persistent anaemia and high HbA2.
Anaemia from blood loss may occur by acute and sudden loss, or by slow and insidious loss. Anaemia from acute blood loss may pose immediate threat to life while anaemia due to chronic loss produces iron deficiency over a period of time.
Aplastic Anaemia And Other Primary Bone Marrow Disorders
‘Bone marrow failure’ is the term used for primary disorders of the bone marrow which result in impaired formation of the erythropoietic precursors and consequent anaemia.
It includes the following disorders:
- Aplastic anaemia, most importantly.
- Other primary bone marrow disorders
- Examples: Myelophthisic anaemia, pure red cell aplasia, and myelodysplastic syndromes.
These disorders are examples of hypoproliferative anaemias in which anaemia is not the only finding but the major problem is pancytopenia i.e. anaemia, leucopenia and thrombocytopenia. While myelodysplastic syndromes other conditions are considered below.
Aplastic Anaemia:
Aplastic anaemia is defined as pancytopenia (i.e. simultaneous presence of anaemia, leucopenia and thrombocytopenia) resulting from aplasia of the bone marrow. The underlying defect in all cases appears to be sufficient reduction in the number of haematopoietic pluripotent stem cells which results in decreased or total absence of these cells for division and differentiation.
Etiology And Classification:
More than half the cases of aplastic anaemia are idiopathic.
Cases with known etiology are classified into 2 main types:
Primary and secondary:
1. Primary Aplastic Anaemia:
Primary aplastic anaemia includes 2 entities: a congenital form called Fanconi’s anaemia and an immunologically-mediated acquired form.
- Fanconi’s anaemia This has an autosomal recessive inheritance and is often associated with other congenital anomalies such as skeletal and renal abnormalities, and sometimes mentalm retardation.
- Immune causes In many cases, suppression of haematopoietic stem cells by immunologic mechanisms may cause aplastic anaemia. The observations in support of autoimmune mechanisms are the clinical response to immunosuppressive therapy and in vitro marrow culture experiments.
2. Secondary Aplastic Anaemia:
Secondary causes are more common than primary and include a variety of industrial, physical, chemical, iatrogenic and infectious causes:
1. Drugs: A number of drugs are cytotoxic to the marrow and cause aplastic anaemia. The association of a drug with aplastic anaemia may be either predictably dose-related or an idiosyncratic reaction.
- Primary Aplastic Anaemia
- Fanconi’s anaemia (congenital)
- Immune-mediated (acquired)
- Example: Hyperimmunoglobulinaemia, thymoma, GVHD in immunodeficiency
- Secondary Aplastic anaemia
- Drugs
- Dose-related aplasia e.g. with antimetabolites (methotrexate), mitotic inhibitors (daunorubicin), alkylating agents (busulfan), nitroso urea, anthracyclines.
- Idiosyncratic aplasia e.g. with chloramphenicol, sulfa drugs, oxyphenbutazone, phenylbutazone, chlorpromazine, gold salts.
- Toxic chemicals, For Example: Benzene derivatives, insecticides, and arsenicals.
- Radiation
- Viral infections For Example: Hepatitis, EB virus infection (infectious mononucleosis), HIV (AIDS), Parvovirus-B19 (PRCA)
- PNH
- Idiopathic
- Drugs
- Dose-related aplasia of the bone marrow occurs with antimetabolites (Example: Methotrexate), mitotic inhibitors (Example: Daunorubicin), alkylating agents (e.g. busulfan), nitroso urea and anthracyclines. In such cases, withdrawal of the drug usually allows recovery of the marrow elements.
- Idiosyncratic aplasia is depression of the bone marrow due to qualitatively abnormal reaction of an individual to a drug when first administered. The most serious and most common example of idiosyncratic aplasia is associated with chloramphenicol. Other such common drugs are sulfa drugs, oxyphenbutazone, phenylbutazone, chlorpromazine, gold salts etc.
2. Toxic chemicals These include examples of industrial, domestic and accidental use of substances such as benzene derivatives, insecticides, arsenicals etc.
3. Infections Aplastic anaemia may occur following viral hepatitis, Epstein-Barr virus infection, AIDS and other viral illnesses.
4. Miscellaneous Lastly, aplastic anaemia has been reported in association with certain other illnesses such as SLE, and with therapeutic X-rays.
Clinical Features:
The onset of aplastic anaemia may occur at any age and is usually insidious.
The clinical manifestations include the following:
- Anaemia and its symptoms like mild progressive weakness and fatigue.
- Haemorrhage from various sites due to thrombocytopenia such as from the skin, nose, gums, vagina, bowel, and occasionally in the CNS and retina.
- Infections of the mouth and throat are commonly present.
- The lymph nodes, liver and spleen are generally not enlarged.
Laboratory Findings:
The diagnosis of aplastic anaemia is made by a thorough laboratory evaluation and excluding other causes of pancytopenia.
The following haematological features are found:
- B Haemoglobin levels are moderately reduced. The blood smear generally shows normocytic normochromic anaemia but sometimes macrocytosis may be present. The reticulocyte count is reduced or zero.
- Leucopenia: The absolute granulocyte count is particularly low (below 1500/µl) with relative lymphocytosis. The neutrophils are morphologically normal but their alkaline phosphatase score is high.
- Thrombocytopenia: Platelet count is always reduced.
- Bone marrow examination: A bone marrow aspirate may yield a ‘dry tap’. A trephine biopsy is generally essential for making the diagnosis which reveals patchy cellular areas in a hypocellular or aplastic marrow due to replacement by fat. There is usually a severe depression of myeloid cells, megakaryocytes and erythroid cells so that the marrow chiefly consists of lymphocytes and plasma cells. Haematopoietic stem cells bearing CD34 marker are markedly reduced or absent.
Principles Of Treatment:
The patients of mild aplasia may show spontaneous recovery, while the management of severe aplastic anaemia is a most challenging task. In general, younger patients show better response to proper treatment.
The broad outlines of the treatment are as under:
- Aplastic anaemia
- Pancytopenia with normal or increased marrow cellularity
- Example:
- Myelodysplastic syndromes
- Hypersplenism
- Megaloblastic anaemia
- Paroxysmal nocturnal haemoglobinuria
- Bone marrow infiltrations
- Example:
- Haematologic malignancies (leukaemias, lymphomas, myeloma)
- Non-haematologic metastatic malignancies
- Storage diseases
- Osteopetrosis
- Myelofibrosis
1. General management It consists of the following:
- Identification and elimination of the possible cause.
- Supportive care consisting of blood transfusions, platelet concentrates, and treatment and prevention of infections.
2. Specific treatment The specific treatment has been attempted with varying success and includes the following:
- Marrow stimulating agents such as androgen may be administered orally.
- Immunosuppressive therapy with agents such as anti-thymocyte globulin and antilymphocyte serum has been tried with 40-50% success rate. Very high doses of glucocorticoids or cyclosporine may yield similar responses. But splenectomy does not have any role in the management of aplastic anaemia.
- Bone marrow transplantation is considered in severe cases under the age of 40 years where the HLA and mixed culture-matched donor is available (Other indications for bone marrow transplantation are: acute leukaemias—AML in first remission and ALL in second remission, thalassaemia major and combined immunodeficiency disease). Complications of bone marrow transplantation such as severe infections, graft rejection and graft-versus-host disease (GVHD) may occur.

Myelophthisic Anaemia:
Development of severe anaemia may result from infiltration of the marrow termed as myelophthisic anaemia.
The causes for marrow infiltrations include the following:
- Haematologic malignancies (Example: leukaemia, lymphoma, myeloma).
- Metastatic deposits from non-haematologic malignancies (e.g. cancer breast, stomach, prostate, lung, thyroid).
- Advanced tuberculosis.
- Primary lipid storage diseases (Gaucher’s and Niemann-Pick disease).
- Osteopetrosis and myelofibrosis may rarely cause myelophthisis.
Laboratory Findings:
The type of anaemia in myelophthisis is generally normocytic normochromic with some fragmented red cells, basophilic stippling and normoblasts in the peripheral blood. Thrombocytopenia is usually present but the leucocyte count is increased with slight shift-toleft of myeloid cells i.e. a picture of leucoerythroblastic reaction consisting of immature myeloid cells and normoblasts is seen in the peripheral blood. Treatment consists of reversing the underlying pathologic process.
Pure Red Cell Aplasia:
Pure red cell aplasia (PRCA) is a rare syndrome involving a selective failure in the production of erythroid elements in the bone marrow but with normal granulopoiesis and megakaryocytopoiesis.
PRCA exists in the following forms:

- Transient self-limited PRCA It is due to temporary marrow failure in aplastic crisis in haemolytic anaemias and in acute B19 parvovirus infection and in transient erythroblastopenia in normal children.
- Acquired PRCA It is seen in middle-aged adults in association with some other diseases, most commonly thymoma; others are connective tissue diseases (SLE, rheumatoid arthritis), lymphoid malignancies (lymphoma, T-cell chronic lymphocytic leukaemia) and solid tumours.
- Chronic B19 parvovirus infections PRCA may occur from chronic B19 parvovirus infection in children and is common and treatable.
- Congenital PRCA (Blackfan-Diamond syndrome) It is a rare chronic disorder detected at birth or in early childhood. It occurs due to mutation in a ribosomal RNA processing gene termed as RPS19. The disorder is corrected by glucocorticoids and marrow transplantation.
Laboratory Findings PRCA is characterised by the following laboratory findings:
- CBC findings Normocytic normochromic anaemia but with normal granulocyte and platelet count.
- Reticulocytopenia
- Bone marrow Eryhroid hypoplasia with absent or rare red cell precursors, giant pronormoblasts having the cytopathic effects of B19 parvovirus infection.
Aplastic Anaemia and Other Primary Bone Marrow Disorders:
- Aplastic anaemia is pancytopenia due to aplasia of bone marrow. It may be primary (inherited) or secondary (acquired). Secondary causes are more common and may be due to drugs, toxic chemicals or viral infections.
- Other primary bone marrow failure disorders are myelophthisic anaemia (from marrow infiltration), pure red cell aplasia (selective failure of marrow erythropoiesis) and myelodysplastic syndromes (discussed later)
Clinical Case 4:
A 60-year old female from a well-to-do family presents to the hospital with progressive dyspnoea, and malaise for past few months. Examination reveals tachycardia and pallor. She denies any obvious unusual bleeding from anywhere. Past medical history is insignificant. Blood examination reveals haemoglobin level is 7 g/dL, and the peripheral blood smear shows pale microcytes.
The next best step in the initial evaluation of this patient should be focused on identifying which of the following?
- Malnutrition
- Haemolysis
- Liver pathology
- Blood loss
Clinical Case 5:
A 3-year old female child is brought by the mother to the hospital with persisting fever, chills and failure to thrive. The facial appearance of the child shows prominence of forehead and cheeks and slightly protruding upper teeth.
On examination, the child has pallor ++, stunted growth, and enlargement of the spleen and liver by two fingers below the costal margin. The child dies in the hospital from overwhelming infection. At autopsy, clumps of erythroid precursor cells were found in the liver and the spleen.
The presence of these precursor cells is most likely related to which of the following conditions?
- Erythropoietin deficiency
- Chronic haemolysis
- Iron deficiency
- Portal hypertension
Leave a Reply