Red Blood Cells and Bleeding Disorders
Question 1. Define anemia.
Answer:
Definition: Reduction of total circulating red cell mass below normal limits
Diagnosis is made by:
- Reduction in hematocrit or
- Reduction in hemoglobin concentration of blood to levels that are below the normal range
Note:
Normal hemoglobin (gm/dl) levels in adults:
13.6–17.2 in males
12.0–15.0 in females
Read and Learn More Preparatory Manual of Pathology Question and Answers
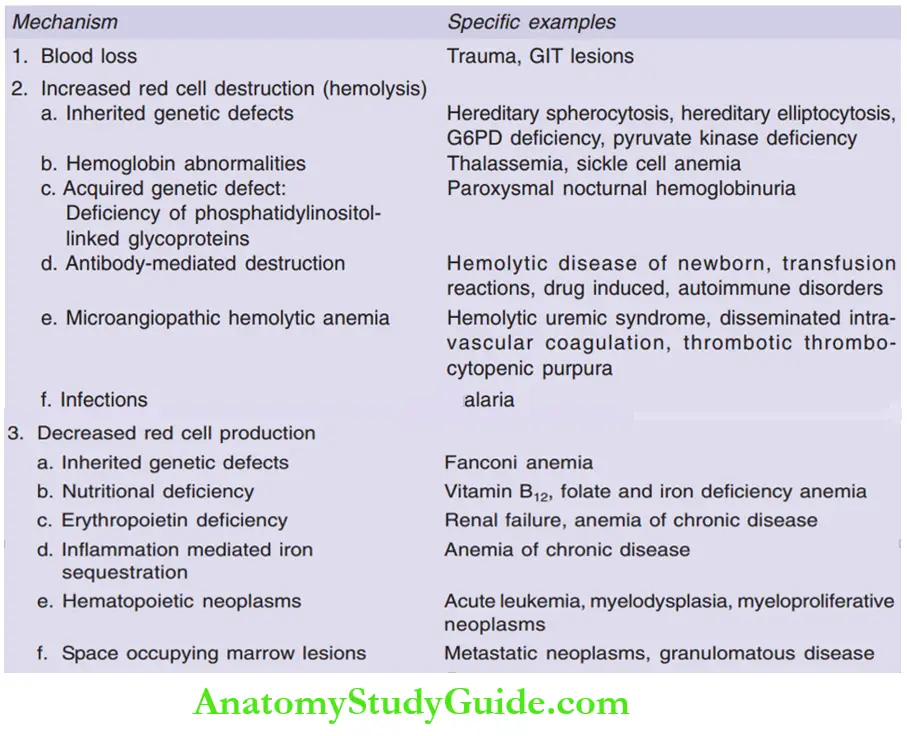
Question 2. Write the classification of anemia.
Answer:
Question 3. Write a note on red cell indices.
Answer:
Red cell indices
Mean cell volume (MCV): Average volume of a red cell expressed in femtoliters (fl)
Mean cell hemoglobin (MCH): Average content (mass) of hemoglobin per red cell, expressed in picograms
Mean cell hemoglobin concentration (MCHC): Average concentration of hemoglobin in a given volume of packed red cells, expressed in grams per deciliter
Red cell distribution width (RDW): Coefficient of variation of red cell volume
Adult reference ranges

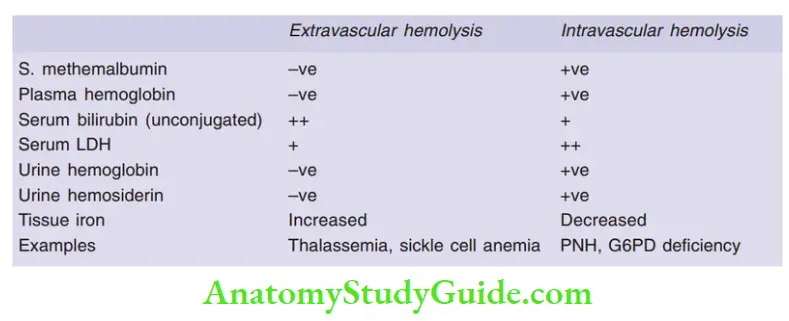
Question 4. Enumerate the differences between extravascular hemolysis and intravascular hemolysis.
Answer:
Question 5. Discuss the pathogenesis and morphology of hereditary spherocytosis.
Answer:
Hereditary spherocytosis (HS)
- Autosomal dominant disorder
- Characterized by inherited defects in the red cell membrane
- Red cells become spherical, less deformable, and vulnerable to splenic sequestration and destruction
Pathogenesis
- RBC membrane components include α-spectrin, β-spectrin, ankyrin, band 4.2 or band 3
- Mutations most commonly affect ankyrin, band 3, spectrin, or band 4.2
- Mutations result in the loss of red cell membrane fragments
- Due to the loss of membrane, the red cell assumes the smallest possible diameter for a given volume, a sphere
Consequences of the mutation
- Spherocytes are less deformable than normal RBCs and are trapped in the splenic cords, where they are phagocytosed by macrophages
- Young HS red cells are normal in shape, but as they age, they shed the membrane fragments
- The life span of the affected red cells is decreased to 10 to 20 days from the normal 120 days
Morphology
- Spherocytes: Smears show small, hyperchromic RBCs, that lack the central zone of pallor
- Increased reticulocyte count, marrow erythroid hyperplasia, hemosiderosis, and mild jaundice
- Cholelithiasis (pigment stones)
- Moderate splenomegaly (congestion of splenic cords and increased phagocytes)
![]()
Question 6. Write a note on the pathogenesis of sickle cell anemia and the factors affecting the rate of sickling.
Answer:
Pathogenesis
- Oxygenated hemoglobin is a free-flowing liquid
- Deoxygenated hemoglobin (HbS) gets converted from a free-flowing liquid into a viscous gel followed by long needle-like fibers, producing a sickle or holly-leaf shape
Factors affecting the rate of sickling
1. Interaction of HbS with the other types of hemoglobin in the cell:
- HbA: More HbA levels, less sickling (seen in sickle cell trait)
- HbF: Inhibits the polymerization of HbS, hence infants do not become symptomatic till 6 months of age
- HbSC disease: Individuals with both HbS and HbC have symptomatic sickling disorder (termed HbSC disease)
2. Mean cell hemoglobin concentration (MCHC):
- Conditions that increase MCHC increase sickling, for example, dehydration
- Conditions that reduce MCHC reduce sickling
3. Intracellular pH:
- Decrease in pH, reduces the oxygen affinity of hemoglobin (as a result fraction of deoxygenated HbS increases), thereby increasing the sickling
4. Transit time of red cells through microvascular beds:
- Sickling is confined to the vessel walls where the blood flow is slow (slow transit time)
- Blood flow is sluggish in normal spleen, bone marrow, and inflamed vascular beds which are prominently affected in sickle cell disease
Question 7. Discuss the morphology, lab investigations, clinical features, and treatment of sickle cell anemia.
Answer:
Morphology
Peripheral smear
- Increased numbers of irreversibly sickled cells, target cells, and Howell-Jolly bodies (small nuclear remnants) are present
- Increased reticulocyte count
![]()
Bone marrow
- Hypercellular with compensatory erythroid hyperplasia
- Expansion of the marrow leads to bone resorption and secondary new bone formation, resulting in prominent cheekbones and a “crew-cut” appearance of the skull on X-ray
Spleen
- Enlarged due to red pulp congestion, caused by trapping of sickled red cells in cords and sinuses
- Due to chronic erythrocytosis, the spleen becomes infarcted, fibrosis, and shrunken, over time, resulting in auto splenectomy
Infarction
- Can be seen in bones, brain, kidney, liver, retina, and pulmonary vessels
- Vascular stagnation in subcutaneous tissues leads to leg ulcers
Clinical features
1. Vaso-occlusive crises (pain crises):
- Painful bone crises: Dactylitis of the bones of hands or feet
- Acute chest syndrome: Vaso-occlusive crisis involving the lungs, presents as fever, cough, chest pain, and pulmonary infiltrates
- Priapism may lead to erectile dysfunction
- Stroke and retinopathy
2. Sequestration crises:
- Massive entrapment of sickle red cells leads to rapid splenic enlargement, hypovolemia, and shock
- Pneumococcus pneumonia and Haemophilus influenza septicemia are common due to poor splenic function
3. Aplastic crises: Occur due to infection of red cell progenitors by parvovirus B19
Lab diagnosis
- Sickling test: Sickling can be induced by mixing a blood sample (with HbS) with an oxygen-consuming reagent, such as sodium metabisulfite
- Hemoglobin electrophoresis: To demonstrate the presence of HbS and exclude other sickle syndromes, such as HbSC disease
Treatment
- Hydroxyurea
-
- Increases red cell HbF levels
- An anti-inflammatory effect (inhibition of leukocyte production)
- Hematopoietic stem cell transplantation offers a chance for a cure
Question 8. Discuss in detail the pathogenesis, morphology, and clinical course of beta thalassemia major.
Answer:
β-Thalassemia
- The β-globin gene is located on chromosome 11
- Caused by mutations that diminish the synthesis of β-globin chains
Molecular pathogenesis
- β0 mutations (absent β-globin synthesis)
- β+ mutation (reduced β-globin synthesis)
Mutation includes
- Splicing mutations: A most common cause of β+ thalassemia, resulting in absent/ reduced production of β-globin mRNA
- Promoter region mutations: Mutations reduce the transcription of proteins, leading to β+-thalassemia
- Chain terminator mutations: A most common cause of 0-thalassemia, due to frameshift mutations or the introduction of stop codons
Morphology
1. Peripheral smear
- Marked variation in size (anisocytosis) and shape (poikilocytosis) of RBCs, microcytosis, and hypochromic
- Target cells (hemoglobin collects in the center of the cell), basophilic stippling, and fragmented red cells can be seen
- Reticulocyte count is elevated
- Nucleated red cell precursors (normoblasts) are seen in the peripheral blood
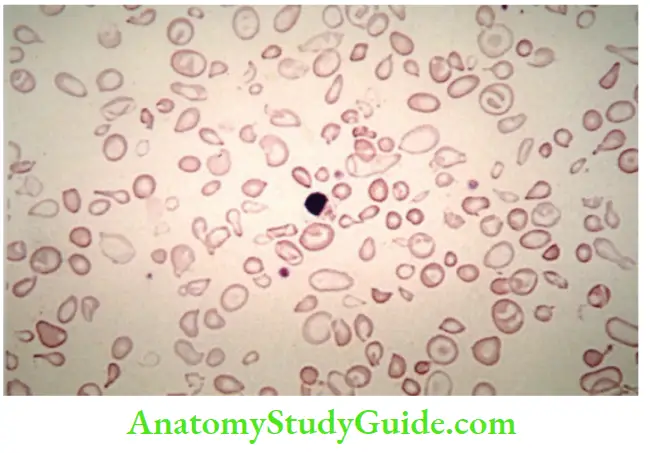
2. Bone marrow and spleen
- Expansion of the hematopoietically active marrow
- Bones of the face and skull erode the existing cortical bone with resultant new bone formation, giving rise to a “crew cut” appearance on X-ray studies
- Spleen weighs as much as 1500 gm
3. Hemosiderosis or secondary hemochromatosis is seen in every patient
Clinical course
- Children, who are not treated with blood transfusion, die at an early age
- Hepatosplenomegaly is seen, due to extramedullary hematopoiesis
- Cardiac disease (an important cause of death) due to progressive iron overload and secondary hemochromatosis
- Patients receiving multiple blood transfusions must be treated with iron chelators to prevent hemochromatosis
- Hematopoietic stem cell transplantation can offer a cure
Question 9. Write a brief note on Bart’s hemoglobin.
Answer:
Bart’s Hemoglobin
- Results in hydrops fetalis
- The most severe form of α-thalassemia, which is caused by the deletion of all four α-globin genes
- In the fetus, excess γ-globin chains form tetramers (Bart’s Hemoglobin)
- Bart’s Hemoglobin has a high affinity for oxygen and hence oxygen delivery to the tissues is markedly decreased
- The fetus shows severe pallor, generalized edema, and massive hepatosplenomegaly, similar to that seen in hemolytic disease of the newborn
- Signs of fetal distress become evident by the third trimester of pregnancy
Question 10. Write a note on paroxysmal nocturnal hemoglobinuria.
Answer:
Paroxysmal nocturnal hemoglobinuria (PNH)
- Hemolytic anemia caused by an acquired genetic defect
- Results due to acquired mutations in the phosphatidylinositol glycan complementation group A gene (PIGA)
- PIGA mutation leads to a deficiency of GPI-linked proteins
- Increased susceptibility of all three cell lines (RBC, WBC, platelets) to complement-mediated lysis
PNH cells are deficient in three GPI-linked proteins
- Decay accelerating factor, or CD55
- Membrane inhibitor of reactive lysis, or CD59
- C8 binding protein
CD59 is a potent inhibitor of C3 convertase that prevents the spontaneous activation of the alternative complement pathway
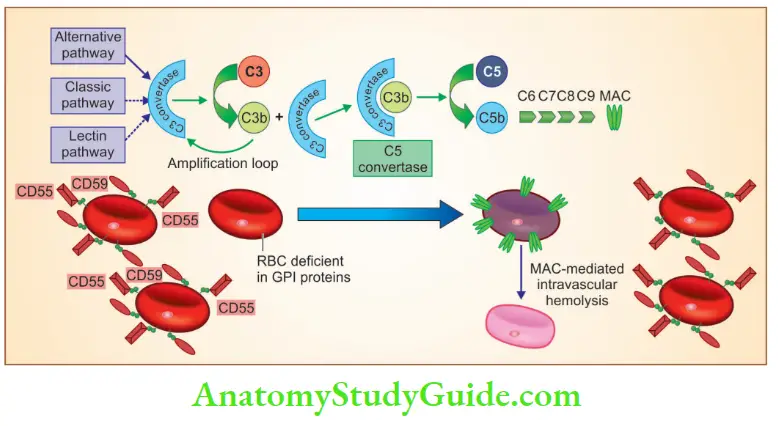
CD59 and CD55 deficient RBCs are destroyed by membrane attack complex with resultant hemolysis.
Clinical features and lab diagnosis
- Manifests as intravascular hemolysis (due to activation of membrane attack complex)
- Hemolysis is paroxysmal and nocturnal (due to a decrease in blood pH during sleep, which increases blood complement activity)
- Thrombosis is the most common cause of death in individuals with PNH
- 5 to 10% of patients develop acute myeloid leukemia or myelodysplastic syndrome
- PNH is diagnosed by flow cytometry
Treatment
- Eculizumab (monoclonal antibody) prevents the conversion of C5 to C5a
- Monoclonal antibody reduces the risk of hemolysis and thrombosis
- Complication: C5 inhibitor therapy leads to an increased risk of serious or fatal meningococcal infections
Question 11. Classify immune hemolytic anemia.
Answer:
Classification of immune hemolytic anemias
1. Warm antibody type (IgG antibodies active at 37°C)
- Primary (idiopathic)
- Secondary: Autoimmune disorders (systemic lupus erythematosus, RA), drugs (penicillin, cephalosporins, α-methyldopa), lymphoid neoplasms (CLL), malignant lymphoma, Hodgkin disease, multiple myeloma, thymoma, AML
2. Cold agglutinin type (IgM antibodies active below 37°C)
- Acute: Mycoplasma infection, infectious mononucleosis
- Chronic: Idiopathic, lymphoid neoplasm
3. Cold hemolysin type (IgG antibodies active below 37°C)
Question 12. Write a note on Coombs’ test.
Answer:
Diagnosis of immune hemolytic anemia requires the detection of antibodies and/or complement on the RBCs of the patient
1. Direct Coombs’ anti-globulin test
- The patient’s red cells are mixed with polyspecific anti-human globulin (AHG) (antiIgG and anti-complement)
- If either immunoglobulin or complement is present on the surface of the red cells, antibodies cause agglutination, appreciated visually as clumping
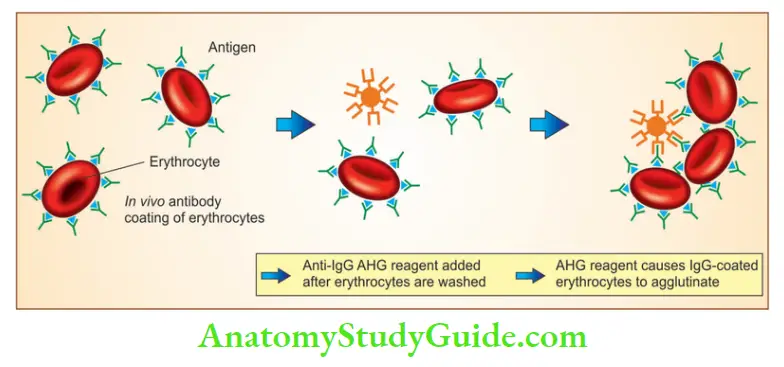
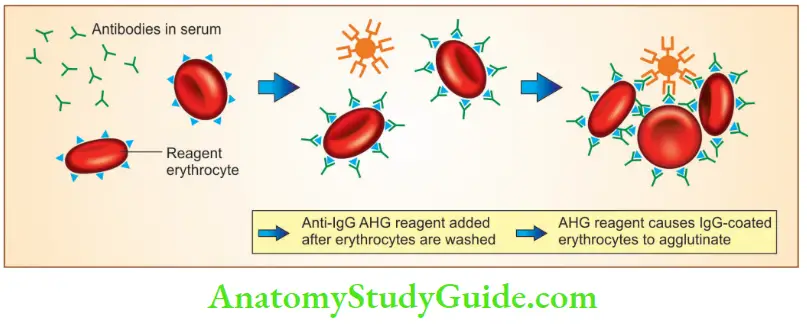
2. Indirect Coombs’ anti-globulin test
- Used to detect antibodies in the patient’s serum
- Positive indirect Coombs’ test indicates alloimmunization (immunization to the antigens from another individual) and/or the presence of autoantibody in the patient’s serum
Question 13. Write a note on the hemolytic uremic syndrome.
Answer:
Hemolytic uremic syndrome (HUS)
- HUS is associated with microangiopathic hemolytic anemia and thrombocytopenia with acute renal failure (in children)
- Intravascular thrombi cause microangiopathic hemolytic anemia
- Thrombocytopenia occurs due to the consumption of platelets
- Coagulation tests in HUS will be normal
Types
1. “Typical” HUS
- Associated with infectious gastroenteritis caused by Escherichia coli strain O157:H7, which elaborates Shiga-like toxin
- The toxin, absorbed from the inflamed gastrointestinal mucosa, enters into the circulation and results in altered endothelial cell function leading to platelet activation and aggregation
2. “Atypical” HUS
- Defects in complement factor H, factor I, or membrane cofactor protein (CD46) that prevent excessive activation of the alternative complement pathway
- Pathogenesis: Excessive complement activation, results in endothelial injury and platelet aggregation
Question 14. Write a note on osmotic fragility.
Answer:
Osmotic fragility test
- Red cells are suspended in decreasing concentrations of hypotonic saline solutions to check their ability to withstand osmotic stress
- In hypotonic solutions, water enters RBCs causing cellular swelling followed by cell lysis
Increased osmotic fragility is seen in:
- Hereditary spherocytosis
- Autoimmune hemolytic anemia
- Hemolytic diseases of newborn
Decreased osmotic fragility is seen in:
- Iron deficiency anemia
- Thalassemia
- Asplenia
- Liver disease
- Reticulocytosis
- Presence of HbS and HbC
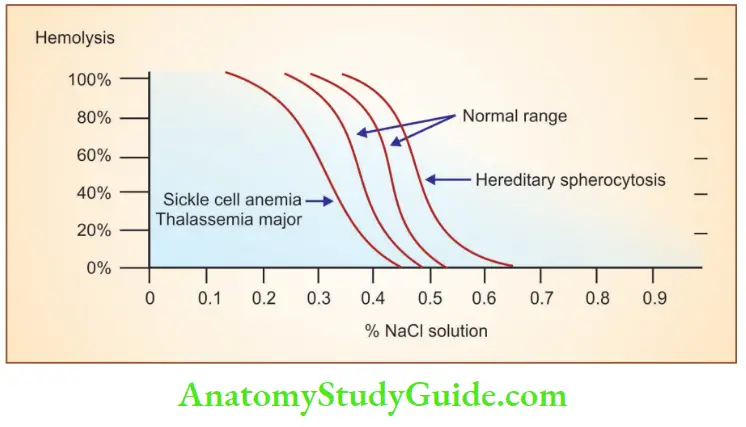
Question 15. List the causes of megaloblastic anemia. Discuss the pathogenesis, morphology, and bone marrow picture of megaloblastic anemia.
Answer:
Megaloblastic anemia occurs due to vitamin B12 deficiency or folic acid deficiency.
1. Causes of vitamin B 12 deficiency
1. Decreased intake
- Inadequate diet, vegetarianism
2. Impaired absorption
- Intrinsic factor deficiency: Pernicious anemia, gastrectomy
- Malabsorption states
3. Diffuse intestinal disease (for example lymphoma, systemic sclerosis)
4. Ileal resection, ileitis
2. Causes of folic acid deficiency
1. Decreased intake
- Inadequate diet, alcoholism, infancy
- Impaired absorption
- Malabsorption states
- Anticonvulsants
- Oral contraceptives
2. Increased requirement
- Pregnancy, infancy, disseminated cancer, markedly increased hematopoiesis
3. Impaired utilization
- Folic acid antagonists
4. Unresponsive to vitamin B12 or folic acid therapy
- DNA synthesis and/or folate metabolism inhibitors (for example methotrexate)
Pathogenesis
- Occurs due to impairment of DNA synthesis
Morphology
Peripheral smear
- Macro-ovalocytes: RBCs are oval and macrocytic, larger than normal, and lack the central pallor of normal red cells
- Marked variation in the size (anisocytosis) and shape (poikilocytosis) of red cells
- Reticulocyte count is low
- In severe anemia, nucleated red cell progenitors appear in the blood
- Neutrophils are larger than normal (macro polymorphonuclear) and show nuclear hypersegmentation (having five or more nuclear lobules)
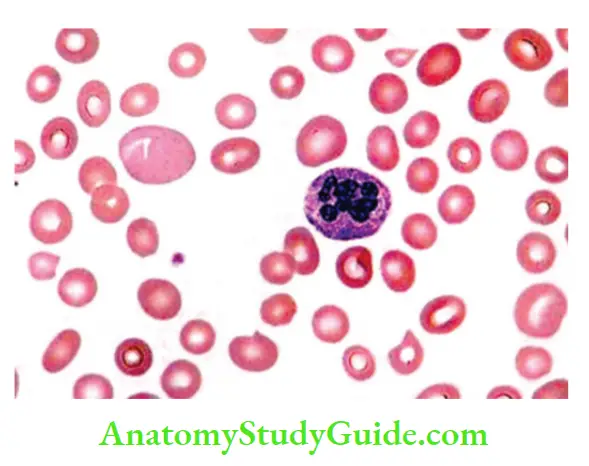
Bone marrow
- Markedly hypercellular marrow with increased hematopoietic precursors
- Megaloblastic changes are detected at all stages of erythroid development
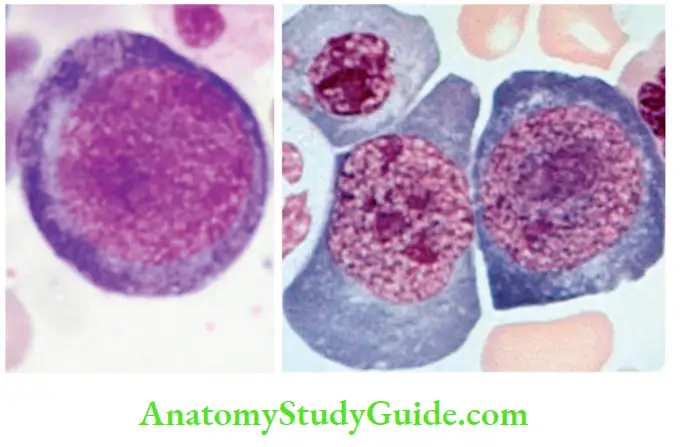
- Megaloblasts are large nucleated erythroid precursors whose nuclear maturation lags behind cytoplasmic maturation
- The nucleus of megaloblast contains loose, open sieve-like chromatin
- Nuclear-to-cytoplasmic asynchrony: Nuclear maturation is delayed, and cytoplasmic maturation proceeds at a normal pace
- Granulocytic precursors show desaturation in the form of giant metamyelocytes and band forms
- Megakaryocytes are abnormally large with bizarre multilobed nuclei
Question 16. Discuss the etiopathogenesis, peripheral smear, and bone marrow findings in iron deficiency anemia.
Answer:
Etiology:
Iron deficiency anemia results from:
- Dietary lack of iron
- Impaired absorption
- Increased requirement
- Chronic blood loss
Note:
- 1 mg of iron must be absorbed from the diet every day
- Daily iron requirement: 7 to 10 mg for adult men, 7 to 20 mg for adult women (only 10% to 15% of ingested iron is absorbed)
- Absorption of inorganic iron is increased by ascorbic acid, citric acid, amino acids, and sugars and is inhibited by tannates (found in tea), carbonates, oxalates, and phosphates
Peripheral smear
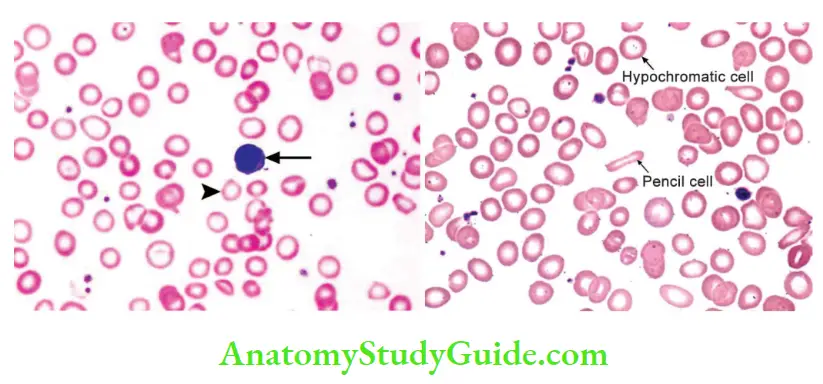
- RBCs appear small (microcytic) and pale (hypochromic) with a zone of pallor in RBC being enlarged
- In severe iron deficiency anemia, hemoglobin may be seen only in the narrow peripheral rim
- Poikilocytosis in the form of small, elongated red cells (pencil cells) can be seen
Bone marrow
- A mild to moderate increase in erythroid progenitors
- Prussian blue stain on bone marrow aspirate smear shows the disappearance of stainable iron from the macrophages
Question 17. Write a note on anemia of chronic disease.
Answer:
Anemia of chronic disease (ACD)
- Red cell production is impaired due to chronic diseases that produce systemic inflammation
Causes
- Osteomyelitis, bacterial endocarditis, lung abscess
- Immune disorders, such as rheumatoid arthritis
- Neoplasms: Carcinomas of the lung and breast and Hodgkin lymphoma
Mechanism
- Inflammatory mediators, IL-6, stimulate the hepatic hepcidin production
- Hepcidin inhibits ferroportin in macrophages and reduces the transfer of iron from the storage pool to the developing erythroid precursors in the bone marrow
- Another mechanism: Cytokine-mediated inhibition of erythropoietin production
Peripheral smear
- RBCs are normocytic and normochromic or microcytic and hypochromic
How to differentiate anemia of chronic disease from iron deficiency anemia?
- Bone marrow: Shows the presence of increased storage iron in marrow macrophages and the presence of sideroblasts
- High serum ferritin levels
Question 18. Write etiology and morphology of bone marrow in aplastic anemia.
Answer:
Aplastic anemia
Characterized by chronic primary hematopoietic failure and pancytopenia
Causes
- Drugs: Alkylating agents, antimetabolites, benzene, chloramphenicol, penicillamine, carbamazepine
- Physical agents: Irradiation, viral infections, hepatitis, cytomegalovirus, Epstein-Barr virus infection, herpes zoster (varicella zoster)
- Inherited: Fanconi anemia, telomerase defects
Morphology
- Markedly hypocellular bone marrow, devoid of hematopoietic cells
- Marrow shows fat cells, fibrous stroma, scattered lymphocytes, plasma cells
- Marrow aspirates give a “dry tap”
- Aplasia is best appreciated in marrow biopsies
Question 19. Enumerate the entities comprising Fanconi anemia.
Answer:
Fanconi anemia:
- Autosomal recessive disorder
Cause
- Occurs due to defects in multiprotein complex (required for DNA repair)
Comprises:
- Congenital anomalies (hypoplasia of the kidney and spleen with bone anomalies, which most commonly involve the thumbs or radius bone)
- Hypoplastic bone marrow
Question 20. Write a note on Bernard-Soulier syndrome.
Answer:
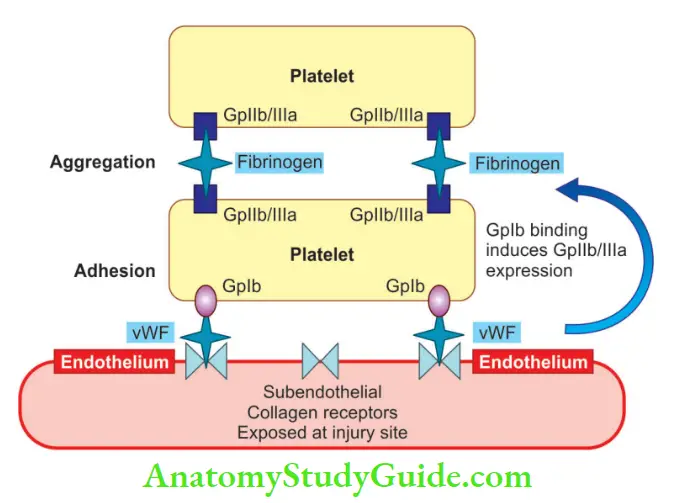
Bernard-Soulier syndrome
- Bleeding due to defective platelet adhesion to sub-endothelial matrix
- Due to a deficiency of platelet membrane glycoprotein complex Ib-IXa, a receptor for vWF
- Gp Ib-IXa is essential for normal platelet adhesion to the sub-endothelial extracellular matrix
- Also known: Deficiency of GpIIb/IIIa results in defective platelet aggregation, a disorder termed Glanzmann thrombasthenia.
Question 21. Mention the function of prothrombin time assay.
Answer:
Prothrombin time (PT) assay
- Assesses the function of the proteins in the extrinsic pathway (factors VII, X, V, II, and fibrinogen)
Question 22. Write a note on von Willebrand’s disease.
Answer:
Von Willebrand disease
- Most common inherited bleeding disorder of humans
- Clinical features: Spontaneous bleeding from mucous membranes (example epistaxis), excessive bleeding from wounds, or menorrhagia
Type 1 vWD
- Autosomal dominant disorder
- Quantitative defects in vWF
- The most common type leads to mild disease
Type 2 vWD
- Autosomal dominant disorder
- Characterized by qualitative defects in vWF
Type 3 vWD
- Autosomal recessive disorder
- Quantitative defects in vWF
- Associated with very low levels of vWF and severe clinical manifestations
Question 23. Write a note on hemophilia A.
Answer:
Hemophilia A (factor VIII deficiency)
- Inherited as an X-linked recessive trait
- Caused by mutations in factor VIII
- Most common hereditary disease associated with life-threatening bleeding
- Affects males and homozygous females
- Patients with 6% to 50% of normal factor VIII levels have mild disease
- Patients with 2% to 5% of normal factor VIII levels have moderately severe disease
- Patients with < 1% of normal factor VIII levels have severe disease and spontaneous hemorrhages into joints (hemarthrosis) are common
- Petechiae are characteristically absent
- Patients have prolonged PTT and a normal PT (due to abnormality of intrinsic coagulation pathway)
- Hemophilia A is treated with infusions of recombinant factor VIII
Hemophilia B (Christmas disease, factor IX deficiency):
- Inherited as an X-linked recessive trait
- PTT is prolonged and the PT is normal
- Treatment: Infusions of recombinant factor IX
Question 24. Write a note on the pathogenesis of disseminated intravascular coagulation.
Answer:
Disseminated intravascular coagulation (DIC)
- Thrombohemorrhagic disorder is characterized by excessive activation of the coagulation pathway with the formation of thrombi in the microvasculature throughout the body
- Cause: Due to systemic activation of thrombin
Two major mechanisms that trigger DIC
- Release of tissue factor (procoagulant) into the circulation
- Widespread injury to the endothelial cells
1. Sources of tissue factor
- Placenta in obstetric complications
- Tissues injured by trauma or burns
- Mucus released from adenocarcinomas
2. Endothelial injury can be seen
- In sepsis: TNF acts as a mediator of endothelial injury
- In systemic lupus erythematosus due to the deposition of antigen-antibody complexes
- Following heat stroke, burns
- Following meningococci, rickettsiae infections
Consequences of DIC
- Widespread deposition of fibrin
- Hemorrhagic diathesis
Question 25. Define thrombocytopenia. Write the causes of thrombocytopenia.
Answer:
- Reference range of platelets: 1,50,000–4,50,000/μL
- Thrombocytopenia: When the platelet count is less than 100,000 platelets/μL
Causes of thrombocytopenia
1. Decreased platelet production
- Selective impairment of platelet production:
- Drug-induced: Alcohol, thiazides, cytotoxic drugs
- Infections: Measles, human immunodeficiency virus (HIV)
- Nutritional deficiencies:
- Vitamin B12 and folate deficiency (megaloblastic anemia)
- Bone marrow failure:
- Aplastic anemia
- Bone marrow replacement:
- Leukemia, disseminated cancer, granulomatous disease Ineffective hematopoiesis:
- Myelodysplastic syndromes
2. Decreased platelet survival
- Immunologic destruction:
- Primary autoimmune
- Chronic immune thrombocytopenic purpura
- Acute immune thrombocytopenic purpura
- Secondary autoimmune
- Systemic lupus erythematosus, B-cell lymphoid neoplasms
- Alloimmune: Post-transfusion and neonatal
- Drug-associated: Quinidine, heparin, sulfa compounds
- Infections: HIV, infectious mononucleosis (transient, mild), dengue fever
- Non-immunologic destruction:
- Disseminated intravascular coagulation
3. Sequestration
- Hypersplenism
4. Dilution
- Transfusions
Question 26. Write a note on idiopathic thrombocytopenic purpura.
Answer:
Acute immune thrombocytopenic purpura
- Affects children and the disease is self-limited (disease resolves in 6 months)
- Symptoms appear 1 to 2 weeks after self-limited viral illness which triggers the development of auto-antibodies
- Glucocorticoids are given only if the thrombocytopenia is severe
Chronic immune thrombocytopenic purpura (ITP)
Pathogenesis
- Auto-antibodies directed against platelet membrane glycoprotein IIb-IIIa or Ib-IX, and are of IgG type
Morphology
1. Spleen
- Normal in size
- Congestion of sinusoids with enlargement of splenic follicles, associated with prominent reactive germinal centers
2. Bone marrow
- To rule out other causes of thrombocytopenia
- Increased number of megakaryocytes, with large, non-lobulated, single nuclei
3. Blood
- Peripheral blood reveals abnormally large platelets
Clinical features
- Most commonly in women less than 40 years of age
- The female-to-male ratio is 3: 1
- Petechiae: Cutaneous bleeding (pinpoint hemorrhage) which when confluent, can give rise to ecchymoses
- Easy bruising, nosebleeds, bleeding from the gums, melena, hematuria, or excessive menstrual flow
- Subarachnoid hemorrhage and intracerebral hemorrhage
- Splenomegaly and lymphadenopathy are uncommon
- PT and APTT are normal
Treatment
- Glucocorticoids (inhibit phagocyte function)
- Splenectomy normalizes the platelet count (in individuals with severe thrombocytopenia)
- Intravenous immunoglobulin or anti-CD20 antibody (rituximab) is effective in patients who relapse after splenectomy
Question 27. Write a note on complications of blood transfusion.
Answer:
Complications of blood transfusion
1. Febrile non-hemolytic reaction
- Most common complication
- The patient presents with fever and chills, mild dyspnea
- Occurs within 6 hours of a transfusion of red cells or platelets
2. Allergic drug reactions
3. Hemolytic reactions
- Acute hemolytic reactions: Due to preformed IgM antibodies against donor RBCs
- Delayed hemolytic reactions: Caused by antibodies that recognize red cell antigens that the recipient was sensitized to previously, for example, through a prior blood transfusion
4. Transfusion-related acute lung injury (TRALI)
- The severe, frequently fatal complication in which factors in a transfused blood product trigger the activation of neutrophils in the lung microvasculature
5. Infectious complications
- HIV, hepatitis C, hepatitis B, malaria, and syphilis
- Transmission of these diseases has now decreased, due to donor screening for these infections
Question 28. What is hematocrit?
Answer:
Hematocrit or packed cell volume
- The volume occupied by red blood cells when a sample of anticoagulated blood is centrifuged
- Indicates the relative proportion of red cells to plasma
Uses
- Detection of the presence and absence of anemia or polycythemia
- For estimation of red cell indices
Methods of estimation
- Wintrobe method
- Microhematocrit method
Question 29. Write bone marrow changes in lead poisoning.
Answer:
Peripheral smear
- Microcytic, hypochromic anemia
- Accompanied by mild hemolysis
- Punctate basophilic stippling of red cells
Bone marrow
- Ring sideroblasts
- Red cell precursors with iron-laden mitochondria, detected by Prussian blue stain
Question 30. Write a note on Bence Jones proteins.
Answer:
Bence Jones proteins
- Monoclonal immunoglobulin light chains, synthesized by neoplastic plasma cells
- Excess production of these chains is seen in multiple myeloma and primary amyloidosis
- Because of their low molecular weight, these are excreted in urine
- When the urine sample is heated, Bence Jones proteins precipitate at temperatures between 40° and 60°C, and the precipitate disappears on heating up to 85°–100°C, and when cooled, there is the reappearance of the precipitate.
- The investigation of choice for its detection in urine is protein electrophoresis
Question 31. What is leucocyte alkaline phosphatase scoring?
Answer:
Leucocyte alkaline phosphate (LAP) score
- It is used in patients with elevated WBC to differentiate reactive leucocytosis from chronic myelogenous leukemia
- Reduced LAP score is seen in CML, aplastic anemia, pernicious anemia
- Elevated LAP score is also seen in leukemoid reaction (or reactive leucocytosis), myelofibrosis, essential thrombocytosis, and polycythemia vera
Leave a Reply