The Kidney and Lower Urinary Tract
Normal Structure Of The Kidney
Anatomy
Table of Contents
The kidneys are bean-shaped paired organs, each weighing about 150 gm in the adult male and about 135 gm in the adult female.
- The hilum of the kidney is situated at the midpoint on the medial aspect where the artery, vein, lymphatics and ureter are located.
- The kidney is surrounded by a thin fibrous capsule which is adherent at the hilum.
- The cut surface of the kidney shows 3 main structures: well-demarcated peripheral cortex, inner medulla and the innermost renal pelvis.
Read And Learn More: Systemic Pathology Notes
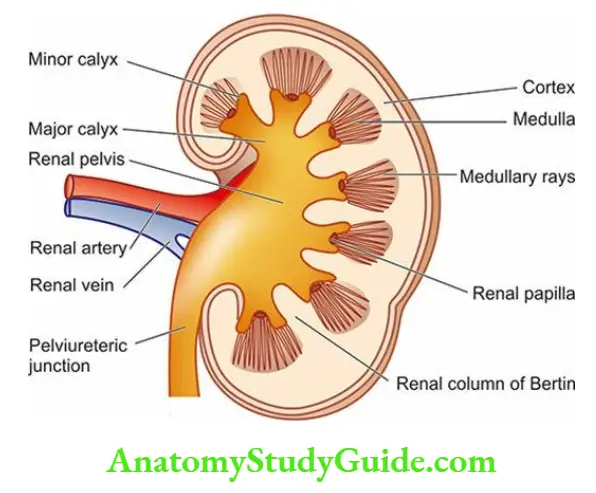
- The renal cortex forms the outer rim of the kidney and is about 1 cm in thickness. It contains all the glomeruli and about 85% of the nephron tubules.
- Remaining 15% of nephrons consisting of collecting tubules, collecting ducts, loops of Henle and vasa recta send their loops into the medulla and are therefore called juxtamedullary nephrons.
- This latter part of the cortex forms faint striations called medullary rays, a misnomer since these structures are located in the cortex but are destined for the medulla.
- Columns of renal cortical tissue that extend into the space between adjacent pyramids are called the renal column (septa) of Bertin; they contain the interlobar arteries.
- The renal medulla is composed of 8-18 cone-shaped renal pyramids.
- The base of a renal pyramid lies adjacent to the outer cortex and forms the corticomedullary junction, while the apex of each called the renal papilla contains the opening of each renal pyramid for passage of urine collected from collecting ducts and goes down into minor calyces.
- The renal pelvis is the funnel-shaped collection area of the urine for drainage into the ureter.
- The minor calyces (8-18 in number in a normal kidney) collect urine from renal papillae and drain into major calyces (2-3 in a normal kidney).
Renal Blood Supply
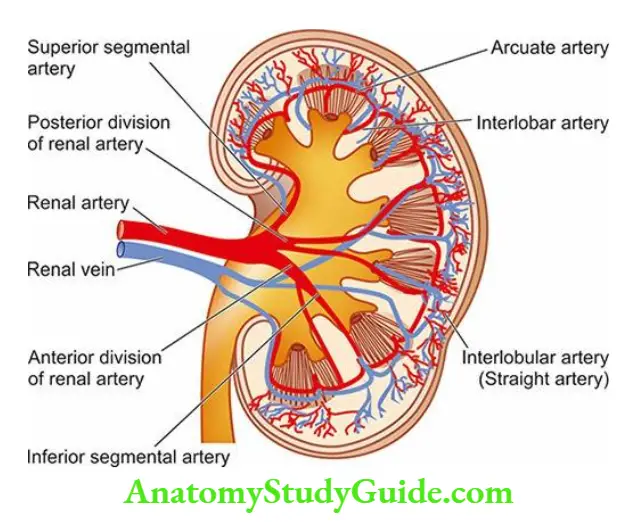
- Each kidney is supplied with blood by a main renal artery which arises from the aorta at the level of the 2nd lumbar vertebra.
- It usually divides into anterior and posterior divisions at the hilum although occasionally these divisions may even arise directly from the aorta.
- The anterior and posterior divisions divide into segmental branches from which interlobar arteries arise which course between the lobes.
- Along their course, they give off the arcuate arteries which arch between the cortex and medulla.
- The arcuate arteries, in turn, give off interlobular arteries which lie in the cortex perpendicular to the capsular surface in the part overlying the pyramids and, therefore, are also called straight arteries.
- It is from the interlobular arteries that the afferent arterioles take their origin, each one supplying a single glomerulus.
- From the glomerulus emerge the efferent arterioles.
- Up to this stage, the arteries and arterioles are end vessels.
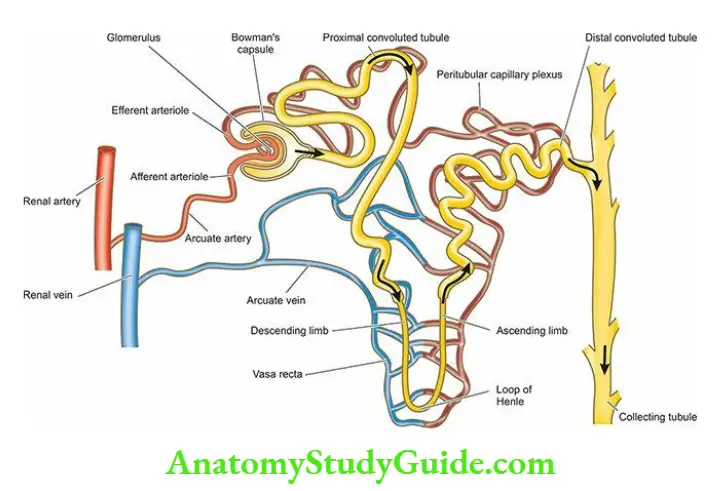
- The efferent arterioles leaving the glomerulus supply peritubular capillary plexus which anastomoses with the capillary plexus of another nephron.
- The juxtamedullary glomeruli, however, give off a series of parallel vessels called vasa recta which descend to the inner medulla supplying the loop of Henle and collecting ducts and anastomose at all levels throughout the medulla with the ascending vasa recta.
- These drain into arcuate veins and then into the veins that accompany the corresponding arteries and finally through a single renal vein into the inferior vena cava.
- Lymphatic drainage likewise occurs through lymphatics associated with the intrarenal vasculature leaving the kidney at the hilum and draining to lateral aortic lymph nodes.
The following important inferences can be drawn from the peculiarities of the renal vasculature:
-
- The renal cortex receives about 90% of the total renal blood supply and the pressure in the glomerular capillaries is high. Therefore, the renal cortex is more prone to the effects of hypertension.
- The renal medulla, on the other hand, is poorly perfused and any interference in blood supply to it results in medullary necrosis.
- The divisions and subdivisions of the renal artery up to arterioles are end-arteries and have no anastomoses. Thus, occlusion of any of the branches results in infarction of the renal parenchyma supplied by it.
- Since the tubular capillary beds are derived from the efferent arterioles leaving the glomeruli, diseases affecting the blood flow through the glomerular tuft have significant effects on the tubules as well.
Histology
- The parenchyma of each kidney is composed of approximately one million microstructures called nephrons.
- A nephron, in turn, consists of 5 major parts, each having a functional role in the formation of urine the glomerular capsule (glomerulus and Bowman’s capsule), the proximal convoluted tubule (PCT), the loop of Henle, the distal convoluted tubule (DCT), and the collecting ducts.
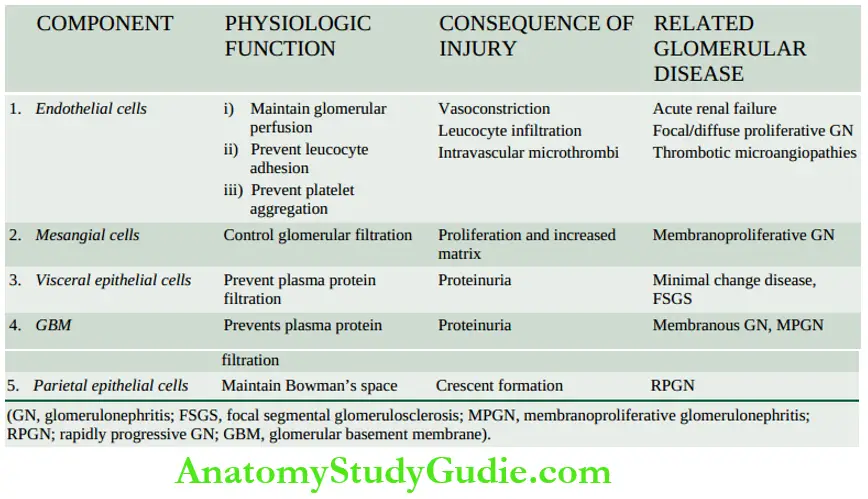
- From the point of view of diseases of the kidneys, 4 components of renal parenchyma require further elaboration: renal vasculature, glomeruli, tubules and interstitium.
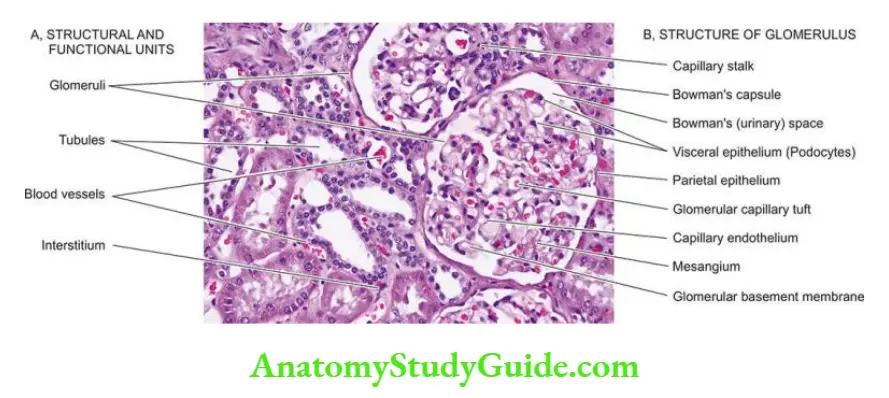
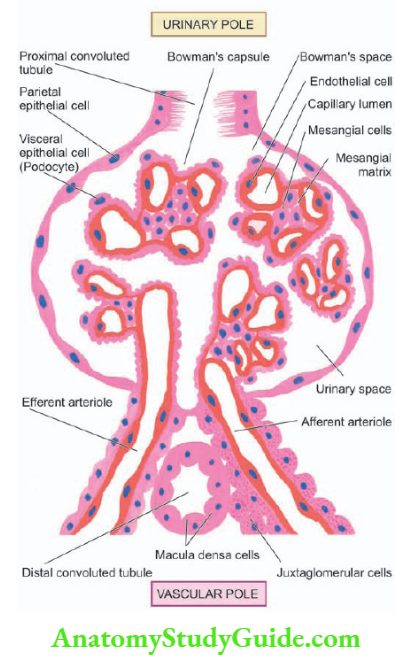
1. Glomerulus The glomerulus consists of the invagination of the blind end of the proximal tubule and contains a capillary tuft fed by the afferent arteriole and drained by the efferent arteriole.
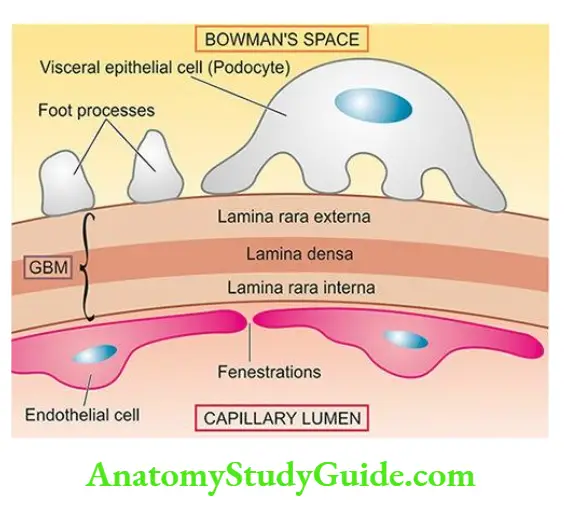
- These capillaries are lined by discontinuous endothelial cells. The capillary tuft is covered by visceral epithelial cells (podocytes) which are continuous with those of the parietal epithelium at the vascular pole.
- The transition to proximal tubular cells occurs at the urinary pole of the glomerulus. The visceral and parietal epithelial cells are separated by the urinary space or Bowman’s space, into which glomerular filtrate passes.
- Subdivisions of capillaries derived from the afferent arterioles result in the formation of lobules (up to 8 in number) within a glomerulus.
- Each lobule of a glomerular tuft consists of a centrilobular supporting stalk composed of mesangium containing mesangial cells (≤ 3 per lobule) and mesangial matrix.
- The mesangium is continuous at the hilum with the lacis cells of the juxtaglomerular apparatus.
- Besides their role as supportive cells, mesangial cells are involved in the production of mesangial matrix and glomerular basement membrane they function in endocytosis of leaked macromolecules and also possibly in the control of glomerular blood flow through contractile elements present in these cells.
- The major function of the glomerulus is complex filtration from the capillaries to the urinary space.
- The glomerular filtrate is quite similar in composition to plasma but lacks proteins and cells. Normally, the glomerular filtration rate (GFR) is about 125 ml/minute.
- The barrier to glomerular filtration consists of the following 3 components:
-
- Fenestrated endothelial cells lining the capillary loops.
- Glomerular basement membrane (GBM) on which the endothelial cells rest. It further consists of 3 layers the central lamina dens, bounded by lamina interna on the endothelial side of the capillary and lamina externa on the visceral epithelial side of the capillary.
- Filtration slit pores between the foot processes of the visceral epithelial cells (podocytes) external to GBM.
- The barrier to filtration of macromolecules of the size and molecular weight of albumin and larger depends upon the following:
-
-
- A normal lamina densa.
- Maintenance of negative charge on both laminae is rare.
- A healthy covering of glomerular epithelial cells.
-
Juxtaglomerular apparatus: The juxtaglomerular apparatus (JGA) is situated at the vascular pole of the glomerulus and is made up of 3 parts
- The juxtaglomerular cells are modified granular smooth muscle cells in the media of the afferent arteriole and contain the hormone, renin.
- The macula dens are comprised of a specialised region of the distal tubule when it returns to the vascular pole of its parent glomerulus. The tubular cells here are taller and narrower than elsewhere with the nuclei lying close together.
- The lacis cells or non-granular cells occupy the space between the macula dens and the arterioles and merge with the glomerular mesangium.
- The JGA is intimately concerned with sodium metabolism and is the principal source of renin production. The mechanism of the release of renin and its role in hypertension.
3. Tubules: The tubules of the kidney account for the greatest amount of the renal parenchyma.
The structure of renal tubular epithelium varies in different parts of the nephron and is correlated with the functional capacity of that part of the tubule.
- Proximal convoluted tubule (PCT) This is the first part arising from the glomerulus and is a highly specialised part functionally.
- It is lined by cuboidal cells with a brush border composed of microvilli and contains numerous mitochondria, Golgi apparatus and endoplasmic reticulum.
- The major functions of PCT are active reabsorption of filtered sodium, potassium, glucose, amino acids, proteins, vitamins, bicarbonate, phosphate, calcium and uric acid, and passive reabsorption of 80% of filtered water.
- Loop of Henle The PCT drains into the straight part of the loop of Henle that consists of thin descending, and thin and thick ascending limbs, both of which have different structures and functions.
- The descending limb is a continuation of PCT while ascending limb continues further into the distal convoluted tubule (DCT).
- The descending segment of the loop is lined by simple epithelium while the ascending limb is lined by columnar cells.
- The major function of the loop of Henle is active reabsorption of sodium, potassium and chloride, and passive diffusion of water resulting in concentrated filtrate of urine.
- Distal convoluted tubule (DCT) The DCT represents a transition from a thick ascending limb from the point where the ascending limb meets the vascular pole of the glomerulus of its origin, to the early collecting ducts.
- The lining cells in DCT are cuboidal. The epithelial cells at the point of the beginning of DCT are taller, narrower and more closely packed to form the macula dens of JGA as already described.
- The DCT further contributes to urinary concentration and acidification, while the macula dens of JGA are the source of renin and has a role in sodium metabolism.
- Collecting ducts The system of collecting ducts is the final pathway by which urine reaches the tip of the renal papilla. The cells lining the collecting ducts are cuboidal but lack the brush border.
- Collecting ducts reabsorb water under the control of ADH, and secrete H+ and K+ ions.
4. Interstitium: In health, the renal cortical interstitium is scanty and consists of a small number of fibroblast-like cells.
- But the medullary interstitium is more plentiful and contains stellate interstitial cells which are considered to produce an anti-hypertensive agent and are involved in the metabolism of prostaglandins.
Normal Structure of the Kidney:
- Each normal kidney weighs about 150 gm in the adult male and about 135 gm in the adult female.
- The cut surface of the kidney shows the peripheral cortex, inner medulla and the innermost renal pelvis.
- Histologically, renal parenchyma is composed of 4 main components: renal vasculature, glomeruli, tubules and interstitium.
- Glomeruli have lobules having central stalks containing mesangial cells and matrix. Lobules have a tuft of capillaries lined by endothelial cells and supported by visceral epithelial cells (podocytes).
- Bowman’s space is lined by parietal epithelial cells. The glomerular basement membrane has lamina dens in the centre and is bounded on either side by lamina rar.
Renal Function Tests
In general, the kidney performs the following vital functions in the body:
- Excretion of waste products resulting from protein metabolism.
- Regulation of acid-base balance by excretion of H+ ions (acidification) and bicarbonate ions.
- Regulation of salt-water balance by hormones secreted both intra- and extra-really.
- Formation of renin and erythropoietin and thereby playing a role in the regulation of blood pressure and erythropoiesis respectively.
- In order to assess renal function, a number of tests have been devised which give information regarding the following parameters:
-
- Renal blood flow
- Glomerular filtration
- Renal tubular function
- Urinary outflow unhindered by any obstruction.
Renal function tests are broadly divided into 4 groups:
- Urine analysis
- Concentration and dilution tests
- Blood chemistry
- Renal clearance tests
In addition, a renal biopsy is performed to confirm the diagnosis of renal disease. Renal biopsy is ideally fixed in alcoholic Bouin’s solution and examined by routine morphology combined with special stains and for further studies as under:
1. Urine Analysis
- Physical examination (output, colour, specific gravity, pH, osmolality)
- Chemical constituents (protein, glucose, red cells, haemoglobin)
- Bacteriologic examination
- Microscopy
2. Concentration And Dilution Tests
- Concentration test (fluid deprivation test)
- Dilution test (excess fluid intake test)
3. Blood Chemistry
- Urea
- Blood urea nitrogen (BUN)
- Creatinine
- β2-microglobulin
4. Renal Clearance Test
- Inulin or mannitol clearance test
- Creatinine clearance
- Urea clearance
- Para-amino hippuric acid (PAH) clearance
- Periodic acid-Schiff stain for highlighting glomerular basement membrane.
- Silver impregnation to outline the glomerular and tubular basement membrane.
- Immunofluorescence to localise the antigens, complements and immunoglobulins.
- Electron microscopy to see the ultrastructure of glomerular changes.
1. Urine Analysis: The simplest diagnostic tests for renal function is the physical, chemical, bacteriologic and microscopic examination of the urine.
- The physical examination includes 24-hour urinary output, colour, specific gravity and osmolality.
- Normally urine is clear, pale or straw-coloured due to pigment urochrome and 700- 2500 ml (average 1200 ml) of urine are passed in 24 hours, mostly during day time.
- Specific gravity is used to measure the concentrating and diluting power of the kidneys.
- Chemical tests are carried out to detect the presence of protein, glucose, red cells and haemoglobin to assess the permeability of the glomerular membrane.
- A number of convenient dipstick tests are available for testing these chemical substances and pH. These consist of paper strips impregnated with appropriate reagents and indicator dyes.
- The bacteriologic examination of the urine is done by a proper and aseptic collection of midstream specimens of urine.
- Urine microscopy is undertaken on a fresh unstained sample. Various components observed on microscopic examination of the urine in renal disease are red cells, pus cells, epithelial cells, crystals and urinary casts.
- The casts are moulded into cylindrical shapes by a passage along tubules in which they are formed.
- They are the result of the precipitation of proteins in the tubule that includes not only albumin but also the tubular secretion of the Tamm Horsfall protein.
- The latter is a high molecular weight glycoprotein normally secreted by ascending loop of Henle and DCT and probably has body defence function normally. Its secretion is increased in glomerular and tubular diseases.
- Casts may be hyaline type consisting of only proteins indicating a noninflammatory aetiology of glomerular filtration of proteins, leucocyte casts inflammatory in origin, or red cell casts from haematuria.
2. Concentration And Dilution Tests: Concentration and dilution tests are designed to evaluate the functional capacity of the renal tubules.
- The ability of the nephron to concentrate or dilute urine is dependent upon both the functional activity of the tubular cells in the renal medulla and the presence of antidiuretic hormone (ADH).
- Failure to achieve adequate urinary concentration can be due to either defects within the renal medulla (nephrogenic diabetes insipidus), or due to the lack of ADH (central diabetes insipidus).
- Traditionally, urinary concentration is determined by the specific gravity of the urine (normal range 1.003 to 1.030, average 1.018) which in cases of tubular disease remains constant at approximately 1.010 regardless of changing levels of plasma hydration.
- However, the determination of urinary-specific gravity provides only a rough estimate of the osmolarity of the urine.
- The tubular disease can be diagnosed in its early stage by water deprivation (concentration) or water excess (dilution) tests.
- In the concentration test, an artificial fluid deprivation is induced in the patient for more than 20 hours.
- If the nephron is normal, water is selectively reabsorbed resulting in the excretion of urine of high solute concentration (specific gravity of 1.025 or more).
- However, if the tubular cells are nonfunctional, the solute concentration of the urine will remain constant regardless of the stress of water deprivation.
- In the dilution test, an excess of fluid is given to the patient. Normally, renal compensation should result in the excretion of urine with high water content and lower solute concentration (specific gravity of 1.003 or less).
- If the renal tubules are diseased, the concentration of solutes in the urine will remain constant irrespective of the excess water intake.
- In the concentration test, an artificial fluid deprivation is induced in the patient for more than 20 hours.
3. Blood Chemistry: Impairment of renal function results in elevation of end-products of protein metabolism.
- This includes an increased accumulation of certain substances in the blood, chiefly urea (normal range 20-40 mg/dl), blood urea nitrogen (BUN) (normal range 10-20 mg/dl) and creatinine (normal range 0.6-1.2 mg/dl).
- An increase of these end-products in the blood is called azotaemia.
- High levels of creatinine are associated with high levels of β2-microglobulin in the serum as well as urine, a low-molecular-weight protein filtered excessively in the urine due to glomerular disease or due to increased production by the liver.
4. Renal Clearance Tests: A clearance test is employed to assess the rate of glomerular filtration and renal blood flow.
- The rate of this filtration can be measured by determining the excretion rate of a substance which is filtered through the glomerulus but subsequently is neither reabsorbed nor secreted by the tubules.
- The glomerular filtration rate (normal 120 ml/minute in an average adult) is usually equal to the clearance of that substance and is calculated from the following equation.

C is the clearance of the substance in ml/minute;
U is the concentration of the substance in the urine;
V is the volume of urine passed per minute; and
P is the concentration of the substance in the plasma.
The substances which are used for clearance tests include inulin, mannitol, creatinine and urea.
- In inulin or mannitol clearance tests, an intravenous infusion of the substance inulin or mannitol is given to maintain constant plasma concentration and accurately timed urine samples are collected.
- Inulin, a mixture of fructose polymers, is considered the ideal substance for the clearance test since it is filtered from the glomerulus and is excreted unchanged in the urine.
- In creatinine clearance tests, there is no need for intravenous infusion of creatinine since creatinine is normally released into plasma by muscle metabolism and a very small fraction of this substance is secreted by the tubules.
- The clearance of creatinine is determined by collecting urine over a 24-hour period and a blood sample is withdrawn during the day.
- In spite of disadvantages like poor reproducibility and secretion of creatinine by the tubules, the ‘endogenous’ creatinine clearance test is an easy and routinely employed method of estimating GFR.
- In the urea clearance test, the sensitivity is much less than the creatinine or inulin clearance because the plasma concentration of urea is affected by a number of factors (for example dietary protein, fluid intake, infection, trauma, surgery, and corticosteroids) and is partly reabsorbed by the tubules.
- Like in creatinine clearance, there is no need for intravenous infusion of urea.
- Para-amino hippuric acid (PAH) clearance test is employed to measure renal blood flow (unlike the preceding tests which measure GFR).
- PAH when infused intravenously is both filtered at the glomerulus as well as secreted by the tubules and its clearance is measured by determining its concentration in arterial blood and urine.
- Normally, renal blood flow is about 1200 ml per minute in an average adult.
Renal Function Tests
- The simplest diagnostic tests for renal function are the physical, chemical, bacteriologic and microscopic examination of the urine.
- Concentration and dilution tests are designed to evaluate the functional capacity of the renal tubules.
- Impaired renal functions results in the elevation of end-products of protein metabolism, namely blood urea, blood urea nitrogen BUN and creatinine.
- A clearance test is employed to assess the rate of glomerular filtration and renal blood flow. These include clearance tests for inulin, mannitol, creatinine, urea and para amino acid.
- A renal biopsy is performed to establish the specific diagnosis of renal disease.
Renal Failure
Pathophysiology Of Renal Disease
Traditionally, diseases of the kidneys initially evolve from the predominant involvement of one of the morphologic components (glomeruli, tubules, interstitium or blood vessels), but eventually, all components are affected leading to end-stage kidneys.
Accordingly, the major groups of renal diseases are as under:
- Glomerular diseases: These are most often immunologically mediated and may be acute or chronic.
- Tubular diseases: These are more likely to be caused by toxic or infectious agents and are often acute.
- Interstitial diseases: These are likewise commonly due to toxic or infectious agents and quite often involve interstitium as well as tubules (tubulointerstitial diseases).
- Vascular diseases: These include changes in the nephron as a consequence of increased intra-glomerular pressure such as in hypertension or impaired blood flow.
- Regardless of the cause, a renal disease usually results in the evolution of one of the two major pathological syndromes acute renal failure and chronic renal failure (or chronic kidney disease, CKD).
- The term ‘azotaemia’ is used for biochemical abnormality characterised by elevation of the blood urea nitrogen (BUN) and creatinine levels, while ‘uraemia’ is defined as an association of these biochemical abnormalities with clinical signs and symptoms.
- The pathophysiological aspects of acute and chronic renal failure are briefly discussed below.
Acute Renal Failure (ARF)
Acute renal failure (ARF) is a syndrome characterised by the rapid onset of renal dysfunction, chiefly oliguria or anuria and a sudden increase in metabolic waste products (urea and creatinine) in the blood with the consequent development of uraemia.
Etiopathogenesis The causes of ARF may be classified as pre-renal, intra-renal and post-renal in nature.
- Pre-renal causes: Pre-renal diseases are those which cause a sudden decrease in blood flow to the nephron.
- Renal ischaemia ultimately results in functional disorders or depression of GFR, or both.
- These causes include inadequate cardiac output and hypovolaemia or vascular disease causing reduced perfusion of the kidneys.
- Intra-renal causes: Intra-renal disease is characterised by disease of renal tissue itself.
- These include vascular disease of the arteries and arterioles within the kidney, diseases of glomeruli, acute tubular necrosis due to ischaemia, or the effect of a nephrotoxin, acute tubulointerstitial nephritis and pyelonephritis.
- Post-renal causes: Post-renal disease is characteristically caused by obstruction to the flow of urine anywhere along the renal tract distal to the opening of the collecting ducts.
- This may be caused by a mass within the lumen or from the wall of the tract, or from external compression anywhere along the lower urinary tract ureter, bladder neck or urethra.
- It is important to note that ARF originating in pre- and post-renal disease such as renal ischaemia or renal infection, eventually leads to intra-renal disease.
- Thus, full-blown ARF reflects some degree of nephron damage.
Clinical Features The clinical features will depend to a large extent on the underlying cause of ARF and on the stage of the disease at which the patient presents. However, one of the following three major patterns usually emerges.
- Syndrome of acute nephritis: This is most frequently associated with acute post-streptococcal glomerulonephritis and rapidly progressive glomerulonephritis.
- Renal dysfunction results from the extensive proliferation of epithelial cells in the glomeruli with a consequent mild increase in glomerular permeability and a decrease in GFR.
- The characteristic features are mild proteinuria, haematuria, oedema and mild hypertension.
- Fluid retention in acute nephritis syndrome appears to be due to both diminished GFR and increased salt and water reabsorption in the distal nephron.
- Syndrome accompanying tubular pathology: When the ARF is caused by the destruction of the tubular cells of the nephron as occurs in acute tubular necrosis, the disease typically progresses through 3 characteristic stages from oliguria to diuresis to recovery.
- Oliguric phase: The initial oliguric phase lasting on an average from 7 to 10 days is characterised by a urinary output of less than 400 ml per day.
- The decline in the formation of the urine leads to the accumulation of waste products of protein metabolism in the blood and resultant azotaemia, metabolic acidosis, hyperkalaemia, hypernatraemia and hypervolaemia due to secondary effects of circulatory overload and pulmonary oedema.
- The specific gravity of the urine is low but the concentration of sodium in urine tends to be elevated.
- Diuretic phase: With the onset of healing of tubules, there is an improvement in urinary output.
- This is believed to occur due to the drawing of water and sodium by preceding high levels of creatinine and urea as they move through the nephron so as to be excreted.
- Since tubular cells have not regained normal functional capacity, the urine is of low or fixed specific gravity.
- The phase of recovery: Full recovery with the healing of tubular epithelial cells occurs in about half the cases, while others terminate in death.
- The process of healing may take up to one year with restoration of normal tubular function.
- Pre-renal syndrome The ARF occurring secondary to disorders in which neither the glomerulus nor the tubules are damaged, results in pre-renal syndrome.
-
- Typically, this pattern is seen in marginal ischaemia caused by renal arterial obstruction, hypovolaemia, hypotension or cardiac insufficiency.
- Due to depressed renal blood flow, there is a decrease in GFR causing oliguria, azotaemia (elevation of BUN and creatinine) and possible fluid retention and oedema.
- Since the tubular cells are functioning normally, the nephron retains its ability to concentrate the glomerular filtrate according to adaptive needs.
-
- Oliguric phase: The initial oliguric phase lasting on an average from 7 to 10 days is characterised by a urinary output of less than 400 ml per day.
Chronic Renal Failure (CRF)
- Chronic renal failure is a syndrome characterised by progressive and irreversible deterioration of renal function due to slow destruction of renal parenchyma, eventually terminating in death when a sufficient number of nephrons have been damaged.
- Acidosis is the major problem in CRF with the development of biochemical azotaemia and clinical uraemia syndrome.
Etiopathogenesis
- All chronic nephropathies can lead to CRF. The diseases leading to CRF can generally be classified into two major groups: those causing glomerular pathology, and those causing tubulointerstitial pathology.
- Though this classification is useful to facilitate study, the disease rarely remains confined to either glomeruli or tubulointerstitial tissue alone. In the final stage of CRF, all parts of the nephron are involved.
-
- Diseases causing glomerular pathology: A number of glomerular diseases associated with CRF have their pathogenesis in immune mechanisms.
- Glomerular destruction results in changes in the filtration process and leads to the development of the nephrotic syndrome characterised by proteinuria, hypoalbuminaemia and oedema.
- The important examples of chronic glomerular diseases causing CRF are covered under two headings primary and systemic.
- Primary glomerular pathology The major cause of CRF is chronic glomerulonephritis, usually initiated by various types of glomerulonephritis such as membranous glomerulonephritis, membranoproliferative glomerulonephritis, lipoid nephrosis (minimal change disease) and anti-glomerular basement membrane nephritis.
- Systemic glomerular pathology Certain conditions originate outside the renal system but induce changes in the nephrons secondarily.
- Major examples of this type are systemic lupus erythematosus, serum sickness nephritis and diabetic nephropathy.
- Diseases causing glomerular pathology: A number of glomerular diseases associated with CRF have their pathogenesis in immune mechanisms.
- Diseases causing tubulointerstitial pathology Damage to tubulointerstitial tissues results in alterations in the reabsorption and secretion of important constituents leading to the excretion of large volumes of dilute urine.
- Tubulointerstitial diseases can be categorised according to initiating aetiology into 4 groups: vascular, infectious, toxic and obstructive.
- Vascular causes Long-standing primary or essential hypertension produces characteristic changes in renal arteries and arterioles referred to as nephrosclerosis.
- Nephrosclerosis causes progressive renal vascular occlusion terminating in ischaemia and necrosis of renal tissue.
- Infectious causes A good example of chronic renal infection causing CRF is chronic pyelonephritis. The chronicity of the process results in progressive damage to the increasing number of nephrons leading to CRF.
- Toxic causes Some toxic substances induce slow tubular injury, eventually culminating in CRF.
- The most common example is an intake of high doses of analgesics such as phenacetin, aspirin and acetaminophen (chronic analgesic nephritis).
- Other substances that can cause CRF after prolonged exposure are lead, cadmium and uranium.
- Obstructive causes Chronic obstruction in the urinary tract leads to progressive damage to the nephron due to fluid back-pressure.
- Examples of this type of chronic injury are stones, blood clots, tumours, strictures and enlarged prostate.
Regardless of the initiating cause, CRF evolves progressively through 4 stages:
- Decreased renal reserve At this stage, damage to renal parenchyma is marginal and the kidneys remain functional.
- The GFR is about 50% of normal, BUN and creatinine values are normal and the patients are usually asymptomatic except at times of stress.
- Renal insufficiency At this stage, about 75% of functional renal parenchyma has been destroyed.
- The GFR is about 25% of normal accompanied by elevation in BUN and serum creatinine.
Polyuria and nocturia occur due to tubulointerstitial damage. - Sudden stress may precipitate the uraemic syndrome.
- The GFR is about 25% of normal accompanied by elevation in BUN and serum creatinine.
- Renal failure At this stage, about 90% of functional renal tissue has been destroyed. The GFR is approximately 10% of normal.
- Tubular cells are essentially nonfunctional.
- As a result, the regulation of sodium and water is lost resulting in oedema, metabolic acidosis, hypocalcaemia, and signs and symptoms of uraemia.
- End-stage kidney (chronic kidney disease) The GFR at this stage is less than 5% of normal and results in the complex clinical picture of uraemic syndrome with progressive primary (renal) and secondary systemic (extra-renal) symptoms.
Clinical Features
- Clinical manifestations of full-blown CRF culminating in uraemic syndrome are described under 2 main headings primary (renal) uraemic manifestations and secondary (systemic or extra-renal) uraemic manifestations.
Primary uraemic (renal) manifestations: Primary symptoms of uraemia develop when there is a slow and progressive deterioration of renal function. The resulting imbalances cause the following manifestations
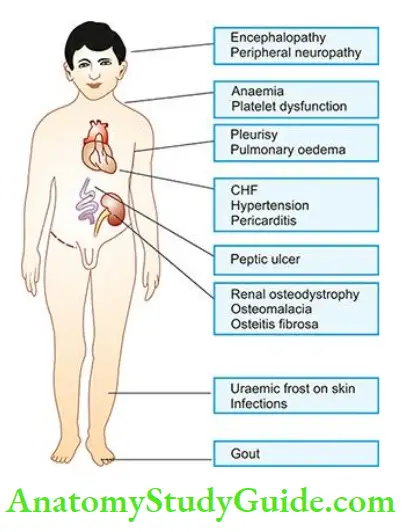
- Metabolic acidosis: As a result of renal dysfunction, the acid-base balance is progressively lost. Excess of hydrogen ions occurs, while bicarbonate level declines in the blood, resulting in metabolic acidosis.
- The clinical symptoms of metabolic acidosis include compensatory Kussmaul breathing, hyperkalaemia and hypercalcaemia.
- Hyperkalaemia: A decreased GFR results in excessive accumulation of potassium in the blood since potassium is normally excreted mainly in the urine.
- Hyperkalaemia is further worsened by metabolic acidosis. The clinical features of hyperkalaemia are cardiac arrhythmias, weakness, nausea, intestinal colic, diarrhoea, muscular irritability and flaccid paralysis.
- Sodium and water imbalance: As GFR declines, sodium and water cannot pass sufficiently into Bowman’s capsule leading to their retention.
- The release of renin from the juxtaglomerular apparatus further aggravates sodium and water retention.
- The main symptoms referable to sodium and water retention are hypervolaemia and circulatory overload with congestive heart failure.
- Hyperuricaemia: Decreased GFR results in excessive accumulation of uric acid in the blood. Uric acid crystals may be deposited in joints and soft tissues resulting in gout.
- 5. Azotaemia: The waste products of protein metabolism fail to be excreted resulting in elevation in the blood levels of urea, creatinine, phenols and guanidines causing biochemical abnormality, azotaemia.
- The secondary manifestations of uraemia are related to the toxic effects of these metabolic waste products.
Secondary uraemic (extra-renal) manifestations: A number of extra-renal systemic manifestations develop secondarily following fluid-electrolyte and acid-base imbalances. These include the following:
- Anaemia: Decreased production of erythropoietin by diseased kidneys results in a decline in erythropoiesis and anaemia. Besides, gastrointestinal bleeding may further aggravate anaemia.
- Integumentary system: Deposit of urinary pigment such as urochrome in the skin causes sallow-yellow colour. The urea content in the sweat as well as in the plasma rises.
- On evaporation of the perspiration, urea remains on the facial skin as powdery ‘uraemic frost’.
- Cardiovascular system: Fluid retention secondarily causes cardiovascular symptoms such as increased workload on the heart due to hypervolaemia and eventually congestive heart failure.
- Respiratory system: Hypervolaemia and heart failure cause pulmonary congestion and pulmonary oedema due to back pressure.
- Radiologically, uraemic pneumonitis shows a characteristic central, butterfly pattern of oedema and congestion in the chest radiograph.
- Digestive system: Azotaemia directly induces mucosal ulcerations in the lining of the stomach and intestines. Subsequent bleeding can aggravate the existing anaemia.
- Gastrointestinal irritation may cause nausea, vomiting and diarrhoea.
- Skeletal system: The skeletal manifestations of renal failure are referred to as renal osteodystrophy.
Two major types of skeletal disorders may occur:
- Osteomalacia occurs from a deficiency of a form of vitamin D which is normally activated by the kidney.
- Since vitamin D is essential for the absorption of calcium, its deficiency results in inadequate deposits of calcium in bone tissue.
- Osteitis fibrosis occurs due to elevated levels of parathormone. How parathormone excess develops in CRF is complex.
- As the GFR is decreased, increasing levels of phosphates accumulate in the extracellular fluid which, in turn, causes a decline in calcium levels.
- Decreased calcium level triggers the secretion of parathormone which mobilises calcium from bone and increases renal tubular reabsorption of calcium thereby conserving it.
- However, if the process of resorption of calcium phosphate from bone continues for sufficient time, hypercalcaemia may be induced with deposits of excess calcium salts in joints and soft tissues and the weakening of bones (renal osteodystrophy).
Renal Failure
- Renal diseases, irrespective of cause, may result in acute renal failure (ARF) and chronic renal failure (CRF).
- ARF may result from prerenal, intrarenal and postrenal causes. Clinically, ARF depending upon the stage, may result in syndrome of acute nephritis, or syndrome of tubular pathology.
- CRF is characterised by progressive and irreversible deterioration of renal function due to the slow destruction of renal parenchyma, eventually terminating in death.
- CRF may be caused by diseases of glomeruli or tubulointerstitium. Clinically, CRF has uraemic manifestations. These may be primary or secondary and involve multiple organs and systems.
Congenital Malformations
Approximately 10% of all persons are born with potentially significant malformations of the urinary system.
- These range in severity from minor anomalies which may not produce clinical manifestations to major anomalies which are incompatible with extrauterine life.
- About half of all patients with malformations of the kidneys have coexistent anomalies either elsewhere in the urinary tract or in other organs.
Malformations of the kidneys are classified into 3 broad groups:
- Abnormalities in the amount of renal tissue: These include anomalies with deficient renal parenchyma (for example unilateral or bilateral renal hypoplasia) or with excess renal tissue (for example. organomegaly, supernumerary kidneys).
- Anomalies of position, form and orientation: These are renal ectopia (pelvic kidney), renal fusion (horseshoe kidney) and persistent foetal lobulation.
- Anomalies of differentiation: This group consists of the more important and common morphologic forms covered under the heading of ‘cystic diseases of the kidney’ described in detail below.
Cystic Diseases Of Kidney
Cystic lesions of the kidney may be congenital or acquired, non-neoplastic or neoplastic. The majority of these lesions are congenital non-neoplastic.
- Cystic lesions in the kidney may occur at any age, extending from foetal life (detected on ultrasonography) to old age.
- Their clinical presentation may include abdominal mass, infection, respiratory distress (due to accompanied pulmonary hypoplasia), haemorrhage, and neoplastic transformation.
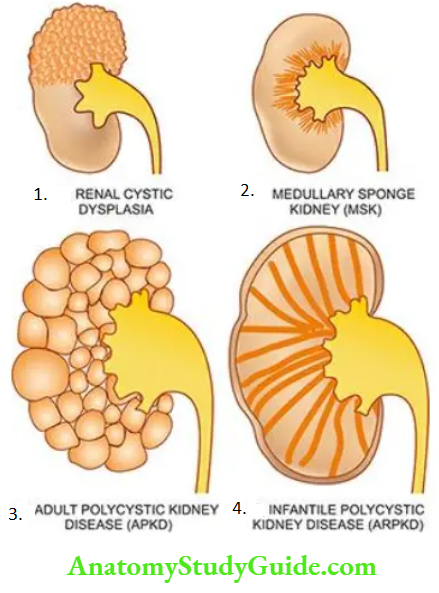
- Potter divided developmental renal cystic lesions into three types 1,2 and 3. A simple classification including all cystic lesions of the kidney is given in and illustrated.
- Non-neoplastic cysts are discussed below while neoplastic cystic lesions of the kidney are described later.
Multicystic Renal Dysplasia
- The term ‘multicystic renal dysplasia’ or Potter type II is used for disorganised meta-nephrogenic differentiation with the persistence of structures in the kidney which are not represented in normal nephrogenesis.
- Renal dysplasia is the most common form of cystic renal disease in the newborn and infants. The condition may occur sporadically or may be familial and part of a syndrome or other anomalies.
- It is commonly associated with obstructive abnormalities of the ureter and lower urinary tract such as obstruction of pelvic ureteric junction (PUJ), ureteral atresia and urethral obstruction.
Classification of cystic lesions of the kidney.
Non-Neoplastic Cystic Lesions
- Renal multicystic dysplasia (Potter type 2)
- Polycystic kidney disease (PKD)
- Adult (autosomal dominant) polycystic kidney disease (ADPKD) (Potter type 3)
- Infantile (autosomal recessive) polycystic kidney disease (ARPKD) (Potter type 1)
- Medullary cystic disease
- Medullary sponge kidney (MSK)
- The nephronophthisis-medullary cystic disease complex
- Simple renal cysts
- Acquired renal cysts (1. dialysis-associated cystic disease, 2. hydatid cyst, 3. tuberculosis, 4. renal cell carcinoma, v. traumatic intrarenal haematoma)
- Para-renal cysts (1. Pyelocalyceal, 2. hilar lymphangiectasia, 3. retroperitoneal, 4. perinephric pseudocysts from trauma)
- Neoplastic Cystic Lesions
- Cystic nephroma
- Cystic partially-differentiated nephroblastoma (CPDN)
- Multifocal cystic change in Wilms’ tumour
Morphologic Features:
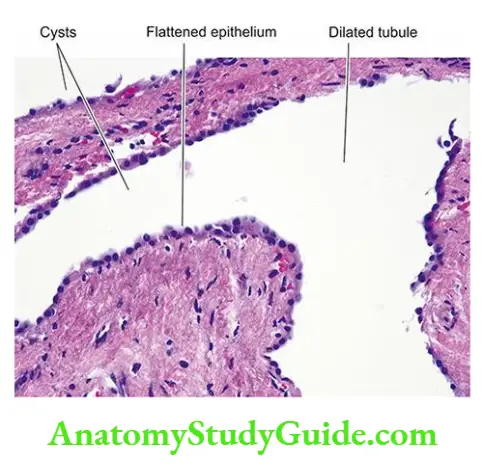
Renal dysplasia may be unilateral or bilateral. The dysplastic process may involve the entire renal mass or a part of it. Grossly, the dysplastic kidney is almost always cystic.
- The kidney or its affected part is replaced by a disorderly mass of multiple cysts resembling a bunch of grapes. Normal renal parenchyma is almost totally obscured by the mass while calyces and pelvis may not be recognised.
- The ureter is invariably abnormal, being either absent or atretic. Histologically, the characteristic feature is the presence of undifferentiated mesenchyme that contains smooth muscle, cartilage and immature collecting ducts.
- The cysts in the mass represent dilated tubules lined by flattened epithelium which are surrounded by concentric layers of connective tissue. Glomeruli and tubules are scanty, primitive or absent.
Clinical Features Unilateral renal dysplasia is frequently discovered in newborns or infants as a flank mass.
- Often, renal dysplasia is associated with other congenital malformations and syndromes such as ventricular septal defect, trachea-oesophagal fistula, lumbosacral meningomyelocele and Down’s syndrome.
- The prognosis of unilateral renal dysplasia following removal of the abnormal kidney is excellent while bilateral renal dysplasia results in death in infancy unless a renal transplant is done.

Polycystic Kidney Disease
Polycystic disease of the kidney (PKD) is a disorder in which a major portion of the renal parenchyma is converted into cysts of varying sizes. The disease occurs in two forms:
- An adult type inherited as an autosomal dominant disease; and
- An infantile type is inherited as an autosomal recessive disorder.
Adult Polycystic Kidney Disease
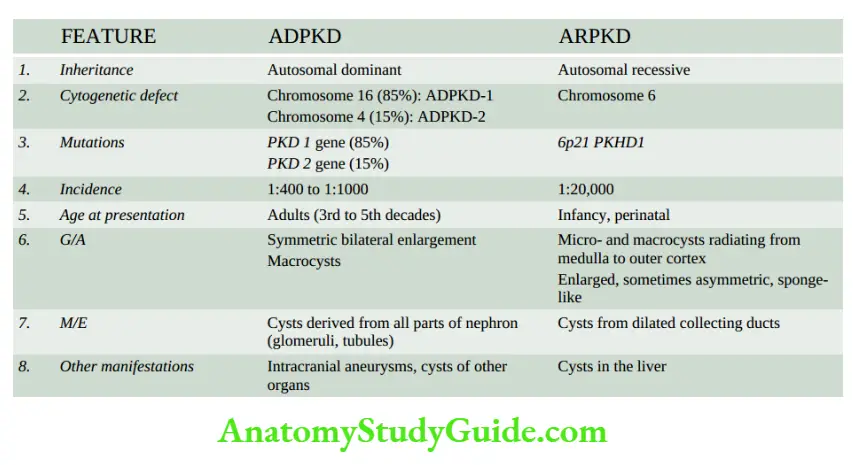
- Adult (autosomal dominant) polycystic kidney disease (ADPKD) is relatively common (incidence 1:400 to 1:1000) and is the cause of end-stage renal failure in approximately 4% of haemodialysis patients.
- The pattern of inheritance is autosomal dominant with a mutation in the PKD gene mutation in PKD-1 gene(encodes protein polycystin1) located on chromosome 16 in over 85% of cases (ADPKD-1) while the remainder 15% of cases have a mutation in the PKD-2 gene (encodes protein polycystin 2) located on chromosome 4 (ADPKD-2).
- A family history of similar renal disease may be present. True adult polycystic renal disease is always bilateral and diffuse.
- Though the kidneys are abnormal at birth, renal function is retained, and symptoms appear in adult life, mostly between the age of 30 and 50 years.
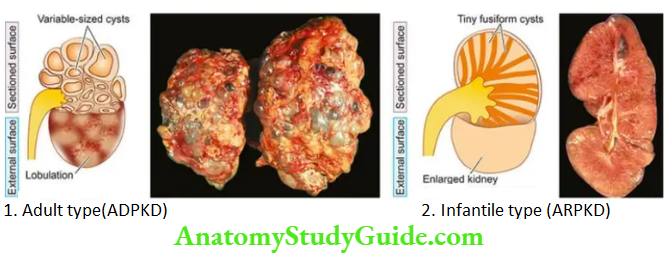
Morphologic Features Grossly, kidneys in ADPKD are always bilaterally enlarged, usually symmetrically, heavy (weighing up to 4 kg) and give a lobulated appearance on the external surface due to underlying cysts.
- The cut surface shows cysts throughout the renal parenchyma varying in size from tiny cysts to 4-5 cm in diameter. The contents of the cysts vary from clear straw-yellow fluid to reddish-brown material.
- The renal pelvis and calyces are present but are greatly distorted by the cysts and may contain concretions.
- The cysts, however, do not communicate with the pelvis of the kidney a feature that helps to distinguish polycystic kidney from hydronephrosis of the kidney on the sectioned surface.
- Histologically, the cysts arise from all parts of the nephron.
- It is possible to find some cysts containing recognisable glomerular tufts reflecting their origin from Bowman’s capsule, while others have epithelial lining like that of distal or proximal tubules or collecting ducts.
- The intervening tissue between the cysts shows some normal renal parenchyma.
- With the advancement of the age of the patient, acquired lesions such as pyelonephritis, nephrosclerosis, fibrosis and chronic inflammation are seen with increasing frequency.
Clinical Features The condition may become clinically apparent at any age but most commonly manifests in 3rd to 5th decades of life.
- The most frequent and earliest presenting feature is a dull ache in the lumbar region.
- In others, the presenting complaints are haematuria or passage of blood clots in urine, renal colic, hypertension, urinary tract infection and progressive CRF with polyuria and proteinuria.
- Adpkd is considered a systemic disease. About a third of patients with ADPKD have cysts of the liver. Other associated congenital anomalies seen less frequently are cysts in the pancreas, spleen, lungs and other organs.
- Approximately 15% of patients have one or more intracranial berry aneurysms of the circle of Willis. Any acquired renal disease is more prone to occur in polycystic kidneys.
Infantile Polycystic Kidney Disease
- The infantile (autosomal recessive) form of polycystic kidney disease (ARPKD) is distinct from the adult form and is less common (incidence 1:20,000 births).
- It is transmitted as an autosomal recessive trait and a family history of a similar disease is usually not present. The condition occurs due to a mutation in chromosome 6 6p21, PKHD1 (polycystic kidney and hepatic disease.
- It is invariably bilateral. The age at presentation may be perinatal, neonatal, infantile or juvenile, but frequently serious manifestations are present at birth and result in death from renal failure in early childhood.
Morphologic Features Grossly, the kidneys are bilaterally enlarged with a smooth external surface and retained normal reniform shape.
- The Cut surface reveals small, fusiform or cylindrical cysts radiating from the medulla and extending radially to the outer cortex. This gives the sectioned surface of the kidney a sponge-like appearance.
- No normal renal parenchyma is grossly recognised. Pelvis, calyces and ureters are normal.
Histologically, the total number of nephrons is normal.
- Since the cysts are formed from the dilatation of collecting tubules, all the collecting tubules show cylindrical or saccular dilatations and are lined by cuboidal to low columnar epithelium.
- Many of the glomeruli are also cystically dilated.
Clinical Features The clinical manifestations depend on the age of the child. In severe form, the gross bilateral cystic renal enlargement may interfere with delivery. In infancy, renal failure may manifest early.
- Almost all cases of infantile polycystic kidney disease have been associated with multiple epithelium-lined cysts in the liver or the proliferation of portal bile ductules.
- In older children, associated hepatic changes develop into what is termed congenital hepatic fibrosis which may lead to portal hypertension and splenomegaly.
- The contrasting features of the two main forms of polycystic kidney disease are presented
Medullary Cystic Disease
Cystic disease of the renal medulla has two main types:
- Medullary sponge kidney, a relatively common and innocuous condition; and
- Nephronophthisis-medullary cystic disease complex is a common cause of chronic renal failure in the juvenile age group.
Medullary Sponge Kidney
- The medullary sponge kidney consists of multiple cystic dilatations of the papillary ducts in the medulla. It has an autosomal dominant transmission.
- The condition occurs in adults and may be recognised as an incidental radiographic finding in asymptomatic cases, or the patients may complain of colicky flank pain, dysuria, haematuria and passage of sandy material in the urine.
- Renal function remains largely normal or may be mildly impaired in long-standing disease with secondary complications of infection and calculus formation.
Morphologic Features Grossly, the kidneys may be enlarged, normal or shrunken in size depending upon the extent of secondary pyelonephritis.
- On the cut surface, the characteristic feature is the presence of several, small (less than 0.5 cm diameter), cystically dilated papillary ducts, which may contain spherical calculi.
- Microscopically, the cysts are lined by tall columnar, cuboidal, transitional or squamous epithelium. The renal cortex may show secondary pyelonephritis but cortical cysts are never a component of the medullary sponge kidney.
Nephronophthisis-Medullary Cystic Disease Complex
- This form of medullary cystic disease, also called juvenile nephronophthisis or uraemic sponge kidney, is a progressive renal disease.
- It is classified into infantile, juvenile and adolescent types depending on the age at presentation, the juvenile form being the most common.
- It is the most common form of genetic cause of end-stage renal disease in children and adolescents. The condition has an autosomal recessive inheritance. Familial occurrence is common.
- The clinical manifestations are due to impaired urinary concentration consequent upon the medullary lesions
and consist of polyuria, polydipsia and enuresis. - Other features include renal osteodystrophy, growth retardation, anaemia and progressive renal failure leading to uraemia.
Morphologic Features Grossly, the kidneys are moderately reduced in size and granular and have narrow cortices. The Cut surface reveals minute cysts, the majority of which are present at the corticomedullary junction.
- Microscopically, the cysts are lined by flattened or cuboidal epithelium. There is widespread nonspecific chronic inflammatory infiltrate and interstitial fibrosis.
- Many glomeruli are hyalinised but tubular atrophy is more pronounced due to marked thickening of the tubular basement membrane.
Simple Renal Cysts
Simple renal cysts or cortical cysts are a very common postmortem finding. They are seen in about half of all persons above the age of 50 years.
- Since these cysts are rare in infants and children, they appear to be acquired rather than congenital lesions.
- Simple cysts of the kidneys are rarely responsible for symptoms.
- However, symptoms may result from rupture, haemorrhage or infection.
- The association between simple cysts and hypertension is common.
- Morphologic Features Grossly, simple renal cysts are usually solitary but may be multiple.
- They are commonly located in the cortex.
- Their size varies from a few millimetres to 10 cm in diameter.
- The wall of the cyst is characteristically yellowish-white and translucent.
- The cyst usually contains clear straw-coloured fluid which may become rust-coloured due to haemorrhage.
- Microscopically, the lining of the cyst is by flattened epithelium.
- The cyst wall contains a variable amount of collagenised fibrous tissue which may occasionally have deposits of haemosiderin or calcium salts
Cystic Diseases of Kidney
-
- Multicystic renal dysplasia is a common condition in newborns and is a disorganised meta-nephrogenic differentiation with the persistence of structures in the kidney.
- Polycystic disease of the kidney is a disorder in which a major portion of the renal parenchyma is converted into cysts of varying sizes. It has 2 types: adult and infantile
- Adult (autosomal dominant) polycystic kidney disease (ADPKD) is relatively common while the infantile (autosomal recessive) form (ARPKD) is less common.
- Cystic disease of the renal medulla has two main types: medullary sponge kidney causing small cystic dilatations of papillary ducts of the medulla and nephronophthisis medullary cystic disease complex.
- Simple renal cysts or cortical cysts are a very common postmortem finding.
Glomerular Diseases
Definition And Classification
Glomerular diseases encompass a large and clinically significant group of renal diseases. Glomerulonephritis (GN) or Bright’s disease is the term used for diseases that primarily involve renal glomeruli.
It is convenient to classify glomerular diseases into 2 broad groups:
- Primary glomerulonephritis in which the glomeruli are the predominant site of involvement.
- Secondary glomerular diseases include certain systemic and hereditary diseases which secondarily affect the glomeruli.
-
- Though this division is widely followed, it is somewhat arbitrary since many primary forms of glomerulonephritis have systemic effects, and many systemic diseases may initially present with glomerular involvement.
- Many classifications of different types of glomerulonephritis have been described, but the most widely accepted classification is based on clinical presentation and pathologic changes in the glomeruli given in.
Clinical Manifestations
- The clinical presentation of glomerular disease is quite variable but in general four features proteinuria, haematuria, hypertension and disturbed excretory function, are present in varying combinations depending upon the underlying condition.
- A firm diagnosis, however, can be established by examination of renal biopsy under light, electron and immunofluorescence microscopy.
Clinicopathologic classification of glomerular diseases.
- Primary Glomerulonephritis
-
- Acute GN
- Post-streptococcal
- Non-streptococcal
- Rapidly progressive GN
- Minimal change in disease
- Membranous GN
- Membrano-proliferative GN
- Focal and diffuse proliferative GN
- Focal segmental glomerulosclerosis (FSGS)
- IgA nephropathy
- Chronic glomerulonephritis
- C3 glomerulopathy
- Acute GN
- Secondary Systemic Glomerular Diseases
-
- Lupus nephritis (SLE)
- Diabetic nephropathy
- Amyloidosis
- Polyarteritis nodosa
- Wegener’s granulomatosis
- Goodpasture syndrome
- Henoch-Schönlein purpura
- Systemic infectious diseases (bacterial example bacterial endocarditis, syphilis, leprosy; viral e.g. HBV, HCV, HIV; parasitic example falciparum malaria, filariasis)
- Idiopathic mixed cryoglobulinaemia
- Hereditary Nephritis
-
- Alport’s syndrome
- Fabry’s disease
- Nail-patella syndrome
- (GN, glomerulonephritis; SLE, systemic lupus erythematosus; HBV, hepatitis B virus; HCV, hepatitis C virus)
A number of clinical syndromes are recognised in glomerular diseases. The following six major glomerular syndromes are commonly found in different glomerular diseases
- nephritic and nephrotic syndromes;
- acute and chronic renal failure;
- asymptomatic proteinuria and haematuria.
These are briefly described below.
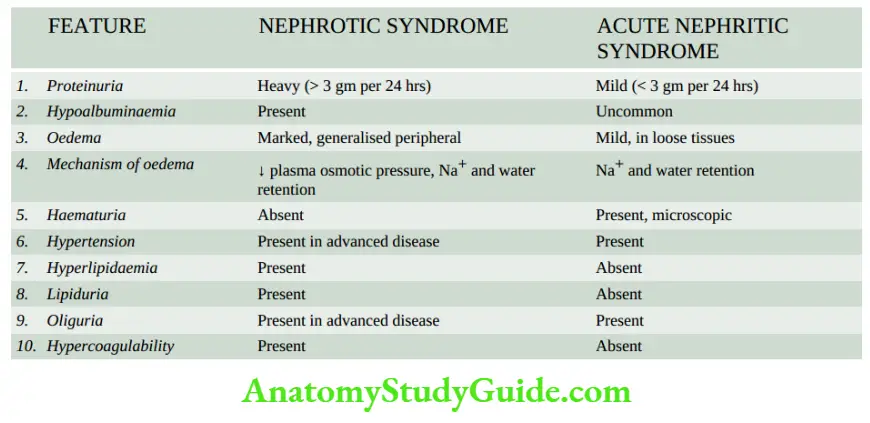
Acute Nephritic Syndrome: This is the acute onset of microscopic haematuria, mild proteinuria, hypertension, oedema and oliguria following an infective illness about 10 to 20 days earlier.
- Haematuria is generally slight giving the urine a smoky appearance and erythrocytes are detectable by microscopy or by chemical testing for haemoglobin. The appearance of red cell casts is another classical feature of acute nephritic syndrome.
- Proteinuria is mild (less than 3 gm per 24 hrs) and is usually non-selective (nephritic range proteinuria).
- Hypertension is variable depending upon the severity of the glomerular disease but is generally mild.
- Oedema in nephritic syndrome is usually mild and results from sodium and water retention.
- Oliguria is variable and reflects the severity of glomerular involvement.
The underlying causes of an acute nephritic syndrome may be primary glomerulonephritic diseases (classically acute glomerulonephritis and rapidly progressive glomerulonephritis) or certain systemic diseases.
Nephrotic Syndrome is a constellation of features in different diseases having varying pathogenesis; it is characterised by findings of massive proteinuria, hypoalbuminaemia, oedema, hyperlipidaemia, lipid, and hypercoagulability.
- Heavy proteinuria (protein loss of more than 3 gm per 24 hrs) is the chief characteristic of nephrotic syndrome (nephrotic range proteinuria).
- In children, protein loss is correspondingly less. A small amount of protein (20 to 150 mg/day) normally passes through the glomerular filtration barrier and is reabsorbed by the tubules.
- But in case of increased glomerular permeability to plasma proteins, excess protein is filtered out exceeding the capacity of tubules for reabsorption and, therefore, appears in the urine.
- Another feature of protein loss is its ‘selectivity’. Highly-selective proteinuria consists mostly of the loss of low molecular weight proteins, while poorly-selective proteinuria is the loss of high molecular weight proteins in the urine.
- In nephrotic syndrome, proteinuria mostly consists of loss of albumin (molecular weight 66,000) in the urine
- Hypoalbuminaemia: is produced primarily consequent to urinary loss of albumin, and partly due to increased renal catabolism and inadequate hepatic synthesis of albumin.
- Often, the plasma albumin level is 1 to 3 gm/dl (normal 3.5 to 5.5 gm/dl) and there is reversed albumin-globulin ratio.
- The concentration of other proteins in the plasma such as immunoglobulins, clotting factors and antithrombin may fall rendering these patients more vulnerable to infections and thrombotic and thromboembolic complications.
- Oedema: nephrotic syndrome appears due to a fall in colloid osmotic pressure consequent upon hypoalbuminaemia.
- Sodium and water retention further contribute to oedema. Nephrotic oedema is usually peripheral but in children, facial oedema may be more prominent.
- Hyperlipidaemia: is a frequent accompaniment of nephrotic syndrome. The exact mechanism of its genesis is not clear.
- It is hypothesised that the liver faced with the stress of massive protein synthesis in response to heavy urinary protein loss, also causes increased synthesis of lipoproteins.
- There are increased blood levels of total lipids, cholesterol, triglycerides, VLDL and LDL but a decrease in HDL. A low blood level of HDL is partly due to its loss in the urine.
- Lipiduria: occurs following hyperlipidaemia due to excessive leakiness of the glomerular filtration barrier.
- Hypercoagulability: Patients with nephrotic syndrome may develop spontaneous arterial or venous thrombosis, renal vein thrombosis and pulmonary embolism due to various factors.
- These include increased urinary loss of antithrombin, hyperfibrinogenaemia from the increased synthesis in the liver, decreased fibrinolysis, increased platelet aggregation and altered levels of protein C and S.
- The causes of nephrotic syndrome are diverse and are listed in The morphology of individual types is described later. But it must be mentioned here that.
- In children, primary glomerulonephritis is the cause in the majority of cases of nephrotic syndrome; the most frequent being lipoid nephrosis (65%).
- In adults, on the other hand, systemic diseases (diabetes, amyloidosis and SLE) are more frequent causes of nephrotic syndrome.
- The most common primary glomerular disease in adults is membranous glomerulonephritis (40%).
- Features of the nephrotic and acute nephritic syndrome have been contrasted.
Causes of acute nephritic syndrome.
- Primary Glomerulonephritis
-
- Acute GN
- Post-streptococcal
- Non-streptococcal
- Rapidly progressive GN
- Membranoproliferative GN
- Focal and diffuse proliferative GN
- IgA nephropathy
- Acute GN
- Systemic Diseases
- Sale
- Polyarteritis nodosa
- Wegener’s granulomatosis
- Henoch-SchÖnlein purpura
- Cryoglobulinaemia
(GN, glomerulonephritis; SLE, systemic lupus erythematosus)
Acute Renal Failure: As already described above, acute renal failure (ARF) is characterised by a rapid decline in renal function.
- ARF has many causes including glomerular disease, principally rapidly progressive GN and acute diffuse proliferative GN.
Chronic Renal Failure: Glomerular causes of chronic renal failure (CRF) have already been described.
- These cases have advanced renal impairment progressing over the years and are detected by significant proteinuria, haematuria, hypertension and azotaemia.
- Such patients generally have small contracted kidneys due to chronic glomerulonephritis.
Asymptomatic Proteinuria: The presence of proteinuria unexpectedly in a patient may be unrelated to renal disease (for example exercise-induced, extreme lordosis and orthostatic proteinuria), or may indicate an underlying mild glomerulonephritis.
- Association of asymptomatic haematuria, hypertension or impaired renal function with asymptomatic proteinuria should raise a strong suspicion of underlying glomerulonephritis.
Causes of nephrotic syndrome.
- Primary Glomerulonephritis
- Minimal change disease (most common in children)
- Membranous GN (most common in adults)
- Membranoproliferative GN
- Focal segmental glomerulosclerosis
- Focal and diffuse proliferative GN
- IgA nephropathy
- Systemic Diseases
- Diabetes mellitus
- Amyloidosis
- Sie
- Systemic Infections
- Viral infections (HBV, HCV, HIV)
- Bacterial infections (bacterial endocarditis, syphilis, leprosy)
- Protozoa and parasites (P. falciparum malaria, filariasis)
- Hypersensitivity Reactions
- Drugs (heavy metal compounds like gold and mercury, other drugs like penicillamine, trimethadione and tolbutamide, heroin addiction)
- Bee stings, snake bites, poison ivy
- Malignancy
- Carcinomas
- Myeloma
- Hodgkin’s disease
- Pregnancy
- Toxaemia of pregnancy
- Circulatory Disturbances
- Renal vein thrombosis
- Constrictive pericarditis
- Hereditary Diseases
- Alport’s disease
- Fabry’s disease
- Nail-patella syndrome
- (GN, glomerulonephritis; SLE, systemic lupus erythematous; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus).
Asymptomatic Haematuria: Asymptomatic microscopic haematuria is common in children and young adolescents and has many diverse causes such as diseases of the glomerulus, renal interstitium, calyceal system, ureter, bladder, prostate, urethra, and underlying bleeding disorder, congenital abnormalities of the kidneys or neoplasia.
- Glomerular haematuria is indicated by the presence of red blood cells, red cell casts and haemoglobin in the urine.
- Glomerular haematuria is frequently associated with asymptomatic proteinuria.
Glomerular Diseases Classification and Clinical Manifestations
- Glomerular diseases are defined as conditions which primarily involve glomeruli.
- Glomerular diseases are classified into primary (glomerulonephritis of various types) and secondary (systemic diseases with secondary involvement of glomeruli).
- Clinically, various glomerular diseases fall into one or more of 6 clinical syndromes nephritic and nephrotic syndromes; acute and chronic renal failure; asymptomatic proteinuria and haematuria.
- Acute nephritic syndrome is the acute onset of microscopic haematuria, mild proteinuria, hypertension, oedema and oliguria following an infective illness about 10 to 20 days earlier.
- Nephrotic syndrome is characterised by findings of massive proteinuria, hypoalbuminaemia, oedema, hyperlipidaemia, lipid, and hypercoagulability.
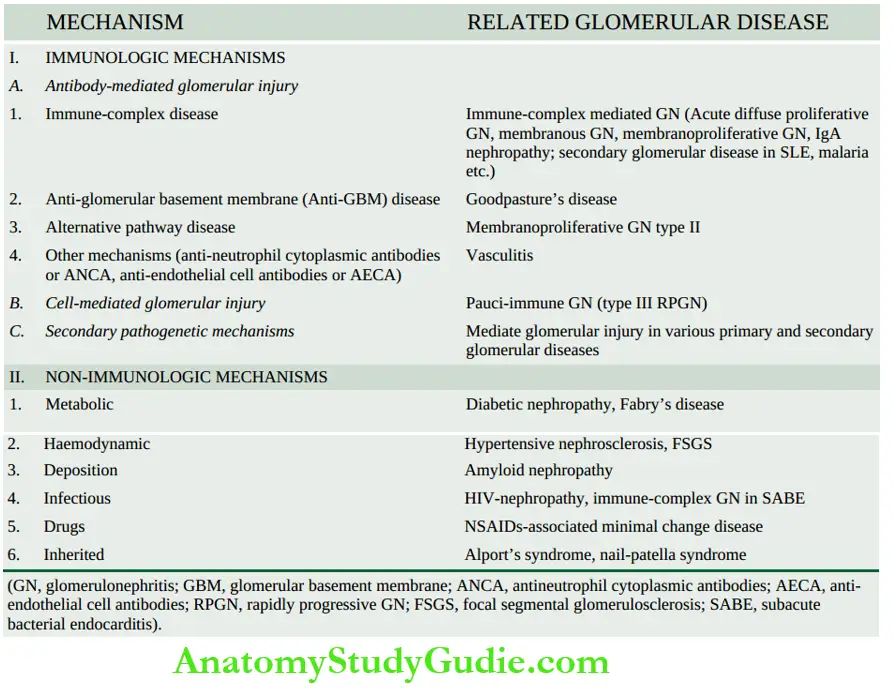
Pathogenesis Of Glomerular Injury
Most forms of primary GN and many of the secondary glomerular diseases in human beings have immunologic pathogenesis.
- This view is largely based on immunofluorescence (IF) studies of GN in humans which have revealed glomerular deposits of immunoglobulins and complement in patterns that closely resemble those of experimental models.
- The consequences of injury at different sites within the glomerulus in various glomerular diseases can be assessed when compared with the normal physiologic role of the main cells involved.
- endothelial, mesangial, visceral epithelial, and parietal epithelial cells as well as of the GBM as summed up in.
- Immunologic mechanisms underlying glomerular injury are primarily antibody-mediated (immune-complex disease).
- There is evidence to suggest that cell-mediated immune reactions in the form of delayed-type hypersensitivity can also cause glomerular injury in some conditions.
- In addition, a few secondary mechanisms and some non-immunologic mechanisms are involved in the pathogenesis of some forms of glomerular diseases in human beings.
Immunologic Mechanisms
Experimental studies and observations in humans have revealed that immunologic mechanisms, most importantly antigen-antibody complexes, underlie most forms of glomerular injury.
- The general principles of these mechanisms in different forms of glomerular diseases are discussed here, while specific features pertaining to individual types of GN are described separately later.
Antibody-Mediated Glomerular Injury
- Immune Complex Disease Majority of cases of glomerular disease result from deposits of immune complexes (antigen-antibody complexes).
- The immune complexes are represented by irregular or granular glomerular deposits of immunoglobulins (IgG, IgM and IgA) and complement (mainly C3).
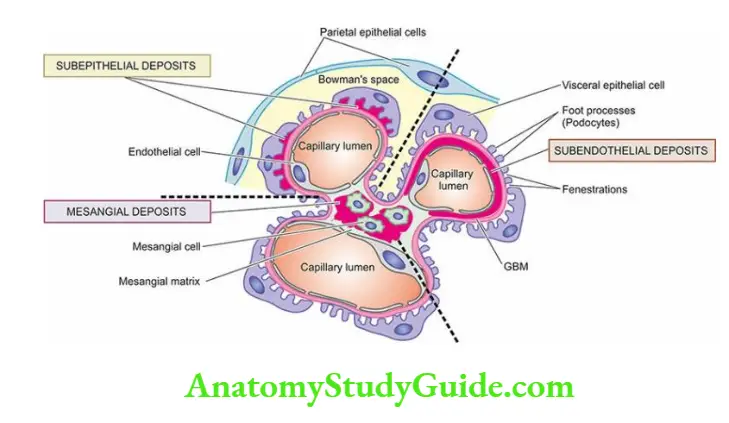
- Based on the experimental models and studies in human beings, the following 3 patterns of glomerular deposits of immune complexes in various glomerular diseases have been observed as illustrated in
-
- Exclusive mesangial deposits are characterised by a very mild form of glomerular disease.
- Extensive subendothelial deposits along the GBM are accompanied by severe hypercellular sclerosing glomerular lesions.
- Subepithelial deposits are seen between the outer surface of the GBM and the podocytes.
- Deposits may be located at one or more of the above sites in any case of glomerular injury.
- It was widely believed earlier that glomerular deposits result from circulating immune complexes. Now, it has been shown that glomerular deposits are formed by one of the following two mechanisms
- Local immune complex deposits: Classic experimental model to understand human in situ immune complex GN is Heymann nephritis.
- In this, rats were injected with homologous kidney homogenates that resulted in a chronic glomerular disease manifested by heavy proteinuria.
- It was due to autoimmunity induced by antibodies to intrinsic non-glomerular antigens resulting in the formation of in situ immune deposits on a glomerular basement membrane.
- The intrinsic antigen in the experiment was found in epithelial brush borders of proximal convoluted tubules and has been named megalin.
-
- A similar phenomenon is seen in human membranous glomerulonephritis. In humans, the corresponding autologous non-basement membrane antigen is identified as gp330 (glycoprotein with a mass of 330 kD) or non-glomerular antigens planted on glomeruli (for example certain drugs, endotoxins, parasitic products etc).
- It is located on the podocytes and coated on pits of proximal tubular epithelial cells. Antibodies are formed against such planted antigens.
- The main antigen-antibody reaction takes place at the soles of the foot processes of podocytes and the immune complexes get deposited at the lamina rar externa of the basement membrane.
- Correspondingly, in membranous glomerulonephritis, granular IgG deposits are found along the subepithelial side of the basement membrane.
- Similarly, electron-dense deposits are found on the epithelial side of the basement membrane.
- Currently, this mechanism is considered responsible for most cases of immune complex GN.
- Circulating immune complex deposits: This mechanism used to be considered important for glomerular injury earlier but now it is believed that circulating immune complexes cause glomerular damage under certain circumstances only.
-
- These situations are their presence in high concentrations for prolonged periods, or when they possess special properties that cause their binding to glomeruli, or when host mechanisms are defective and fail to eliminate immune complexes.
- The antigens evoking antibody response may be endogenous (for example in SLE) or may be exogenous (for example Hepatitis B virus, Treponema pallidum, Plasmodium falciparum and various tumour antigens).
- The antigen-antibody complexes are formed in the circulation and then trapped in the glomeruli where they produce glomerular injury after combining with complement.
-
-
-
- Immune complex GN by either of the above mechanisms is observed in the following human diseases:
- Primary GN examples acute diffuse proliferative GN, membranous GN, membranoproliferative GN, IgA nephropathy and some cases of rapidly progressive GN and focal GN.
- Systemic diseases example glomerular disease in SLE, malaria, syphilis, hepatitis, HenochSchÖnlein purpura and idiopathic mixed cryoglobulinaemia.
-
- Anti-Gbm Disease: Less than 5% of cases of human GN are associated with anti-GBM antibodies. The constituent of GBM acting as an antigen appears to be a component of collagen IV of the basement membrane.
- The experimental model of anti-GBM disease is Masugi nephritis (nephrotoxic serum nephritis) produced in rats by injection of heterologous antibodies against GBM prepared in rabbits by immunisation with rat kidney glomerular tissue.
- Anti-GBM disease is classically characterised by interrupted linear deposits of anti-GBM antibodies (mostly IgG; rarely IgA and IgM) and complement (mainly C3) along the glomerular basement membrane.
- These deposits are detected by immunofluorescence microscopy or by electron microscopy.
- Anti-GBM disease is characteristically exemplified by glomerular injury in Goodpasture syndrome in some cases of rapidly progressive GN.
- About half to two-thirds of the patients with renal lesions in Goodpasture’s syndrome have pulmonary haemorrhage mediated by crossreacting autoantibodies against the alveolar basement membrane.
- Alternative Pathway Disease: As apparent from the above mechanisms, the complement system, in particular C3, contributes to glomerular injury in most forms of GN.
- Deposits of C3 are associated with the early components C1, C2 and C4 which are evidence of classic pathway activation of complement.
- But in alternative pathway activation, there is decreased serum C3 level, decreased serum levels of factor B and properdin, and normal serum levels of C1, C2 and C4 but C3 and properdin are found deposited in the glomeruli without immunoglobulin deposits, reflecting activation of the alternative pathway of complement.
- Such patients have circulating anti-complementary nephritic factor (C3NeF) which is an IgG antibody and acts as an autoantibody to the alternate C3 convertase, leading to persistent alternate pathway activation.
- C3 glomerulopathy is the term used for conditions where there are exclusive deposits of C3 (without immunoglobulin) or C3 is two times more intense than others on IF.
- It includes C3 glomerulonephritis and dense deposit disease.
- The deposits in alternative pathway disease are characteristically electron-dense under electron microscopy; glomerular lesions in such cases are referred to as dense-deposit disease when deposits are intramembranous, while in C3GN the deposits are subendothelial and mesangial.
- Alternative pathway disease occurs in most cases of type II membranoproliferative GN, some patients of rapidly progressive GN, acute diffuse proliferative GN, IgA nephropathy and in SLE.
- Other Mechanisms Of Antibody-Mediated Injury: A few autoantibodies have been implicated in some patients of focal segmental glomerulosclerosis and a few other types of GN. These antibodies include the following:
-
- Anti-neutrophil cytoplasmic antibodies (ANCA) About 40% of cases of rapidly progressive GN are deficient in immunoglobulins in glomeruli (pause-immune GN) and are positive for ANCA against neutrophil cytoplasmic antigens in their circulation.
- ANCA causes endothelial injury by the generation of reactive oxygen radicals. ANCA-mediated vasculitis is also seen in Wegener’s granulomatosis and Churg-Strauss syndrome.
- Anti-endothelial cell antibodies (AECA) Autoantibodies against endothelial antigens have been detected in circulation in several inflammatory vasculitis and glomerulonephritis.
- These antibodies increase the adhesiveness of leucocytes to endothelial cells.
- Anti-neutrophil cytoplasmic antibodies (ANCA) About 40% of cases of rapidly progressive GN are deficient in immunoglobulins in glomeruli (pause-immune GN) and are positive for ANCA against neutrophil cytoplasmic antigens in their circulation.
Cell-mediated Glomerular Injury (Delayed-type Hypersensitivity):
- There is evidence to suggest that cell-mediated immune reactions may be involved in causing glomerular injury, particularly in cases with deficient immunoglobulins (for example in pauci-immune type glomerulonephritis in RPGN).
- Cytokines and other mediators released by activated T-cells stimulate cytotoxicity, recruitment of more leucocytes and fibrogenesis.
- CD4+ T lymphocytes recruit more macrophages while CD8+ cytotoxic T lymphocytes and natural killer cells cause further glomerular cell injury by antibody-dependent cell toxicity.
- Soluble factor derived from T lymphocytes is implicated in proteinuria in minimal change disease and focal GS. However, cell-mediated injury is yet less clear than antibody-mediated glomerular injury.
Secondary Pathogenetic Mechanisms (Mediators of Immunologic Injury):
Secondary pathogenetic mechanisms are a number of mediators of immunologic glomerular injury operating in humans and in experimental models. These include the following:
- Neutrophils: Neutrophils are conspicuous in certain forms of glomerular disease such as in acute diffuse proliferative GN, and may also be present in membranoproliferative GN and lupus nephritis.
- Neutrophils can mediate glomerular injury by activation of complement as well as by the release of proteases, arachidonic acid metabolites and oxygen-derived free radicals.
- These agents cause degradation of GBM and cell injury.
- Mononuclear Phagocytes: Many forms of human and experimental proliferative GN are associated with glomerular infiltration by monocytes and macrophages.
- Accumulation of mononuclear phagocytes is considered an important constituent of hypercellularity in these forms of GN aside from the proliferation of mesangial and endothelial cells.
- Activated macrophages release a variety of biologically active substances which take part in glomerular injury.
- Complement System: The pathogenetic role of classical and alternate pathways of activation of complement has already been highlighted above.
- Besides the components of complement which mediate glomerular injury via neutrophils already mentioned, C5bC6789 (MAC, an acronym for membrane attack complex, also called terminal complex) is capable of inducing damage to GBM directly.
- Platelets: Platelet aggregation and release of mediators play a role in the evolution of some forms of GN. Increased intrarenal platelet consumption has been found to occur in some forms of glomerular disease.
- Mesangial Cells: There is evidence to suggest that mesangial cells present in the glomeruli may be stimulated to produce mediators of inflammation and take part in glomerular injury.
- Coagulation System: The presence of fibrin in early crescents in certain forms of human and experimental GN suggests the role of the coagulation system in glomerular damage.
- Fibrinogen may leak into Bowman’s space and act as a stimulus for cell proliferation.
- Crescents usually transform into scar tissue under the influence of fibronectin which is regularly present in crescents in human glomerular disease.
Non-Immunologic Mechanisms
Though most forms of GN are mediated by immunologic mechanisms, a few examples of glomerular injury by non-immunologic mechanisms are found
- Metabolic glomerular injury example. in diabetic nephropathy (due to hyperglycaemia), and Fabry’s disease (due to sulfatidosis).
- Haemodynamic glomerular injury example systemic hypertension, and intraglomerular hypertension in focal segmental glomerulosclerosis (FSGS).
- Deposition diseases example amyloidosis.
- Infectious diseases example-HBV, HCV, HIV, E. coli-derived nephrotoxin
- Drugs example minimal change disease due to NSAIDs.
- Inherited glomerular diseases example Alport’s syndrome, and nail-patella syndrome.
-
- The evolution of end-stage renal failure in glomerular injury is explained on the basis of adaptive glomerular hypertrophy of unaffected glomeruli that results in increased glomerular blood flow and increased glomerular capillary pressure inducing intraglomerular hypertension.
- These events lead to increased deposition of mesangial matrix and proliferation of mesangial cells, endothelial and epithelial cell injury, and eventually to progressive glomerulosclerosis and end-stage renal failure.
Pathogenesis of Glomerular Injury
- Most forms of glomerular diseases in human beings have immunologic pathogenesis— antibody-mediated or cell-mediated.
- Antibody-mediated glomerular injury is often an immune complex disease, either local (in situ) or circulating deposits. Anti-GBM disease and alternate pathway activation of complement are the other antibody mechanisms. A few autoantibodies (ANCA, AECA) are also implicated in certain glomerular diseases with vasculitis.
- Cell-mediated immune reactions by delayed-type hypersensitivity can also cause glomerular injury.
- Secondary mechanisms involve the role of neutrophils, monocytes, complement, platelets, mesangial cells and activation of the coagulation system.
- Non-immunologic mechanisms explain glomerular injury in certain forms of metabolic, infiltrative and inherited glomerular diseases.
Specific Types Of Primary Glomerulonephritis
Classification of different forms of glomerular diseases into primary and secondary is already presented.
- Features of individual types are described below and a summary of major forms of primary glomerulonephritis is given at the end of this discussion.
Acute Glomerulonephritis (Synonyms: Acute Diffuse Proliferative Gn, Diffuse Endocapillary Gn)
- Acute GN is known to follow acute infection and characteristically presents as acute nephritic syndrome.
- Based on an etiologic agent, acute GN is subdivided into 2 main groups acute post-streptococcal GN and acute non-streptococcal GN, the former being more common.
Acute Post-streptococcal GN
- Acute post-streptococcal GN, though uncommon and sporadic in Western countries, is a common form of GN in developing countries, mostly affecting children between 2 to 14 years of age but 10% of cases are seen in adults above 40 years of age.
- The onset of the disease is generally sudden after 1-2 weeks of streptococcal infection, most frequently of the throat (for example streptococcal pharyngitis) and sometimes of the skin (for example streptococcal impetigo).
Etiopathogenesis The relationship between streptococcal infection and this form of GN is now well established.
- Particularly nephritogenic are types 12, 4, 1 and Red Lake of group A β- haemolytic streptococci (compare the etiologic agent with that of RHD, ).
The glomerular lesions appear to result from the deposition of immune complexes in the glomeruli. The evidence cited in support is as under:
-
- There is epidemiological evidence of preceding streptococcal sore throat or skin infection about 1-2 weeks prior to the attack.
- The latent period between streptococcal infection and the onset of clinical manifestations of the disease is compatible with the period required for the building up of antibodies.
- Streptococcal infection may be identified by culture or may be inferred from elevated titres of antibodies against streptococcal antigens. These include the following:
-
-
- anti-streptolysin O (ASO),
- anti-deoxyribonuclease B (anti-DNAse B),
- anti-streptokinase (ASKase),
- anti-nicotine adenine dinucleotides (anti-NADase) and
- anti-hyaluronidase (AHase).
-
-
-
-
- There is usually hypocomplementaemia indicating involvement of complement in the glomerular deposits.
- It has also been possible to identify the antigenic component of streptococci which is the cytoplasmic antigen, endostreptosin.
-
-
Morphologic Features Grossly, the kidneys are symmetrically enlarged, weighing one and a half to twice the normal weight.
- The cortical, as well as sectioned surface, show petechial haemorrhages giving the characteristic appearance of a flea-bitten kidney.
Light microscopic findings are as under:
- Glomeruli: The glomeruli are affected diffusely. They are enlarged and hypercellular.
- The diffuse hypercellularity of the tuft is due to the proliferation of mesangial, endothelial and occasionally epithelial cells (acute proliferative lesions) as well as by infiltration of leucocytes, chiefly polymorphs and sometimes monocytes (acute exudative lesions).
- There may be small deposits of fibrin within the capillary lumina and in the mesangium.
- Tubules: Tubular changes are not very striking.
- There may be swelling and hyaline droplets in tubular cells, and tubular lumina may contain red cell casts.
- Interstitium: There may be some degree of interstitial oedema and leucocytic infiltration.
- Vessels: Changes in arteries and arterioles are seldom present in acute GN.
- Electron microscopic findings, besides confirming the light microscopic findings, demonstrate the characteristic electron-dense irregular deposits (‘humps’) on the epithelial side of the GBM.
- These deposits represent the immune complexes.
- Immunofluorescence microscopy: reveals that the irregular granular deposits (lumpy bumpy) along the GBM consist principally of IgG and complement C3.
Clinical Features Typically, the patient is a young child, presenting with acute nephritic syndrome, having sudden and abrupt onset following an episode of sore throat or skin infection 1-2 weeks prior to the development of symptoms.
- The features include microscopic or intermittent haematuria, red cell casts, mild non-selective proteinuria (less than 3 gm per 24 hrs), hypertension, periorbital oedema and variably oliguria.
- Less often, the presentation may be as nephrotic syndrome. In adults, the features are atypical and include sudden hypertension, oedema and azotaemia.
- The development of hypertension, in either case, is a poor prognostic sign.
- The prognosis varies with the age of the patient.
- Children almost always (95%) recover completely with the reversal of proliferative glomerular changes.
- Complications arise more often in adults and occasionally in children.
- These include the development of rapidly progressive GN, chronic GN, uraemia and chronic renal failure.
Acute Non-streptococcal GN
- About one-third of cases of acute GN are caused by organisms other than haemolytic streptococci.
- These include other bacteria (for example staphylococci, pneumococci, meningococci, Salmonella and Pseudomonas), viruses (for example hepatitis B virus, mumps, infectious mononucleosis and varicella), parasitic infections (for example malaria, toxoplasmosis and schistosomiasis) and syphilis.
- The appearance of renal biopsy by light microscopy, EM and immunofluorescence microscopy is similar to that seen in acute post-streptococcal GN.
- The prognosis of non-streptococcal GN is not as good as that of streptococcal GN.
Rapidly Progressive Glomerulonephritis(Synonyms: Rpgn, Crescentic Gn, Extracapillary Gn)
- Rapidly progressive glomerulonephritis (RPGN) presents with an acute reduction in renal function resulting in acute renal failure in a few weeks or months.
- It is characterised by the formation of ‘crescents’ (crescentic GN) outside the glomerular capillaries (extra capillary GN).
- Crescents are formed from the proliferation of parietal epithelial cells lining Bowman’s capsule with contribution from visceral epithelial cells and the invading mononuclear cells.
- The stimulus for crescent formation appears to be the presence of fibrin in the capsular space.
- RPGN occurs most frequently in adults, with a slight male preponderance.
- The prognosis of RPGN in general is dismal.
Etiopathogenesis A number of primary glomerular and systemic diseases are characterised by the formation of crescents. Based on the etiologic agents and pathogenetic
- The mechanism, patients with RPGN are divided into 3 groups:
- Type 1 RPGN: anti-GBM disease (intense linear deposits of IgG along the GBM).
- Type 2 RPGN: immune complex disease (diffuse granular deposits and discrete electron-dense deposits).
- Type 3 RPGN: Pauci-immune RPGN (no or only insignificant deposits).
The following three serologic markers are used for categorising RPGN:
-
- Serum C3 level,
- An anti-GBM antibody, and
- Anti-neutrophil cytoplasmic antibody (ANCA).
Type 1 RPGN: Anti-GBM disease: A number of systemic diseases such as Goodpasture’s syndrome, SLE, vasculitis, Wegener’s granulomatosis, Henoch-Schonlein purpura and idiopathic mixed cryoglobulinaemia are associated with crescentic GN.
Goodpasture’s syndrome is the characteristic example of anti-GBM disease and is described below Goodpasture’s syndrome is characterised by acute renal failure due to RPGN and pulmonary haemorrhages.
- The condition is more common in males in 3rd decade of life.
- The disease results from damage to the glomeruli by anti-GBM antibodies which cross-react with the alveolar basement membrane and hence, produce renal as well as pulmonary lesions (type 2 hypersensitivity reaction).
- The evidence in support is the characteristic linear deposits of anti-GBM antibodies consisting of IgG and complement along the GBM, detection of circulating anti-GBM antibodies and induction of glomerular lesions with the injection of anti-GBM antibodies experimentally in monkeys.
- Pulmonary lesions can be experimentally induced if the lungs are previously injured by a viral or bacterial infection or exposed to hydrocarbons. The Goodpasture’s antigen appears to be a component of collagen type 4.
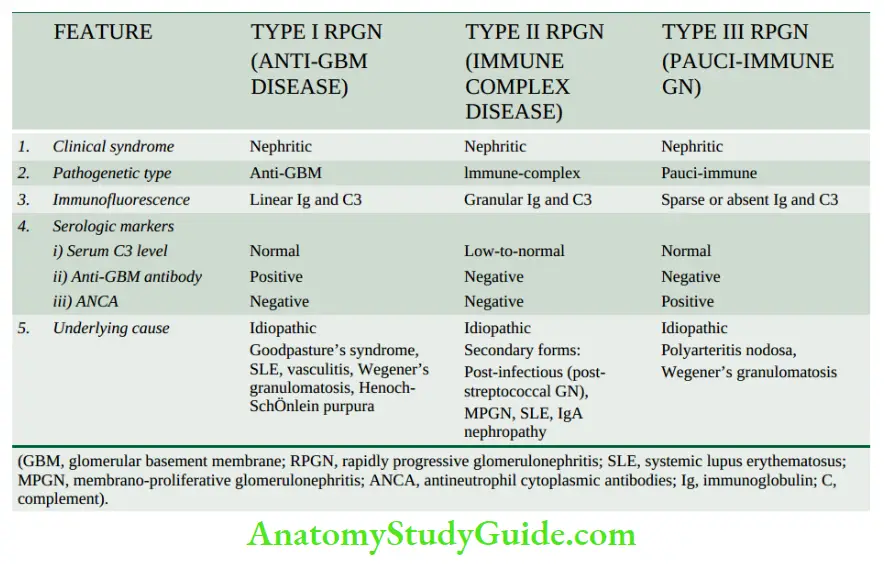
Type 2 RPGN: Immune complex disease: A small proportion of cases of post-streptococcal GN, particularly in adults and sometimes of non-streptococcal origin, develop RPGN.
- The shreds of evidence in support of post-infectious RPGN having immune complex pathogenesis are diffuse granular deposits and discrete electron-dense deposits of immune complexes of IgG and C3 along the glomerular capillary walls, lowering of blood complement level and demonstration of circulating complexes.
- Besides, MPGN, SLE, and IgA nephropathy can also produce this pattern.
Type 3 RPGN: Pauci-immune GN: These include cases of Wegener’s granulomatosis and microscopic polyarteritis nodosa. The pathogenesis of pauci-immune GN is yet not fully defined.
- However, the majority of these patients are ANCA-positive, implying a defect in humoral immunity.
- Serum complement levels are normal and the anti-GBM antibody is negative.
- There is little or no glomerular immune deposit (pauci-immune).
Morphologic Features: Grossly, the kidneys are usually enlarged and pale with a smooth outer surface (large white kidney). The cut surface shows a pale cortex and congested medulla.
Light microscopic findings vary according to the cause but, in general, the following features are
present
- Glomeruli: Irrespective of the underlying aetiology, all forms of RPGN show pathognomonic ‘crescents’ on the inside of Bowman’s capsules.
- These are collections of pale staining polygonal cells which commonly tend to be elongated.
- Eventually, crescents obliterate the Bowman’s space and compress the glomerular tuft. Initially, the crescents are cellular, and progress to fibro cellular and fibrous crescents.
- Fibrin deposition is invariably present alongside crescents.
- Besides the crescents, glomerular tufts may show increased cellularity as a result of the proliferation of endothelial and mesangial cells and leucocytic infiltration. Fibrin thrombi are frequently present in the glomerular tufts.
- Tubules: Tubular epithelial cells may show hyaline droplets.
- Tubular lumina may contain casts, red blood cells and fibrin.
- Interstitium: The interstitium is oedematous and may show early fibrosis.
- Inflammatory cells, usually lymphocytes and plasma cells, are commonly distributed in the interstitial tissue.
- Vessels: Arteries and arterioles may show no change, but cases associated with hypertension usually show severe vascular changes.
- Electron microscopic findings vary according to the type of RPGN. Post-infectious RPGN cases show electron-dense subepithelial granular deposits similar to those seen in acute GN, while cases of RPGN in Goodpasture’s syndrome show characteristic linear deposits along the GBM.
-
- Immunofluorescence microscopy shows the following patterns in various types of RPGN
-
-
- The linear pattern of RPGN in Goodpasture’s syndrome (type I RPGN), containing IgG accompanied by C3 along the capillaries.
- The granular pattern of post-infectious RPGN (type II RPGN) consists of IgG and C3 along the capillary wall.
- Scanty or no deposits of immunoglobulin and C3 in pauci-immune GN (type 3 RPGN).
-
Clinical Features Generally, the features of post-infectious RPGN are similar to those of acute GN, presenting as acute renal failure.
- The patients of Goodpasture’s syndrome may present with acute renal failure and/or associated intrapulmonary haemorrhage producing recurrent haemoptysis. The prognosis of all forms of RPGN is poor.
- However, post-infectious cases have somewhat better outcomes and may show recovery.
Minimal Change Disease (Synonyms: Mcd, Lipoid Nephrosis, Foot Process Disease, Nil Deposit Disease)
- Minimal change disease (MCD) is a condition in which the nephrotic syndrome is accompanied by no apparent change in glomeruli by light microscopy.
- Its other synonyms, lipoid nephrosis and foot process disease are descriptive terms for fatty changes in the tubules and electron microscopic appearance of flattened podocytes respectively.
- Minimal change disease accounts for 80% of cases of nephrotic syndrome in children under 16 years of age with preponderance in boys (ratio of boys to girls 2:1).
- In fact, historically, lipoid nephrosis was the first condition associated with nephrotic syndrome.
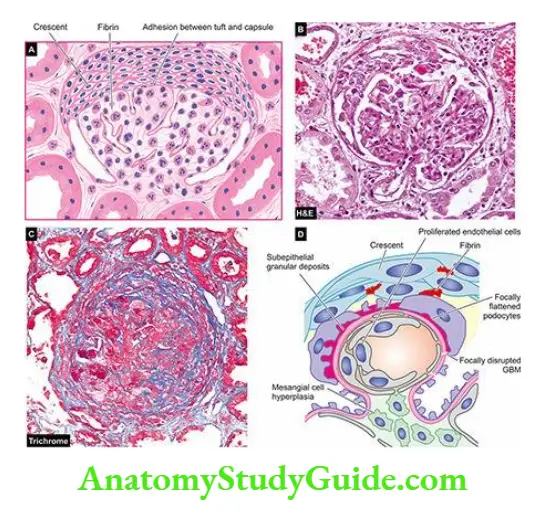
Etiopathogenesis The aetiology of MCD remain elusive.
However, the following two groups have been identified:
- Idiopathic (majority of cases).
- Cases associated with systemic diseases (Hodgkin’s disease, HIV infection) and drug therapy (for example NSAIDs, rifampicin, interferon-α).
The following features point to possible immunologic pathogenesis for MCD:
- Absence of deposits by immunofluorescence microscopy.
- Normal circulating levels of complement but the presence of circulating immune complexes in many cases.
- Universal satisfactory response to steroid therapy.
- Evidence of increased suppressor T cell activity with the elaboration of cytokines (interleukin-8, tumour necrosis factor) which probably cause foot process flattening and altered charge on the GBM.
- Detection of a mutation in the nephrin gene in cases of congenital MCD has focused attention on the genetic basis.
- Nephrotic syndrome in MCD in children is characterised by selective proteinuria containing mainly albumin, and minimal amounts of high molecular weight proteins such as α2- macroglobulin.
The basis for selective proteinuria appears to be as under:
- Reduction of normal negative charge on GBM due to loss of heparan sulfate proteoglycan from the GBM.
- Change in the shape of epithelial cells producing foot process flattening due to reduction of sialoglycoprotein cell coat.
- Adults having MCD, however, have non-selective proteinuria, suggesting a more extensive membrane permeability defect.
Morphologic Features Grossly, the kidneys are of normal size and shape. By light microscopy, the findings are as under:
- Glomeruli The most characteristic feature is no apparent abnormality in the glomeruli except for a slight increase in the mesangial matrix at the most (minimal change disease or nil lesion).
- Tubules There is the presence of fine lipid vacuolation and hyaline droplets in the cells of proximal convoluted tubules and, hence, the older name of the condition is ‘lipoid nephrosis’.
- Interstitium There may be oedema of the interstitium.
- Vessels Blood vessels do not show any significant change.
By electron microscopy, the most characteristic feature of the disease is identified which is the diffuse flattening of foot processes of the visceral epithelial cells (podocytes) and, hence, the name foot process disease or podocytopathy. - Unlike other forms of GN, no deposits are seen and the GBM is normal.
- By immunofluorescence microscopy, no deposits of complement or immunoglobulins are recognised (nil deposit disease).
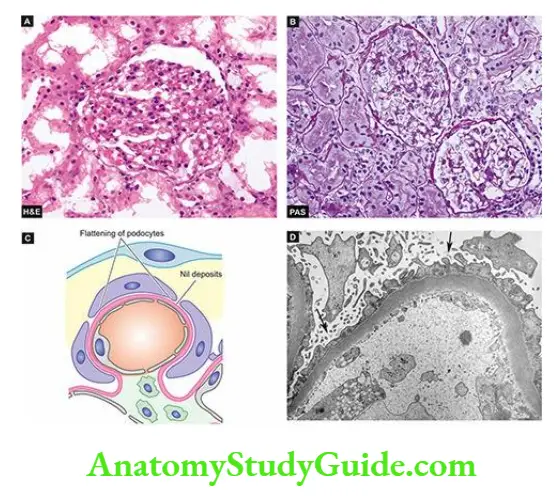
Clinical Features The classical presentation of MCD is of fully-developed nephrotic syndrome with massive and highly selective proteinuria; hypertension is unusual.
- Most frequently, the patients are children under 16 years (peak incidence at 6-8 years of age). The onset may be preceded by an upper respiratory infection, atopic allergy or immunisation.
- The disease characteristically responds to steroid therapy.
- In spite of remissions and relapses, the long-term prognosis is very good and most children become free of albuminuria after several years.
Membranous Glomerulonephritis (Synonym: Epimembranous Nephropathy)
- Membranous GN is characterised by widespread thickening of the glomerular capillary wall and is the most common cause of nephrotic syndrome in adults.
- In the majority of cases (85%), membranous GN is truly idiopathic, while in about 15% of cases, it is secondary to an underlying condition (for example SLE, malignancies, infections such as chronic hepatitis B and C, syphilis, malaria and drugs).
Etiopathogenesis Idiopathic membranous GN is an immune complex disease. The deposits of immune complexes are formed locally because circulating immune complexes are detected in less than a quarter of cases.
- Since leucocytic infiltration is not a feature of membranous GN, damage to the GBM is mediated directly by complement.
- While the nephritogenic antigen against which autoantibodies are formed in idiopathic membranous GN is not known yet, the antigen in cases of secondary membranous GN is either an endogenous (for example DNA in SLE) or exogenous one (for example hepatitis B virus, tumour antigen, treponema antigen, drug therapy with penicillamine).
- Currently, the pathogenesis of membrane alteration in membranous GN is believed to be by MAC (membrane attack complex C35b-C9) terminal complex on podocytes.
Morphologic Features Grossly, the kidneys are enlarged, pale and smooth. Light microscopy shows the following findings:
- Glomeruli: The characteristic finding is diffuse thickening of the glomerular capillary walls with all the glomeruli being affected more or less uniformly.
- As the disease progresses, the deposits are incorporated into an enormously thickened basement membrane, producing ‘duplication’ of GBM which is actually the formation of a new basement membrane.
- These basement membrane changes are best appreciated by silver impregnation stains (black colour) or by periodic acid-Schiff stains (pink colour). There is no cellular proliferation in the glomerular tufts.
- Tubules: The renal tubules remain normal except in the early stage when lipid vacuolation of the proximal convoluted tubules may be seen.
- Interstitium: The interstitium may show fine fibrosis and scanty chronic inflammatory cells.
- Vessels: In the early stage, vascular changes are not prominent, while later hypertensive changes of arterioles may occur.
- Electron microscopy shows characteristic electron-dense deposits in subepithelial locations.
- The basement membrane material protrudes between deposits as ‘spikes.
-
- Immunofluorescence microscopy reveals granular deposits of immune complexes consisting of IgG associated with complement C3.
- In secondary cases of membranous GN, the relevant antigen such as hepatitis B or tumour antigen may be seen.
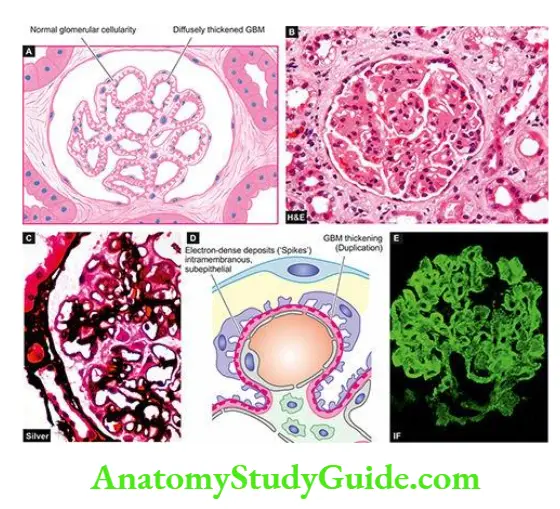
Clinical Features Presentation in the majority of cases is the insidious onset of nephrotic syndrome in an adult. The proteinuria is usually of non-selective type.
- In addition, microscopic haematuria and hypertension may be present at the onset or may develop during the course of the disease.
- The changes in membranous GN are irreversible in the majority of patients.
- Progression to impaired renal function and end-stage renal disease with progressive azotaemia occurs in approximately 50% of cases within a span of 2 to 20 years.
- Renal vein thrombosis has been found to develop in patients with membranous GN due to hypercoagulability.
- The role and beneficial effects of steroid therapy with or without the addition of immunosuppressive drugs are debatable.
Membranoproliferative Glomerulonephritis (Synonyms: Mpgn, Mesangiocapillary Gn)
- Membranoproliferative GN is another important cause of nephrotic syndrome in children and young adults.
- As the name implies, it is characterised by two histologic features an increase in cellularity of the mesangium associated with increased lobulation of the tuft, and irregular thickening of the capillary wall.
Etiopathogenesis Etiology of MPGN is unknown though in some cases there is evidence of preceding streptococcal infection. Based on ultrastructural, immunofluorescence and pathogenetic mechanisms, three types of MPGN are recognised
- Type 1 or classic form: is an example of an immune complex disease and comprises more than 70% of cases. It is characterised by immune deposits in the subendothelial position.
- Immune complex MPGN is seen in association with systemic immune-complex diseases (for example SLE, mixed cryoglobulinaemia, Sjögren’s syndrome), chronic infections (for example bacterial endocarditis, HIV, hepatitis B and C) and malignancies (for example lymphomas and leukaemias).
- Type 2 or dense deposit disease: is an example of an alternative pathway disease and constitutes about 30% of cases. The capillary wall thickening is due to the deposition of electron-dense material in the lamina dens of the GBM.
- Type 2 MPGN is an autoimmune disease in which patients have IgG autoantibody termed C3 nephritic factor.
- Type 2 cases have an association with partial lipodystrophy, an unusual condition of unknown pathogenesis characterised by symmetrical loss of subcutaneous fat from the upper half of the body.
- Type 3 is rare and shows features of type I MPGN and membranous nephropathy in association with systemic diseases or drugs.
Morphologic Features Grossly and by light microscopy, all three types of MPGN are similar.
- Grossly, the kidneys are usually pale in appearance and firm in consistency. By light microscopy, the features are as under:
-
- Glomeruli: Glomeruli show highly characteristic changes. They are enlarged with accentuated lobular patterns. The enlargement is due to a variable degree of mesangial cell proliferation and an increase in the mesangial matrix.
- The GBM is considerably thickened; with silver stains, it shows two basement membranes with a clear zone between them. This is commonly referred to as ‘double contour’, splitting, or ‘tram track’ appearance.
- Tubules: Tubular cells may show vacuolation and hyaline droplets.
- Interstitium: There may be scattered chronic inflammatory cells and some finely granular foam cells in the interstitium.
- Vessels: Hypertensive vascular changes are prominent in cases in which hypertension develops.
- By electron microscopy and immunofluorescence microscopy, the changes are different in the three types of MPGN
- Type 1: It shows electron-dense deposits in subendothelial locations conforming to the immune complex character of the disease.
- These deposits reveal positive fluorescence for C3 and slightly fainter staining for IgG.
- Type 2: The hallmark of type II MPGN is the presence of dense amorphous deposits within the lamina dens of the GBM and in the mesangium.
- Immunofluorescence studies reveal the universal presence of C3 and properdin in the deposits but the immunoglobulins are usually absent.
- Type 3: This rare form has electron-dense deposits within the GBM as well as in subendothelial and subepithelial regions of the GBM. Immunofluorescence studies show the presence of C3, IgG and IgM.
- Glomeruli: Glomeruli show highly characteristic changes. They are enlarged with accentuated lobular patterns. The enlargement is due to a variable degree of mesangial cell proliferation and an increase in the mesangial matrix.
Clinical Features Clinically, there are many similarities between the main forms of MPGN. The most common age at diagnosis is between 15 and 20 years.
- Approximately 50% of the patients present with nephrotic syndrome; about 30% have asymptomatic proteinuria, and 20% have nephritic syndrome at presentation.
- The proteinuria is non-selective. Haematuria and hypertension are frequently present. Hypocomplementaemia is a common feature.
- With time, the majority of patients progress to renal failure, while some continue to have proteinuria, haematuria and hypertension with stable renal function.
- Prognosis of type I is relatively better and the majority of patients survive without clinically significant impairment of GFR, while type II cases run a variable clinical course.
Focal Proliferative Glomerulonephritis (Synonym: Mesangial Proliferative Gn)
- Focal proliferative GN is characterised by pathologic changes in fewer than 50% of glomeruli (focal), often confined to one or two lobules of the affected glomeruli while other glomeruli are normal.
- Focal GN is, thus, a pathologic diagnosis.
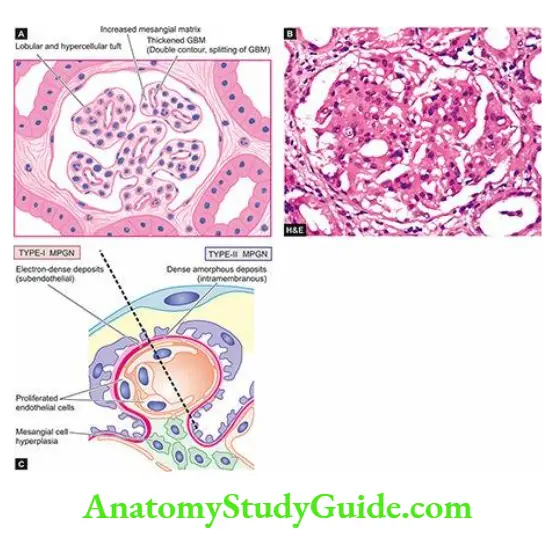
Etiopathogenesis may occur in the following diverse clinical settings:
-
- As an early manifestation of a number of systemic diseases such as SLE (class III), HenochSchÖnlein purpura, subacute bacterial endocarditis, Wegener’s granulomatosis, and polyarteritis nodosa, Goodpasture’s syndrome.
- As a component of a known renal disease such as IgA nephropathy.
- As a primary idiopathic glomerular disease unrelated to systemic or other renal disease.
- The diverse settings under which focal GN is encountered make it unlikely that there are common etiologic agents or pathogenetic mechanisms.
- However, the observation of mesangial deposits of immunoglobulins and complement suggests immune complex disease and participation of the mesangium.
Morphologic Features By light microscopy, the single most important feature in focal GN is the abnormality seen in a certain number of glomeruli and generally confined to one or two lobules of the affected glomeruli i.e. focal and segmental glomerular involvement.
- The pathologic change most frequently consists of focal and segmental cellular proliferation of mesangial cells and endothelial cells but sometimes necrotising changes can be seen.
- The condition must be distinguished from focal segmental glomerulosclerosis.
- By immunofluorescence microscopy, widespread mesangial deposits of immunoglobulins (mainly IgA with or without IgG), complement (C3) and fibrin are demonstrated in most cases of focal GN.
Clinical Features The clinical features vary according to the condition causing it. Haematuria is one of the most common clinical manifestations. Proteinuria is frequently mild to moderate but hypertension is uncommon.
Diffuse Proliferative Glomerulonephritis (Synonym: Dpgn)
DPGN is similar to focal proliferative GN in terms of aetiology and pathogenesis
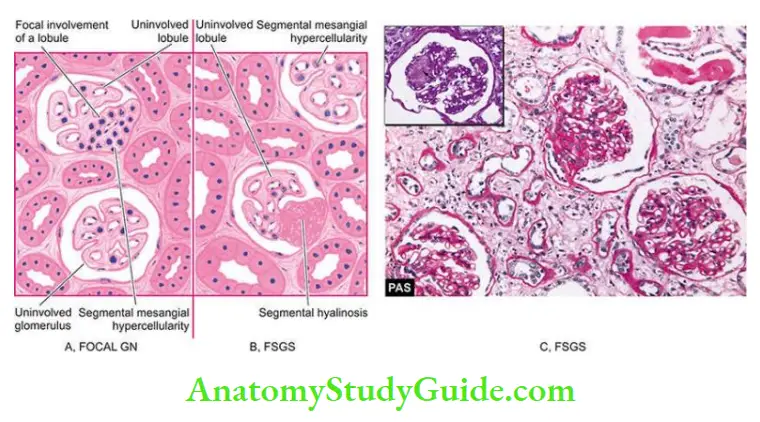
Morphologic Features These are similar to focal proliferative GN but in its advanced form. It differs from focal GN in the following ways:
- The number of glomeruli involved is more than 50% i.e. diffuse proliferation.
- The cells partaking in DPGN are more diffuse type and not mesangial cells alone; it also includes the proliferation of epithelial, endothelial and inflammatory cells.
- DPGN represents class IV lupus nephritis.
Focal Segmental Glomerulosclerosis (Synonyms: Focal Sclerosis, Focal Hyalinosis)
- Focal segmental glomerulosclerosis (FSGS) is a condition in which there is sclerosis and hyalinosis of some glomeruli and portions of their tuft (less than 50% in a tissue section), while the other glomeruli are normal by light microscopy involvement is focal and segmental.
- The incidence of FSGS has increased over the last decades and is currently responsible for about one-third of cases of nephrotic syndrome in adults.
Etiopathogenesis Fsgs was previously believed to be a variant of MCD with accentuation of epithelial damage in the form of hyalinosis and sclerosis. Currently, the condition is divided into 3 groups:
- Idiopathic type: This group comprises the majority of cases. It is found in children and young adults with presentation of nephrotic syndrome.
- It differs from minimal change disease in having non-selective proteinuria, being steroid-resistant, and may progress to chronic renal failure.
- Immunofluorescence microscopy reveals deposits of IgM and C3 in the sclerotic segment.
- With superimposed primary glomerular disease: There may be cases of FSGS with superimposed MCD or IgA nephropathy.
- Those associated with MCD show good response to steroid therapy and progression to chronic renal failure may occur after a long time.
- Secondary type: This group consists of focal segmental sclerotic lesions as a secondary manifestation of certain diseases such as HIV, diabetes mellitus, reflux nephropathy, heroin abuse and analgesic nephropathy.
- The hallmark of the pathogenesis of FSGS is an injury to visceral epithelial cells that results in the disruption of visceral epithelial cells and resultant nephron loss.
Morphologic Features By light microscopy, depending upon the severity of the disease, a variable number of glomeruli are affected focally and segmentally, while others are normal.
- The affected glomeruli show solidification or sclerosis of one or more lobules of the tuft.
- Hyalinosis refers to the collection of eosinophilic, homogeneous, PAS-positive, hyaline material present on the inner aspect of a sclerotic peripheral capillary loop.
- Mesangial hypercellularity is present in an appreciable number of cases. In addition, to glomerular changes, there is interstitial fibrosis and infiltration by mononuclear leucocytes, and tubular epithelial cell atrophy and degeneration.
- Besides the lesions of focal and segmental scarring, a variant of FSGS, collapsing glomerulopathy, has been described in HIV patients.
- It is segmental or global glomerular collapse of the tuft along with the presence of hyperplasia and hypertrophy of podocytes producing a pseudo-crescent and a rapid decline in renal function.
- A few other variants have also been observed that include perihilar, cellular and tip variants.
- By electron microscopy, diffuse loss of foot processes as seen in minimal change disease is evident but, in addition, there are electron-dense deposits in the region of hyalinosis and sclerosis which are believed to be immune complexes.
- By immunofluorescence microscopy, the deposits in the lesions are shown to contain IgM and C3.
Clinical Features The condition may affect all ages including children and has male preponderance.
-
- The most common presentation is in the form of nephrotic syndrome with heavy proteinuria.
- Haematuria and hypertension tend to occur more frequently than in minimal change disease.
- Evidence of renal failure may be present at the onset.
Iga Nephropathy (Synonyms: Berger’S Disease, Iga Gn)
- IgA nephropathy is emerging as the most common form of glomerulopathy worldwide and its incidence has been rising. It is characterised by aggregates of IgA, deposited principally in the mesangium.
- The condition was first described by Berger, a French physician in 1968 (Not to be confused with Buerger’s disease or thromboxane obliterans described by an American pathologist in 1908.
Etiopathogenesis The aetiology of IgA nephropathy remains unclear:
-
- It is idiopathic in most cases.
- Seen as part of Henoch-SchÖnlein purpura.
- Association with chronic inflammation in various body systems (for example chronic liver disease, inflammatory bowel disease, interstitial pneumonitis, leprosy, dermatitis herpetiformis, uveitis, ankylosing spondylitis, Sjögren’s syndrome, monoclonal IgA gammopathy).
Pathogenesis of IgA nephropathy is explained on the basis of the following mechanisms:
-
-
- In view of exclusive mesangial deposits of IgA and elevated serum levels of IgA and IgA immune complexes, IgA nephropathy has been considered to arise from the entrapment of these complexes in the mesangium.
- There is an absence of early components of the complement but the presence of C3 and properdin in the mesangial deposits, which point towards the activation of the alternate complement pathway.
- Since there is a close association between mucosal infections (for example the respiratory, gastrointestinal or urinary tract), it is suggested that IgA deposited in the mesangium could be due to increased mucosal secretion of IgA.
- HLA-B35 association has been reported in some cases. Another possibility is a genetically determined abnormality of the immune system producing an increase in circulating IgA.
-
Morphologic Features By light microscopy, the pattern of involvement varies. These include focal proliferative GN, focal segmental glomerulosclerosis, membranoproliferative GN, and rarely RPGN.
- Generally, it is a milder form of chronic glomerulonephritis, with changes limited to the mesangial area.
- By electron microscopy, finely granular electron-dense deposits are seen in the mesangium.
- By immunofluorescence microscopy, the diagnosis is firmly established by demonstration of mesangial deposits of IgA, with or without IgG, and usually with C3 and properdin.
Clinical Features The disease is common in children and young adults. The clinical picture is usually characterised by recurrent bouts of haematuria that are often precipitated by mucosal infections.
- Mild proteinuria is usually present and occasionally nephrotic syndrome may develop.
Chronic Glomerulonephritis (Synonym: Endstage Kidney, Chronic Kidney Disease)
Chronic GN is the final stage of a variety of glomerular diseases which result in an irreversible impairment of renal function. The conditions which may progress to chronic GN, in descending order of frequency, are as under:
- Rapidly progressive GN (90%)
- Membranous GN (50%)
- Membranoproliferative GN (50%)
- Focal segmental glomerulosclerosis (50%)
- IgA nephropathy (40%)
- Acute post-streptococcal GN (1%).
However, about 20% of cases of chronic GN are idiopathic without evidence of preceding GN of any type
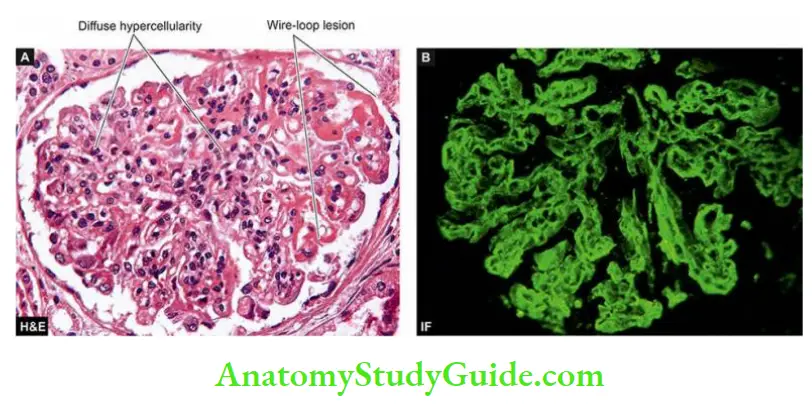
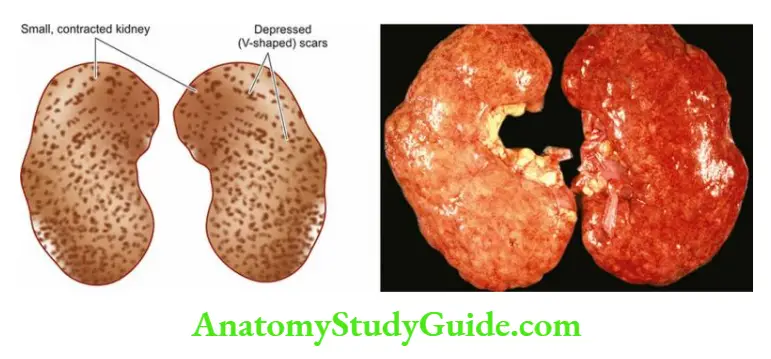
Morphologic Features Grossly, the kidneys are usually small and contracted weighing as low as 50 gm each. The capsule is adherent to the cortex.
- The cortical surface is generally diffusely granular. On the cut section, the cortex is narrow and atrophic, while the medulla is unremarkable.
- Microscopically, the changes vary greatly depending on the underlying glomerular disease. In general, the following changes are seen.
- Glomeruli are reduced in number and most of those present show completely hyalinised tufts, giving the appearance of acellular, eosinophilic masses which are PASpositive. Evidence of underlying glomerular disease may be present.
- Tubules Many tubules completely disappear and there may be atrophy of tubules close to scarred glomeruli. Tubular cells show hyaline droplets, and degeneration and tubular lumina frequently contain eosinophilic, homogeneous casts.
- Interstitium There is fine and delicate fibrosis of the interstitial tissue and varying number of chronic inflammatory cells are often seen.
- Vessels Advanced cases which are frequently associated with hypertension show conspicuous arterial and arteriolar sclerosis.
- Patients of chronic kidney disease on dialysis show a variety of dialysis-associated changes that include acquired cystic disease, the occurrence of adenomas and adenocarcinomas of the kidney, calcification of tufts and deposition of calcium oxalate crystals in tubules.
- Microscopically, the changes vary greatly depending on the underlying glomerular disease. In general, the following changes are seen.
Clinical Features:
The patients are usually adults. The terminal stage of chronic GN is characterised by hypertension, uraemia and progressive deterioration of renal function.
- Besides the primary changes due to chronic renal failure, there are a variety of systemic manifestations of uraemia. These patients eventually die if they do not receive a renal transplant.
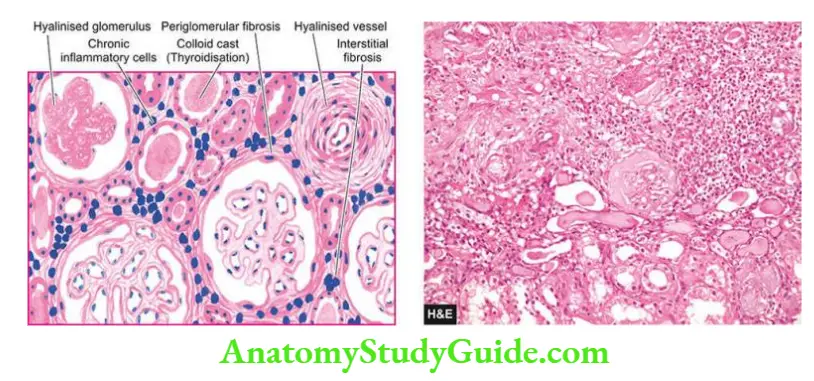
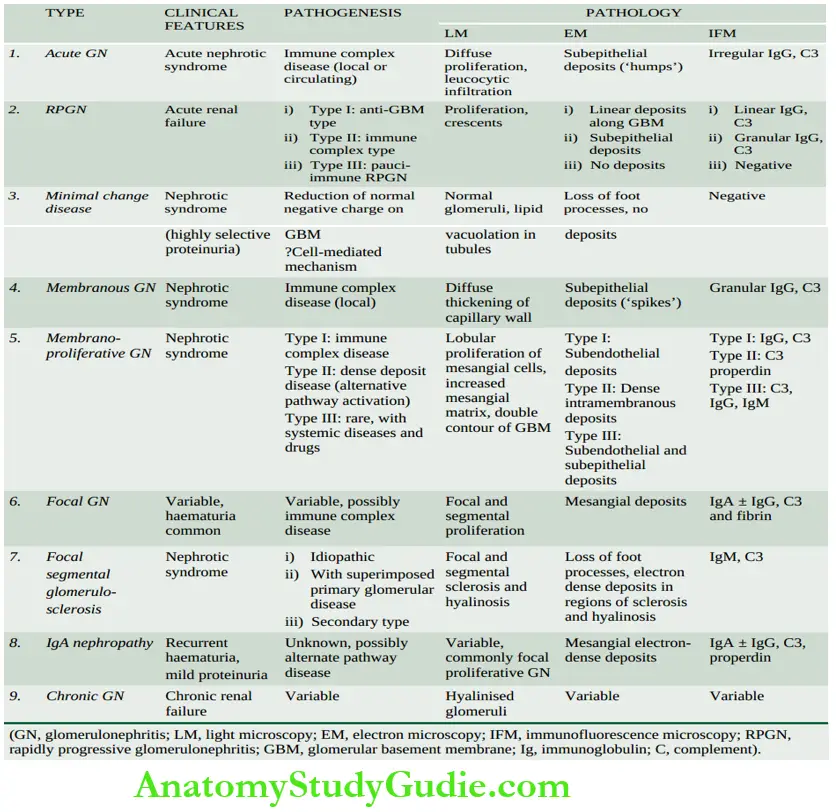
Primary Glomerulonephritis
- Glomerular diseases are divided into 3 groups: primary glomerulonephritis (GN), and secondary glomerular diseases.
- Based on clinical course, etiopathogenesis and morphology, primary GN has several types.
- Acute GN is known to follow acute infection (post-streptococcal) and characteristically presents as an acute nephritic syndrome in children. It has exudative lesions and subepithelial lumpy-dumpy electron-dense deposits.
- RPGN is an acute reduction in renal function resulting in acute renal failure rapidly and is characterised by the formation of ‘crescents’. It may have either linear or granular deposits; a pauci-immune RPGN also exists.
- Minimal change disease is nephrotic syndrome in children accompanied by no apparent change in glomeruli by light microscopy and flattening of podocytes on EM.
- Membranous GN is characterised by widespread thickening of the glomerular capillary wall and is the most common cause of nephrotic syndrome in adults. It has intramembranous subepithelial electron-dense deposits as spikes.
- Membranoproliferative GN is characterised by an increase in mesangial cellularity associated with increased lobulation of the tuft, and irregular thickening of the capillary wall.
- It may have subepithelial electron-dense deposits (type-I) or intramembranous dense amorphous deposits (type-2).
- Focal and diffuse proliferative GN is a pathologic diagnosis depending upon whether <50% or > 50% glomeruli show proliferative changes. Diffuse proliferative GN is similar but is different in aetiology and pathogenesis.
- Focal segmental glomerulosclerosis is sclerosis and hyalinosis of some glomeruli and portions of their tuft.
- IgA nephropathy or Berger’s disease is another common form of GN in which there are mesangial deposits of IgA.
- Chronic GN is the final stage of a variety of glomerular diseases which result in an irreversible impairment of renal function and show diffuse changes affecting the whole nephron, interstitium and blood vessels.
Secondary Glomerular Diseases
Glomerular involvement may occur secondary to certain systemic diseases or a few hereditary diseases.
- In some of these, renal involvement may be the initial presentation, while in others clinical evidence of renal disease appears long after other manifestations have appeared.
- A list of these conditions has already been given in. The important examples are described below.
Lupus Nephritis
- Renal manifestations of systemic lupus erythematosus (SLE) are termed lupus nephritis. Other clinical manifestations, aetiology and pathogenesis of this multi-system autoimmune disease are described in Chapter 5.
- The incidence of renal involvement in SLE ranges from 40 to 75%. The two cardinal clinical manifestations of lupus nephritis are proteinuria and haematuria.
- In addition, hypertension and casts of different types such as red cell casts, fatty casts and leucocyte casts in the urinary sediment are found.
- Pathogenesis of lesions in lupus nephritis is linked to genes related to major histocompatibility complexes and B-cell signalling pathways such as TNF superfamily members.
Morphologic Features According to the WHO, six patterns of mutually-merging renal lesions are seen in lupus nephritis Class
- class 1: Minimal lesions On light microscopy, these cases do not show any abnormality.
- But an examination by electron microscopy and immunofluorescence microscopy shows deposits within the mesangium which consist of IgG and C3.
- Class 2: Mesangial lupus nephritis These cases have mild clinical manifestations. By light microscopy, there is an increase in the number of mesangial cells and the amount of mesangial matrix.
- Ultrastructural and immunofluorescence studies reveal granular mesangial deposits of IgG and C3; sometimes IgA and IgM are also present in the deposits.
- Class 3: Focal segmental lupus nephritis This is characterised by focal and segmental proliferation of endothelial and mesangial cells, together with infiltration by macrophages and sometimes neutrophils.
- Haematoxylin bodies of gross may be present. Subendothelial and subepithelial deposits of IgG, often with IgM or IgA and C3, are seen.
- Class 4: Diffuse proliferative lupus nephritis This is the most severe and the most common form of lupus nephritis. Class IV lesions show the following changes.
-
- Diffuse proliferation of endothelial, mesangial, and sometimes epithelial cells, involving most or all glomeruli.
- Segmental thickening of the GBM in the form of ‘wire loop’ lesions on light microscopy.
-
-
- Corresponding areas of the GBM on electron microscopy show large electron-dense deposits in the mesangium and in the subendothelial region of the GBM which on immunofluorescence are positive for IgG; sometimes also for IgA or IgM, and C3.
-
- Class 5: Membranous lupus nephritis These lesions resemble those of idiopathic membranous GN.
- These consist of diffuse thickening of glomerular capillary wall on light microscopy and show subendothelial deposits of immune complexes containing IgG, IgM and C3 on ultrastructural studies. Mesangial hypercellularity is present in some cases.
- Class 6: Sclerosing lupus nephritis This is a chronic kidney disease of SLE, similar to chronic GN. Most glomeruli are sclerosed and hyalinised and there may be remnants of preceding lesions.
- Although in a given case, the lesions in lupus nephritis fit into one of the classes described above, it is not unusual to find the overlapping and progressive transformation of lupus lesions during the course of the disease.
Diabetic Nephropathy
- Renal involvement is an important complication of diabetes mellitus. Chronic kidney disease with renal failure accounts for deaths in more than 10% of all diabetics.
- Renal complications are more severe, develop early and more frequently in type 1 (earlier called insulin-dependent) diabetes mellitus (30-40% cases) than in type 2 (earlier termed non-insulin-dependent) diabetics (about 20% cases).
- A variety of clinical syndromes are associated with diabetic nephropathy that includes asymptomatic proteinuria, nephrotic syndrome, progressive renal failure and hypertension.
- Cardiovascular disease is 40 times more common in patients with chronic kidney disease in diabetes mellitus than in non-diabetics and more diabetics die from cardiovascular complications than from uraemia.
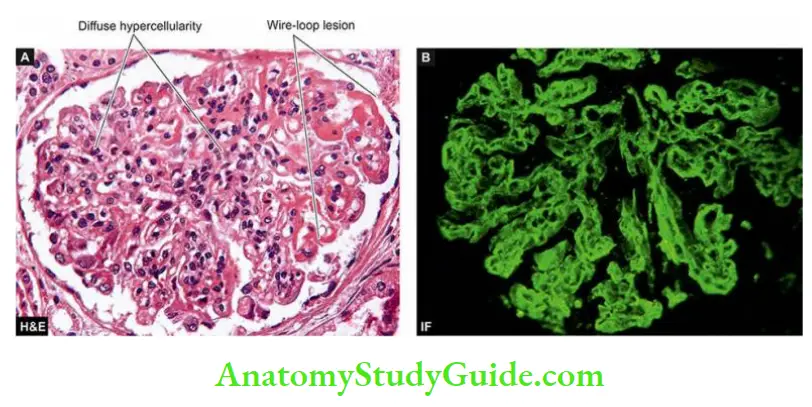
Morphologic Features Diabetic nephropathy encompasses 4 types of renal lesions in diabetes mellitus: diabetic glomerulosclerosis, vascular lesions, diabetic pyelonephritis and tubular lesions (Armanni-Ebstein lesions).
- Diabetic Glomerulosclerosis: Glomerular lesions in diabetes mellitus are particularly common and account for a majority of abnormal findings referable to the kidney.
-
- Pathogenesis of these lesions in diabetes mellitus is explained by the following sequential changes: hyperglycaemia → glomerular hypertension → renal hyperperfusion → deposition of proteins in the mesangium → glomerulosclerosis → renal failure.
- In addition, cellular infiltration in renal lesions in diabetic glomerular lesions is due to growth factors, particularly transforming growth factor-β.
- The strict control of blood glucose levels and control of systemic hypertension in these patients retards progression to diabetic nephropathy.
- Glomerulosclerosis in diabetes may take one of 2 forms diffuse or nodular lesions
- Diffuse glomerulosclerosis: Diffuse glomerular lesions are the most common. There is the involvement of all parts of most of the glomeruli
- The pathologic changes consist of thickening of the GBM and diffuse increase in the mesangial matrix with the mild proliferation of mesangial cells.
- Various insulative lesions such as capsular hyaline drops and fibrin caps may also be present.
- Capsular drop is an eosinophilic hyaline thickening of the parietal layer of Bowman’s capsule and bulges into the glomerular space.
- A Fibrin cap is a homogeneous, brightly eosinophilic material appearing on the wall of a peripheral capillary of a lobule.
- Nodular glomerulosclerosis: Nodular lesions of diabetic glomerulosclerosis are also called as Kimmelstiel-Wilson (KW) lesions or intercapillary glomerulosclerosis.
- These lesions are specific for type 1 diabetes (juvenile-onset diabetes) or islet cell antibody-positive diabetesmellitus.
- The pathologic changes consist of one or more nodules in a few or many glomeruli. Nodule is an ovoid or spherical, laminated, hyaline, acellular mass located within a lobule of the glomerulus.
- The nodules are surrounded peripherally by glomerular capillary loops which may have normal or thickened GBM. The nodules are PAS-positive and contain lipids and fibrin.
- As the nodular lesions enlarge, they compress the glomerular capillaries and obliterate the glomerular tuft.
- As a result of glomerular and arteriolar involvement, renal ischaemia occurs leading to tubular atrophy and interstitial fibrosis and results in grossly small, contracted kidney.
-
- Vascular Lesions Atheroma: of renal arteries is very common and severe in diabetes mellitus. Hyaline arteriolosclerosis affecting the afferent and efferent arterioles of the glomeruli is also often severe in diabetes.
- These vascular lesions are responsible for renal ischaemia that results in tubular atrophy and interstitial fibrosis.
- Diabetic Pyelonephritis Poorly-controlled diabetics are particularly susceptible to bacterial infections.
- Papillary necrosis (necrotising papillitis) is an important complication of diabetes that may result in acute pyelonephritis. Chronic pyelonephritis is 10 to 20 times more common in diabetics than in others.
- Tubular Lesions (Armanni-Ebstein Lesions) In untreated diabetics who have extremely high blood sugar levels, the epithelial cells of the proximal convoluted tubules develop extensive glycogen deposits appearing as vacuoles.
- These are called Armanni-Ebstein lesions.
- The tubules return to normal on control of the hyperglycaemic state.
Hereditary Nephritis
A group of hereditary diseases principally involving the glomeruli are termed hereditary nephritis. These include Alport’s syndrome, Fabry’s disease and nail-patella syndrome.
- Alport’s syndrome: Out of various hereditary nephritis, Alport’s syndrome is relatively more common and has been extensively studied.
- This is an X-linked dominant disorder having a mutation in the α-5 chain of type IV collagen located on the X-chromosome. It affects males more severely than females.
- The syndrome consists of sensorineural deafness and ophthalmic complications (lens dislocation, posterior cataracts and corneal dystrophy) associated with hereditary nephritis.
- The condition is slowly progressive, terminating in chronic kidney disease in the 2nd to 3rd decades of life.
- The common presenting features are persistent or recurrent haematuria accompanied by erythrocyte casts, proteinuria and hypertension.
- By light microscopy, the glomeruli have predominant involvement and show segmental proliferation of mesangial cells with increased mesangial matrix and occasional segmental sclerosis.
- Another prominent feature is the presence of lipid-laden foam cells in the interstitium. As the disease progresses, there is increasing sclerosis of glomeruli, tubular atrophy and interstitial fibrosis.
- Electron microscopy: reveals characteristic basement membrane splitting or lamination in the affected parts of glomeruli. Immunofluorescence studies fail to show deposits of immunoglobulins or complement components.
- Fabry’s disease, another hereditary nephritis is characterised by the accumulation of neutral glycosphingolipids in lysosomes of glomerular, tubular, vascular and interstitial cells.
- Nail-patella syndrome: is a rare hereditary disease having abnormality in the α-1 chain of collagen V on chromosome 9 associated with multiple osseous defects of elbows, knees and nail dysplasia.
- About half the cases develop nephropathy.
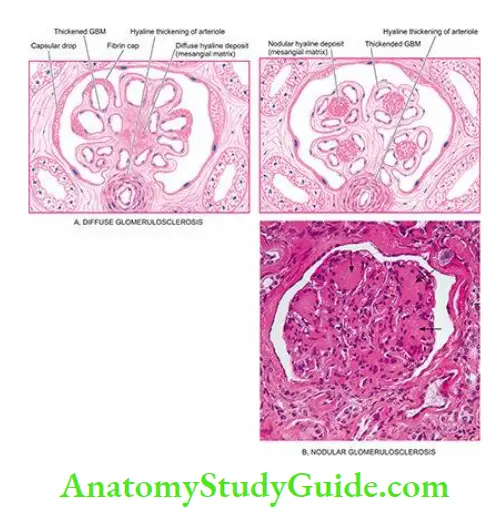
Secondary Glomerular Diseases:
- Renal manifestations of systemic lupus erythematosus are termed lupus nephritis and consist of 6 progressive classes (class 1 to 6).
- Diabetic nephropathy encompasses diabetic glomerulosclerosis, vascular lesions, diabetic pyelonephritis and tubular lesions.
- Diabetic glomerulosclerosis consists of 2 types of lesions: diffuse and nodular; the latter are characteristic Kimmelstiel-Wilson lesions.
- Hereditary nephritis includes Alport’s syndrome, Fabry’s disease and nail-patella syndrome.
Tubular And Tubulointerstitial Diseases
- It is difficult to separate the involvement of the tubules and the interstitium since most forms of tubular diseases also involve the interstitium, while the tubules and interstitium may be involved secondarily as a part of the diseases of other renal parenchymal components.
- For the purpose of the present discussion, this group parenchymal of diseases is discussed under 2 headings:
- Primary tubular diseases include tubular injury by ischaemic or toxic agents. acute tubular necrosis.
- Tubulointerstitial diseases include inflammatory involvement of the tubules and interstitium pyelonephritis (acute and chronic).
Acute Tubular Necrosis:
- Acute tubular necrosis (ATN) is the term used for acute renal failure (ARF) resulting from the destruction of tubular epithelial cells.
- ATN is the most common and most important cause of ARF characterised by sudden cessation of renal function.
- Various other causes of ARF (prerenal, intra-renal and post-renal), as well as the clinical syndrome accompanying ATN (oliguric phase, diuretic phase and phase of recovery), are described already.
- Based on aetiology and morphology, two forms of ATN are distinguished as ischaemic and toxic; however, both forms have somewhat common pathogenesis.
Pathogenesis Of Atn The pathogenesis of both types of ATN resulting in ARF is explained on the basis of the following sequential mechanism and is illustrated in:
-
- Renal tubules are highly susceptible to injury by ischaemia and toxic agents.
- Tubular damage in ischaemic ATN is initiated by arteriolar vasoconstriction induced by the renin-angiotensin system, while in toxic ATN by direct damage to tubules by the agent.
- Debris of the desquamated epithelium due to necrosis causes tubular obstruction and may block urinary outflow with consequent reduction of GFR and also produce casts in the urine.
- These events cause increased intratubular pressure resulting in damage to the tubular basement membrane.
- Due to increased intratubular pressure, there is tubular rupture.
- Damage to tubules is accompanied by leakage of fluid into the interstitium causing interstitial oedema.
- The leakage of tubular fluid into the interstitium increases interstitial pressure.
- Leaked fluid incites host inflammatory response.
- Increased interstitial pressure causes compression of tubules and blood vessels and sets up a vicious cycle of accentuated ischaemia and necrosis.
- Ultimately, it leads to reduced GFR and consequently oliguria.
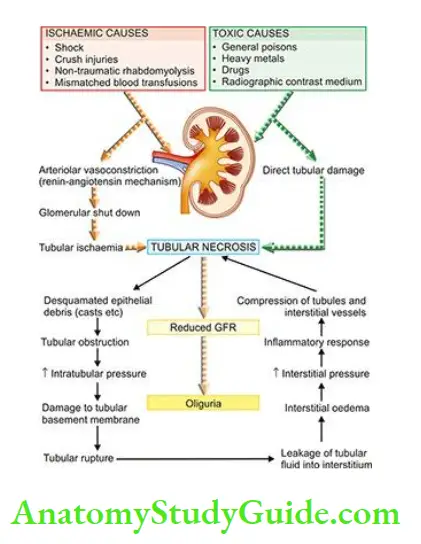
Ischaemic Atn
Ischaemic ATN, also called tubulorrhectic ATN, lower (distal) nephron nephrosis, anoxic nephrosis, or shock kidney, occurs due to hypoperfusion of the kidneys resulting in focal damage to the distal parts of the convoluted tubules.
- Etiology Ischaemic ATN is more common than toxic ATN and accounts for more than 80% of cases of tubular injury. Ischaemia may result from a variety of causes as follows:
-
- Shock (post-traumatic, surgical, burns, dehydration, obstetrical and septic type).
- Crush injuries.
- Non-traumatic rhabdomyolysis induced by alcohol, coma, muscle disease or extreme muscular exertion (myoglobinuric nephrosis).
- Mismatched blood transfusions, black-water fever (hemoglobinuria nephrosis).
Morphologic Features Grossly, the kidneys are enlarged and swollen. On the cut section, the cortex is often widened and pale, while the medulla is dark.
- Histologically, predominant changes are seen in the tubules, while glomeruli remain unaffected. The interstitium shows oedema and mild chronic inflammatory cell infiltrate. Tubular changes are as follows:
-
- Dilatation of the proximal and distal convoluted tubules.
- Focal tubular necrosis at different points along the nephron.
- Flattened epithelium lining the tubules suggests epithelial regeneration.
- Eosinophilic hyaline casts or pigmented haemoglobin and myoglobin casts in the tubular lumina.
- Disruption of the tubular basement membrane adjacent to the cast may occur (tubulorrhexis).
-
-
- The prognosis of ischaemic ATN depends upon the underlying aetiology.
- In general, cases that follow severe trauma, surgical procedures, extensive burns and sepsis have much worse outlook than the others.
-
Toxic Atn
Toxic Atn, also called nephrotoxic ATN or toxic nephrosis or upper (proximal) nephron nephrosis, occurs as a result of direct damage to tubules, more marked in proximal portions, by ingestion, injection or inhalation of a number of toxic agents.
Etiology The toxic agents causing toxic ATN are as under:
- General poisons such as mercuric chloride, carbon tetrachloride, ethylene glycol, mushroom poisoning and insecticides.
- Heavy metals (mercury, lead, arsenic, phosphorus and gold).
- Drugs such as sulfonamides, certain antibiotics (gentamycin, cephalosporin), anaesthetic agents (methoxyflurane, halothane), barbiturates, and salicylates.
- Radiographic contrast material.
Morphologic Features Poisoning with mercuric chloride provides the classic example that produces widespread and readily discernible tubular necrosis (acute mercury nephropathy).
- Grossly, the kidneys are enlarged and swollen. On the cut section, the cortex is pale and swollen, while the medulla is slightly darker than normal.
- Histologically, the appearance varies according to the cause of toxic ATN but, in general, it involves the segment of tubule diffusely (unlike ischaemic ATN where the involvement of the nephron is focal).
-
- In mercuric chloride poisoning, the features are as follows:
- Epithelial cells of mainly proximal convoluted tubules are necrotic and desquamated into the tubular lumina.
- The desquamated cells may undergo dystrophic calcification.
- The tubular basement membrane is generally intact.
- The regenerating epithelium, which is flat and thin with few mitoses, may be seen lining the tubular basement membrane.
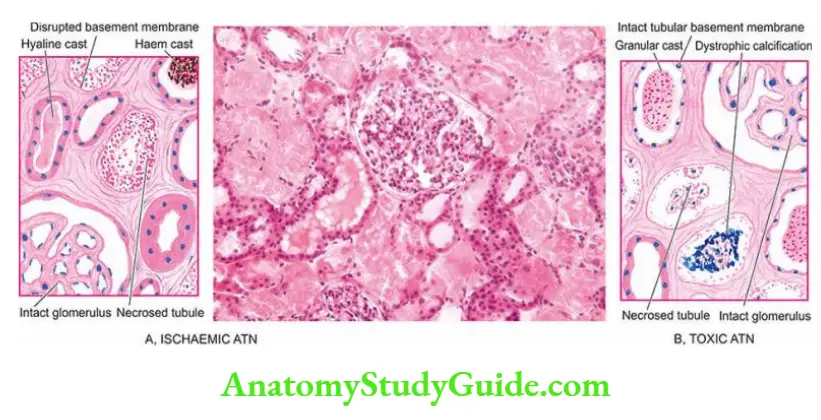
- The prognosis of toxic ATN is good if there is no serious damage to other organs such as the heart and liver. The contrasting features of the two forms of ATN are presented in
Acute Tubular necrosis
- Acute tubular necrosis (ATN) is acute renal failure resulting from the destruction of tubular epithelial cells.
- Based on aetiology and morphology, ATN may be ischaemic or toxic.
- Ischaemic ATN is more common (for example in shock) and produces focal tubular necrosis, disrupted basement membrane and casts in the tubular lumina.
- Toxic ATN may occur due to poisoning and produces more diffuse damage to the proximal tubules.
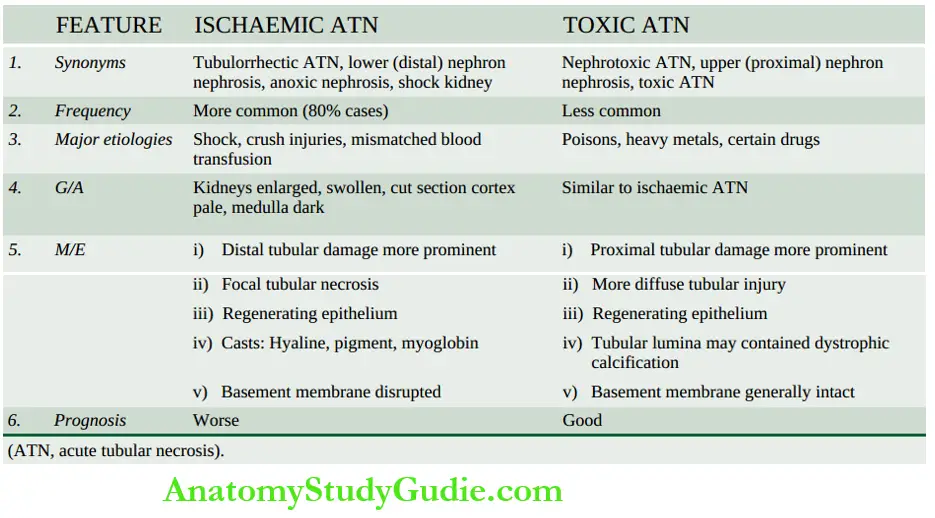
Tubulointerstitial Diseases
The term tubulointerstitial nephritis is used for an inflammatory process that predominantly involves the renal interstitial tissue and is usually accompanied by some degree of tubular damage.
- A number of primary glomerular, tubular, vascular and obstructive diseases are secondarily associated with interstitial reactions.
- However, the term interstitial nephritis is reserved for those cases where there is no primary involvement of glomeruli, tubules or blood vessels.
- The older nomenclature, interstitial nephritis, is currently used synonymously with tubulointerstitial nephritis or tubulointerstitial nephropathy.
- A number of bacterial and non-bacterial, acute and chronic conditions may produce tubulointerstitial nephritis and are listed in The important and common examples among these are discussed below.
Acute Pyelonephritis
Acute pyelonephritis is an acute suppurative inflammation of the kidney caused by pyogenic bacteria.
Etiopathogenesis Most cases of acute pyelonephritis follow an infection of the lower urinary tract.
- The most common pathogenic organism in urinary tract infection (UTI) is Escherichia coli (in 90% of cases), followed in decreasing frequency, by Enterobacter, Klebsiella, Pseudomonas and Proteus.
- The bacteria gain entry into the urinary tract, and then into the kidney by one of the two routes: ascending infection and haematogenous infection:
- Ascending infection This is the most common route of infection. The common pathogenic organisms are inhabitants of the colon and may cause faecal contamination of the urethral orifice, especially in females in the reproductive age group.
- This has been variously attributed to the shorter urethra in females liable to faecal contamination, hormonal influences facilitating bacterial adherence to the mucosa, absence of prostatic secretions which have antibacterial properties, and urethral trauma during sexual intercourse.
- The last named produces what is appropriately labelled as ‘honeymoon pyelitis’.
- Ascending infection may occur in a normal individual but the susceptibility is increased in patients with diabetes mellitus, pregnancy, urinary tract obstruction or instrumentation.
- Bacteria multiply in the urinary bladder and produce asymptomatic bacteriuria found in many of these cases.
- After having caused urethritis and cystitis, the bacteria in susceptible cases ascend further up into the ureters against the flow of urine, extending into the renal pelvis and then the renal cortex.
- The role of vesicoureteral reflux is not a significant factor in the pathogenesis of acute pyelonephritis as it is in chronic pyelonephritis.
- Haematogenous infection Less often, acute pyelonephritis may result from the blood-borne spread of infection.
- This occurs more often in patients with obstructive lesions in the urinary tract and in debilitated or immunosuppressed patients.
- Ascending infection This is the most common route of infection. The common pathogenic organisms are inhabitants of the colon and may cause faecal contamination of the urethral orifice, especially in females in the reproductive age group.
Tubulointerstitial diseases
- 1. Infective
-
- Acute pyelonephritis
- Chronic pyelonephritis
- Tuberculous pyelonephritis
- Other infections (viruses, parasites etc)
- 2. Non-Infective
-
- Acute hypersensitivity interstitial nephritis
- Analgesic abuse (phenacetin) nephropathy
- Myeloma nephropathy
- Balkan nephropathy
- Urate nephropathy
- Gout nephropathy
- Radiation nephritis
- Transplant rejection
- Nephrocalcinosis
- Idiopathic interstitial nephritis
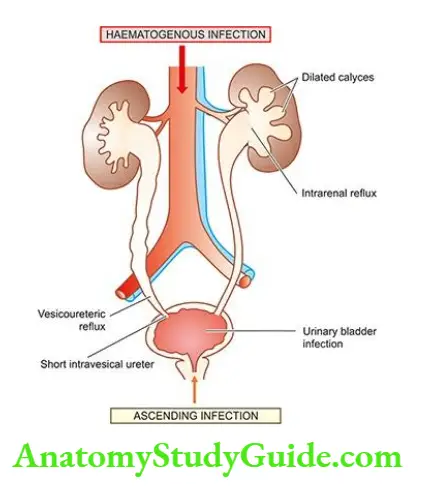
Morphologic Features Grossly, well-developed cases of acute pyelonephritis show enlarged and swollen kidney that bulges on the section.
The cut surface shows small, yellow-white abscesses with a haemorrhagic rim. These abscesses may be several millimetres across and are situated mainly in the cortex.
Microscopically, acute pyelonephritis is characterised by extensive acute inflammation involving the interstitium and causing destruction of the tubules.
- Generally, the glomeruli and renal blood vessels show considerable resistance to infection and are spared.
- The acute inflammation may be in the form of a large number of neutrophils in the interstitial tissue and bursting into tubules or may form focal neutrophilic abscesses in the renal parenchyma.
Clinical Features Classically, acute pyelonephritis has an acute onset with chills, fever, loin pain, lumbar tenderness, dysuria and frequency of micturition.
- Urine will show an abundance of bacteria, pus cells and pus cell casts in the urinary sediment.
- Institution of specific antibiotics, after identification of bacteria by culture followed by sensitivity test, eradicates the infection in the majority of patients.
Complications of acute pyelonephritis are encountered more often in patients with diabetes mellitus or with urinary tract obstruction. Following are the three important complications of acute pyelonephritis

- Papillary necrosis or necrotising papillitis develops more commonly in analgesic abuse nephropathy and in sickle cell disease but may occur as a complication of acute pyelonephritis as well.
- It may affect one or both kidneys.
- Grossly, the necrotic papillae are yellow to grey-white, sharply-defined areas with congested borders and resemble infarction. The pelvis may be dilated.
- Microscopically, necrotic tissue is separated from the viable tissue by a dense zone of polymorphs. The necrotic area shows characteristic coagulative necrosis as seen in renal infarcts.
- Pyonephrosis Rarely, the abscesses in the kidney in acute pyelonephritis are extensive, particularly in cases with obstruction.
- This results in the inability of the abscesses to drain and this transforms the kidney into a multilocular sac filled with pus called pyonephrosis or renal carbuncle.
- Perinephric abscess The abscesses in the kidney may extend through the capsule of the kidney into the perinephric tissue and form a perinephric abscess.
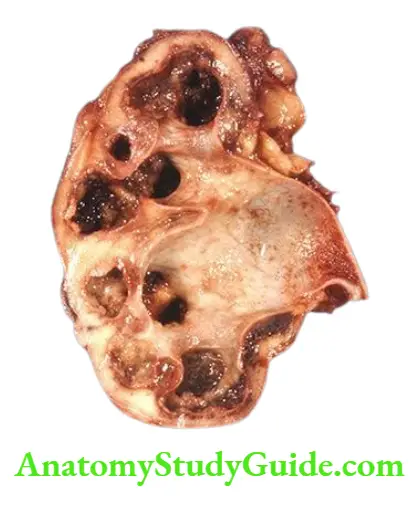
Chronic Pyelonephritis
Chronic pyelonephritis is a chronic tubulointerstitial disease resulting from repeated attacks of inflammation and scarring.
- Etiopathogenesis Depending upon the aetiology and pathogenesis, two types of chronic pyelonephritis are described as reflux nephropathy and obstructive pyelonephritis
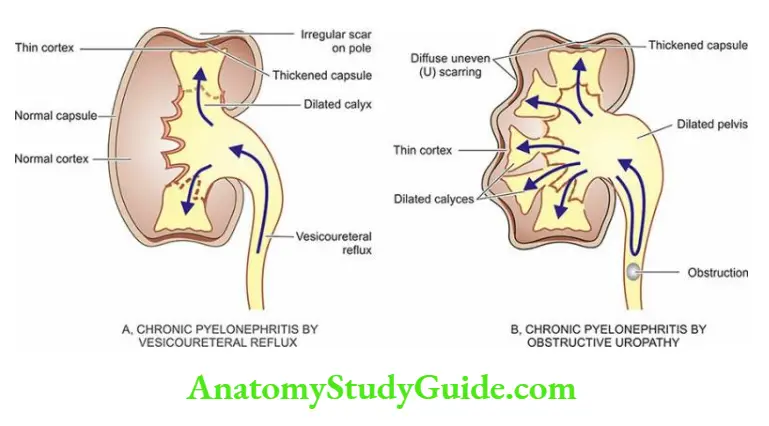
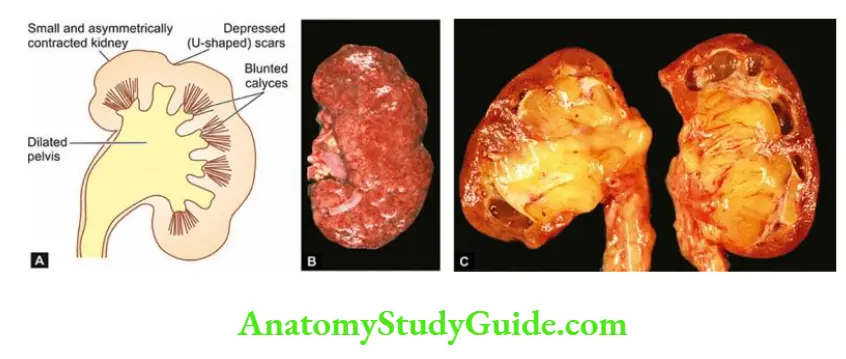
- Reflux nephropathy: Reflux of urine from the bladder into one or both the ureters during micturition is the major cause of chronic pyelonephritis.
- Vesicoureteric reflux is particularly common in children, especially in girls, due to congenital absence or shortening of the intravesical portion of the ureter so that the ureter is not compressed during the act of micturition.
- Reflux results in an increase in pressure in the renal pelvis so that the urine is forced into renal tubules which is eventually followed by damage to the kidney and scar formation.
- Vesicoureteric reflux is more common in patients with urinary tract infections, whether symptomatic or asymptomatic, but reflux of sterile urine can also cause renal damage.
- Obstructive pyelonephritis: Obstruction to the outflow of urine at different levels predisposes the kidney to infection. Recurrent episodes of such obstruction and infection result in renal damage and scarring.
- Rarely, recurrent attacks of acute pyelonephritis may cause renal damage and scarring.
Morphologic Features Grossly, the kidneys show a rather characteristic appearance.
- The kidneys are usually small and contracted (weighing less than 100 gm) showing unequal reduction, which distinguishes it from other forms of contracted kidney.
- The surface of the kidney is irregularly scarred; the capsule can be stripped off with difficulty due to adherence to scars.
- These scars are of variable size and show characteristic U-shaped depressions on the cortical surface. There is generally blunting and dilatation of calyces (calyectasis) and dilated pelvis of the kidney.
Microscopically, predominant changes are seen in the interstitium and the pelvicalyceal system:
- Interstitium: There is a chronic interstitial inflammatory reaction, chiefly composed of lymphocytes, plasma cells and macrophages with pronounced interstitial fibrosis.
- Xanthogranulomatous pyelonephritis is an uncommon variant characterised by a collection of foamy macrophages admixed with other inflammatory cells and giant cells.
- Tubules: The tubules show varying degrees of atrophy and dilatation. Dilated tubules may contain eosinophilic colloid casts producing hybridisation of tubules.
- A few tubules may contain neutrophils.
- Pelvicalyceal system: The renal pelvis and calyces are dilated. The walls of the pelvis and calyces show marked chronic inflammation and fibrosis.
- Lymphoid follicles with germinal centres may be present in the pelvicalyceal walls.
- The lining epithelium may undergo squamous metaplastic change.
- Blood vessels: entrapped in the scarred areas show obliterative endarteritis. There may be changes in hypertensive hyaline arteriolosclerosis.
- Glomeruli: Though the glomerular tuft in the scarred area is usually intact, there is often periglomerular fibrosis. In advanced cases, there may be hyalinisation of glomeruli.
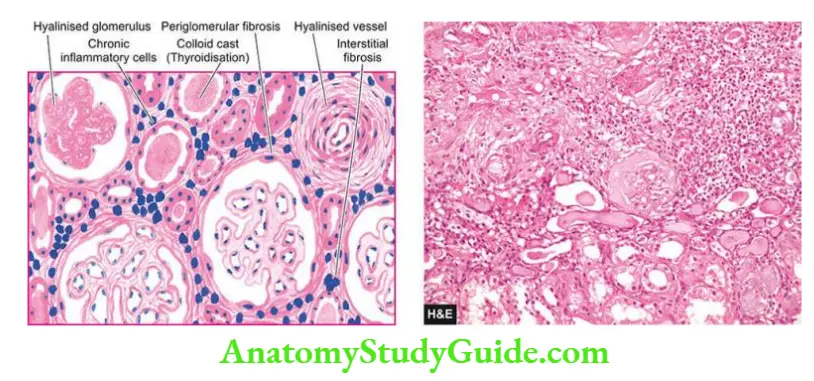
Clinical Features Chronic pyelonephritis often has an insidious onset. The patients present with a clinical picture of chronic renal failure or with symptoms of hypertension.
- Sometimes, the patients may present with features of acute recurrent pyelonephritis with fever, loin pain, lumbar tenderness, dysuria, pyuria, bacteriuria and frequency of micturition.
- Diagnosis is made by intravenous pyelography (IVP). The culture of the urine may give positive results. Longstanding cases of chronic pyelonephritis may develop secondary systemic amyloidosis.
- Features to distinguish chronic pyelonephritis from chronic glomerulonephritis are summed up.
Tuberculous Pyelonephritis
- Tuberculosis of the kidney occurs due to the haematogenous spread of infection from another site, most often from the lungs.
- Less commonly, it may result from ascending infection from tuberculosis of the genitourinary system such as from epididymis or Fallopian tubes.
- The renal lesions in tuberculosis may be in the form of tuberculous pyelonephritis or appear as multiple miliary tubercles.
Morphologic Features Grossly, the lesions in tuberculous pyelonephritis are often bilateral, usually involving the medulla with the replacement of the papillae by caseous tissue.
- Obstruction may result in tuberculous pyonephrosis in which thinned-out renal parenchyma surrounds dilated pelvis and calyces filled with caseous material.
- Histologically, a typical caseating epithelioid cell granulomatous reaction is seen. Acid-fast bacilli can often be demonstrated in the lesions.
Clinical Features Most patients are young to middle-aged adults.
- The clinical presentation is extremely variable but it should always be considered as a possibility in a patient in whom there is persistent sterile pyuria, microscopic haematuria and mild proteinuria after effective antibiotic therapy for urinary tract infection.
- The diagnosis rests on the identification of M. tuberculosis by the repeated culture of urine on LJ media.
Myeloma Nephropathy
Renal involvement in multiple myeloma is referred to as myeloma nephropathy or myeloma kidney. Functional renal impairment in multiple myeloma is a common manifestation, developing in about 50% of patients.
- The pathogenesis of myeloma kidney is related to excess filtration of Bence Jones proteins through the glomerulus, usually kappa (κ) light chains.
- These light chain proteins are precipitated in the distal convoluted tubules in combination with Tamm-Horsfall proteins, the urinary glycoproteins.
- The precipitates form tubular casts which are eosinophilic and often laminated. These casts may induce peritubular interstitial inflammatory reactions.
- Not all light chains are nephrotoxic and their toxicity occurs under the acidic pH of the tubular fluid.
Morphologic Features Grossly, the kidneys may be normal or small and shrunken.
- Histologically, there are some areas of tubular atrophy while many other tubular lumina are dilated and contain characteristic bright pink laminated cracked or fractured casts consisting of Bence-Jones proteins called fractured casts.
- These casts are surrounded by peritubular interstitial inflammatory reactions including the presence of nonspecific inflammatory cells and some multinucleate giant cells induced by tubular casts.
Nephrocalcinosis
Nephrocalcinosis is a diffuse deposition of calcium salts in renal tissue in a number of renal diseases, including hypercalcaemia, hyperphosphataemia and renal tubular acidosis.
- Most commonly, it develops as a complication of severe hypercalcaemia such as due to hyperparathyroidism, hypervitaminosis D, excessive bone destruction in metastatic malignancy, hyperthyroidism, excessive calcium intakes such as in milk-alkali syndrome and sarcoidosis.
- Clinically, patients of hypercalcaemia and nephrocalcinosis may have renal colic, band keratopathy due to calcium deposits in the cornea, visceral metastatic calcification, polyuria and renal failure.
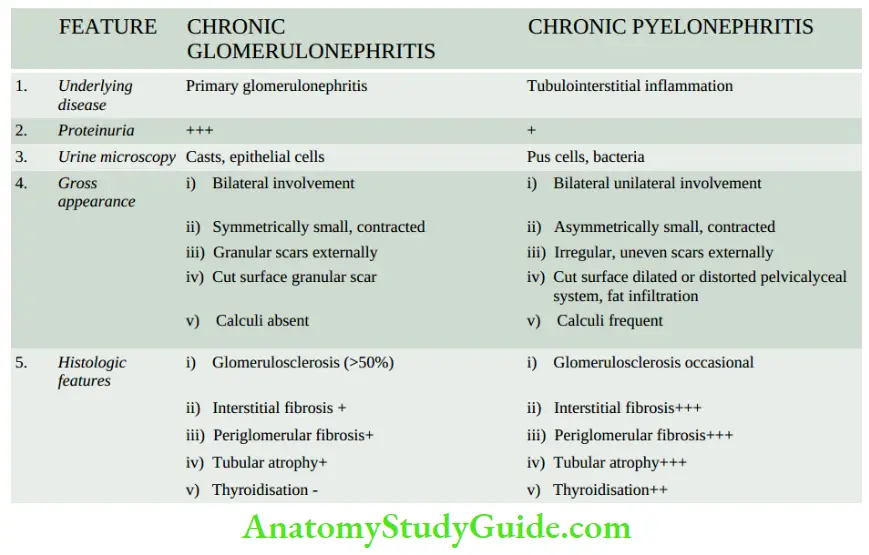
- Morphologic Features Nephrocalcinosis due to hypercalcaemia characteristically shows deposition of calcium in the tubular epithelial cells in the basement membrane, within the mitochondria and in the cytoplasm.
- These concretions may produce secondary tubular atrophy, interstitial fibrosis and nonspecific chronic inflammation in the interstitium.
- As calcification occurs intracellularly, radiological evidence is usually not present until fairly late in the disease. The calcium deposits become first visible as small opacities in the renal papillae.
Tubulointerstitial Diseases
- Tubulointerstitial diseases include several infectious and non-infectious inflammatory conditions involving predominantly tubulointerstitial tissue.
- Acute pyelonephritis is an acute suppurative inflammation of the kidney caused by pyogenic bacteria by either ascending infection or by a haematogenous route.
- Chronic pyelonephritis is a chronic tubulointerstitial disease resulting from repeated attacks of inflammation and scarring.
- It may be due to reflux nephropathy or obstruction to the outflow of urine. Tuberculous pyelonephritis is a variant of chronic pyelonephritis.
- Non-infectious tubulointerstitial diseases include myeloma nephropathy developing in half the cases of multiple myeloma, nephrocalcinosis affecting tubular epithelium due to hypercalcaemia, and tubulointerstitial involvement due to drugs etc.
Obstructive Uropathy
Obstruction in the urinary tract is common and important because it increases the susceptibility to infection and stone formation. Obstruction can occur at any age and in either sex.
- The cause of obstruction may lie at any level of the urinary tract renal pelvis, ureters, urinary bladder and urethra. The obstruction at any of these anatomic locations may be intraluminal, intramural or extramural.
- Important causes are listed and illustrated. The obstruction may be unilateral or bilateral, partial or complete, sudden or insidious.
- Complete bilateral obstruction may result in irreversible renal failure, whereas long-standing chronic partial obstruction may cause various functional abnormalities and anatomic changes.
- There are three important anatomic sequelae of obstruction, namely: hydronephrosis, hydroureter and hypertrophy of the bladder. Before describing these conditions, an account of the most common and important cause of obstructive uropathy, nephrolithiasis, is given first.
Nephrolithiasis
- Nephrolithiasis or urolithiasis is the formation of urinary calculi at any level of the urinary tract.
- Urinary calculi are worldwide in distribution but are particularly common in some geographic locations such as in parts of the United States, South Africa, India and South-East Asia.
- It is estimated that approximately 2% of the population experiences renal stone disease at some time in their life with a male-female ratio of 2:1.
- The peak incidence is observed in the 2nd to 3rd decades of life. Renal calculi are characterised clinically by colicky pain (renal colic) as they pass down along the ureter and manifest by haematuria.

Types Of Urinary Calculi
There are 4 main types of urinary calculi calcium-containing, mixed (struvite), uric acid and cystine stones, and a few rare types.
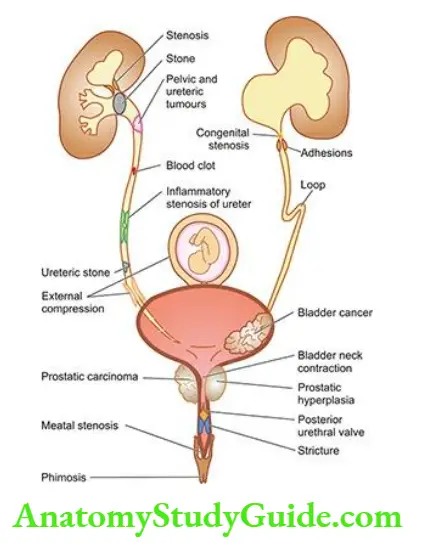
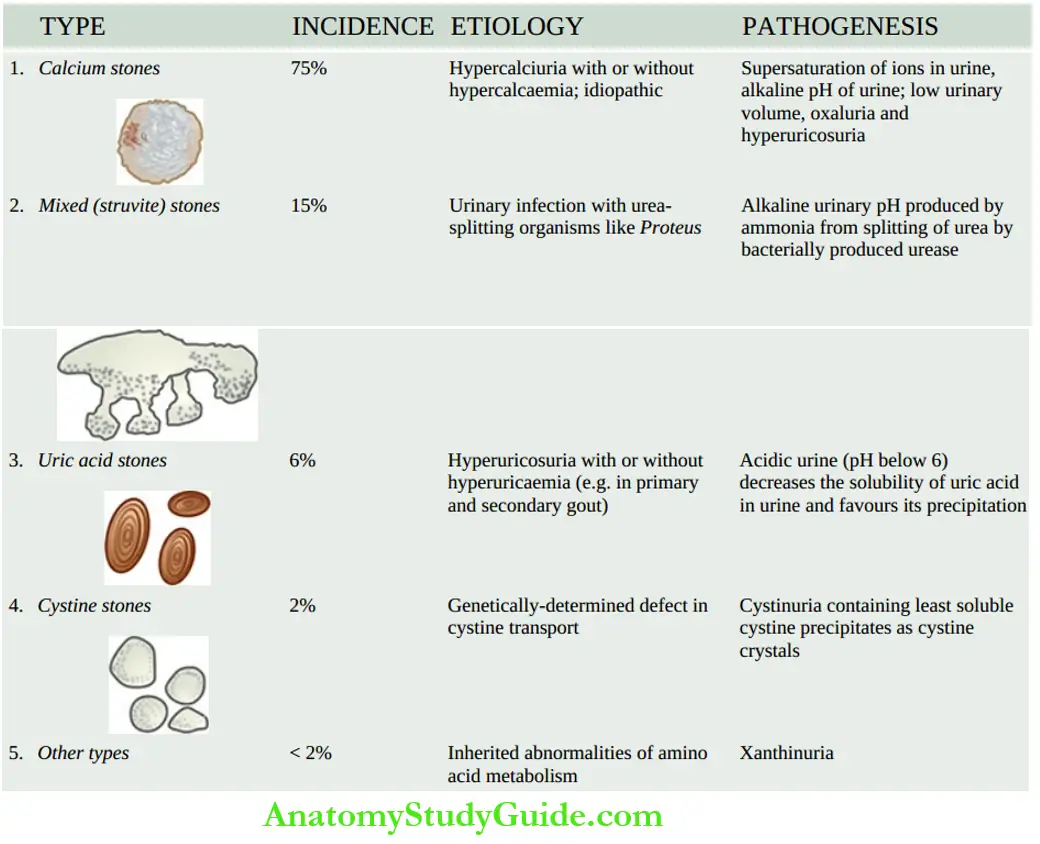
- Calcium Stones are the most common comprising about 75% of all urinary calculi. They may be pure stones of calcium oxalate (50%) or calcium phosphate (5%), or a mixture of calcium oxalate and calcium phosphate (45%).
- The aetiology of calcium stones is variable.
- About 50% of patients with calcium stones have idiopathic hypercalciuria without hypercalcaemia.
- Approximately 10% of cases are associated with hypercalcaemia and hypercalciuria, most commonly due to hyperparathyroidism, or a defect in the bowel (i.e. absorptive hypercalciuria), or in the kidney (i.e. renal hypercalciuria).
- About 15% of patients with calcium stones have hyperuricosuria with a normal blood uric acid level and without any abnormality of calcium metabolism.
- In about 25% of patients with calcium stones, the cause is unknown as there is no abnormality in urinary excretion of calcium, uric acid or oxalate and is referred to as ‘idiopathic calcium stone disease’.
- Pathogenesis The mechanism of calcium stone formation is explained on the basis of an imbalance between the degree of supersaturation of the ions forming the stone and the concentration of inhibitors in the urine.
- A most likely site where the crystals of calcium oxalate and/or calcium phosphate are precipitated is the tubular lining or around some fragment of debris in the tubule acting as a nidus of the stone.
- The stone grows, as more and more crystals are deposited around the nidus.
- A number of other predisposing factors contributing to the formation of calcium stones are alkaline urinary pH, decreased urinary volume and increased excretion of oxalate and uric acid.
- Morphology Calcium stones are usually small (less than a centimetre), ovoid, hard, and with a granular rough surface.
- Their surface may turn dark brown due to old blood pigment deposited in them as a result of repeated trauma caused to the urinary tract by these sharp-edged stones.
- Mixed (Struvite) Stones: About 15% of urinary calculi are made of magnesium ammonium-calcium phosphate, often called struvite; hence mixed stones are also called ‘struvite stones’ or ‘triple phosphate stones’.
- Etiopathogenesis Struvite stones are formed as a result of infection of the urinary tract with urea-splitting organisms that produce urease such as species of Proteus, and occasionally Klebsiella, Pseudomonas and Enterobacter.
- These are, therefore, also known as infection-induced stones. However, E. coli does not form urease.
- Morphology Struvite stones are yellow-white or grey. They tend to be soft and friable and irregular in shape. ‘Staghorn stone’ which is a large, solitary stone that takes the shape of the renal pelvis where it is often formed is an example of struvite stone.
- Uric Acid Stones: Approximately 6% of urinary calculi are made of uric acid. Uric acid calculi are radiolucent, unlike radio-opaque calcium stones.
- Etiology Uric acid stones are frequently formed in cases with hyperuricaemia and hyperuricosuria such as due to primary gout or secondary gout due to myeloproliferative disorders (for example in leukaemias), especially those on chemotherapy, and administration of uricosuric drugs (example salicylates, probenecid).
- Other factors contributing to their formation are acidic urinary pH (below 6) and low urinary volume.
- Pathogenesis The solubility of uric acid at a pH of 7 is 200 mg/dl while at a pH of 5 is 15 mg/dl.
- Thus, as the urine becomes more acidic, the solubility of uric acid in urine decreases and precipitation of uric acid crystals increases favouring the formation of uric acid stones.
- Hyperuricosuria is the most important factor in the production of uric acid stones, while hyperuricaemia is found in about half the cases.
- Morphology Uric acid stones are smooth, yellowish-brown, hard and often multiple. On the cut section, they show a laminated structure.
- Cystine Stones: comprise less than 2% of urinary calculi.
- Etiology Cystine stones are associated with cystinuria due to a genetically-determined defect in the transport of cystine and other amino acids across the cell membrane of the renal tubules and the small intestinal mucosa.
- Pathogenesis The resultant excessive excretion of cystine which is the least soluble of the naturally-occurring amino acids leads to the formation of crystals and eventually cystine calculi.
- Morphology Cystine stones are small, rounded, smooth and often multiple.
- They are yellowish and waxy.
- Other Calculi: Less than 2% of urinary calculi consist of other rare types such as due to inherited abnormality of enzyme metabolism example hereditary xanthinuria developing xanthine stones.
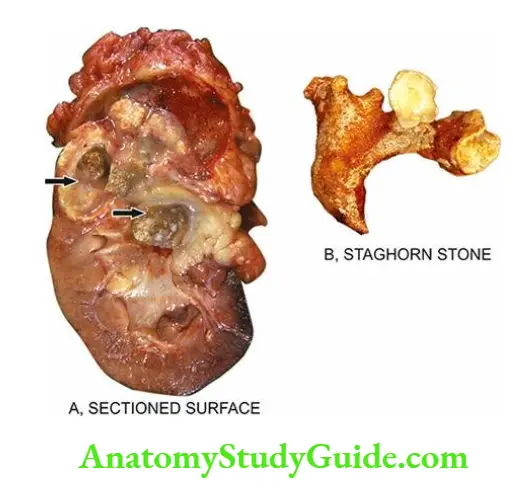
Hydronephrosis
- Hydronephrosis is the term used for the dilatation of the renal pelvis and calyces due to partial or intermittent obstruction to the outflow of urine.
- Hydronephrosis develops if one or both the pelviureteric sphincters are incompetent, as otherwise there will be dilatation and hypertrophy of the urinary bladder but no hydronephrosis.
- Hydroureter nearly always accompanies hydronephrosis. Hydronephrosis may be unilateral or bilateral.
Unilateral Hydronephrosis
This occurs due to some form of ureteral obstruction at the level of pelvic ureteric junction (PUJ). The causes are:
- An intraluminal example is a calculus in the ureter or renal pelvis.
- Intramural examples are congenital PUJ obstruction, atresia of the ureter, inflammatory stricture, trauma, and neoplasm of the ureter or bladder.
- Extramural example obstruction of the upper part of the ureter by the inferior renal artery or vein, pressure on the ureter from outside such as carcinoma cervix, prostate, rectum, colon or caecum and retroperitoneal fibrosis.
Bilateral Hydronephrosis
- This is generally the result of some form of urethral obstruction but can occur from the various causes listed above if the lesions involve both sides. Based on this, hydronephrosis may be of the following types:
- Congenital example atresia of the urethral meatus, congenital posterior urethral valve.
- Acquired examples are bladder tumours involving both ureteric orifices, prostatic enlargement, prostatic carcinoma and prostatitis, bladder neck stenosis, inflammatory or traumatic urethral stricture and phimosis.
Morphologic Features
- The pathologic changes vary depending upon whether the obstruction is sudden and complete, or incomplete and intermittent. The latter situation is more common.
- Grossly, the kidneys may have moderate to marked enlargement. Initially, there is extrarenal hydronephrosis characterised by the dilatation of the renal pelvis medially in the form of a sac.
- As the obstruction persists, there is progressive dilatation of the pelvis and calyces and pressure atrophy of renal parenchyma.
- Eventually, the dilated pelvicalyceal system extends deep into the renal cortex so that a thin rim of the renal cortex is stretched over the dilated calyces and the external surface assumes a lobulated appearance.
- This advanced stage is called intrarenal hydronephrosis.
- An important point of distinction between the sectioned surface of advanced hydronephrosis and polycystic kidney disease is the direct continuity of dilated cystic spaces ( dilated calyces) with the renal pelvis in the former.
- Microscopically, the wall of the hydronephrotic sac is thickened due to fibrous scarring and chronic inflammatory cell infiltration. There is progressive atrophy of tubules and glomeruli along with interstitial fibrosis.
- Stasis of urine in hydronephrosis causes infection (pyelitis) resulting in the filling of the sac with pus, a condition called pyonephrosis.
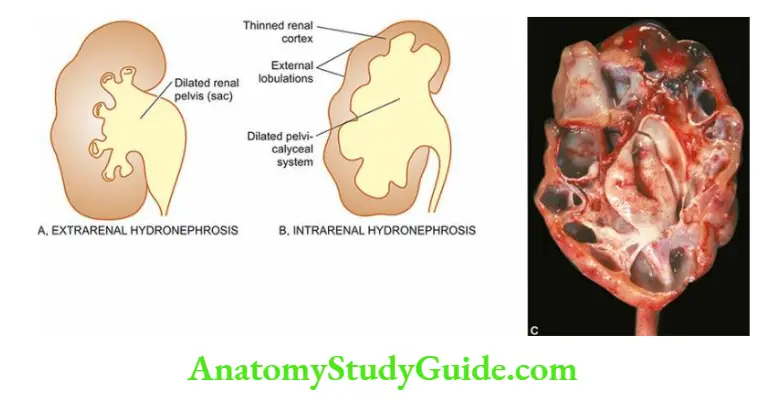
Obstructive Uropathy
- Obstruction in the urinary tract may occur at different levels: intraluminal (example calculi), intramural (example PUJ obstruction), and extramural (for example pregnant uterus).
- Nephrolithiasis or urolithiasis is the formation of urinary calculi at any level of the urinary tract and is the most common cause of obstructive uropathy.
- There are 4 main types of urinary calculi calcium-containing, mixed (struvite), uric acid and cystine stones, and a few rare types.
- There are three important anatomic sequelae of obstruction, namely: hydronephrosis, hydroureter and hypertrophy of the bladder.
- Hydronephrosis develops if one or both of the pelviureteric sphincters are incompetent. Depending upon aetiology, it may be unilateral or bilateral. Hydroureter invariably accompanies it.
Renal Vascular Diseases
Renal blood vessels which enormously perfuse the kidney are affected secondarily in the majority of renal diseases. Renal blood flow is controlled by systemic and local haemodynamic, hormonal and intrinsic intrarenal mechanisms.
Diseases which disturb these controlling mechanisms give rise to primary renal vascular lesions. These diseases are as under:
- Most importantly, hypertensive vascular disease and its consequent renal manifestations in the form of benign and malignant nephrosclerosis.
- Thrombotic microangiopathy
- Renal cortical necrosis
- Renal infarcts
- Renal infarcts are already described in other conditions discussed here.
Hypertension Vascular Disease
Elevated arterial blood pressure is a major health problem, particularly in developed countries.
- Persistent and sustained high blood pressure has damaging effects on the heart, brain (for example cerebrovascular accident or stroke, Chapter 30) and kidneys (benign and malignant nephrosclerosis).
Definition And Classification:
- Hypertension is a common disease in industrialised countries and accounts for 6% of death worldwide.
- Epidemiologic studies have revealed that with an elevation in systolic and diastolic blood pressure (SBP and DBP) above normal in adults, there is a continuously increased risk of cardiovascular disease, stroke and renal disease cardiovascular risk doubles with every 20 mmHg increase in systolic and 10 mmHg increase in diastolic blood pressure above normal levels.
- Criteria for normal blood pressure, prehypertension and hypertension (stage 1 and stage 2) have been recently revised in 2018 by AHA/ACC (American College of Cardiology and American Heart Association). According to these criteria,
- Normal cut-off values for SBP and DBP are taken as <120 and <80 mmHg respectively.
- Arterial blood pressure is considered elevated at an SBP of 120-129 and a DBP of <80 mmHg.
- Arterial or systemic hypertension in adults is defined clinically as persistent elevation of SBP of >130 mmHg, or DBP of >80 mmHg, and is graded into stages 1 and 2 as under
-
-
- Stage 1 hypertension is SBP of 130-139 mmHg, or DBP of 80-89 mmHg.
- Stage 2 hypertension is labelled in cases with corresponding values of SBP as ≥140 mmHg and DBP as ≥90 mmHg.
-
Diastolic pressure is often considered more significant. Since blood pressure varies with many factors such as the age of the patient, exercise, and emotional disturbances like fear and anxiety, it is important to measure blood pressure at least twice during two separate examinations under the least stressful conditions.
Hypertension is generally classified into 2 types:
- Primary or essential hypertension in which the cause of the increase in blood pressure is unknown. Essential hypertension constitutes about 80-95% patients of with hypertension.
- Secondary hypertension is when the increase in blood pressure is caused by diseases of the kidneys, endocrines or some other organs.
- Secondary hypertension comprises the remaining 5-20% of cases of hypertension. According to the clinical course, both essential and secondary hypertension may be benign or malignant.
-
- Benign hypertension: is the moderate elevation of blood pressure and the rise is slow over the years. About 90-95% patients of with hypertension have benign hypertension.
- Malignant hypertension: is marked and sudden increase of SBP >200 mmHg and DBP ≥140 mmHg in a known case of hypertension or in a previously normotensive individual; the patients develop papilloedema, retinal haemorrhages and hypertensive encephalopathy.
- Less than 5% of hypertensive patients develop malignant hypertension, and life expectancy after diagnosis in these patients is generally less than 2 years if not treated effectively.

Etiology And Pathogenesis
The aetiology and pathogenesis of secondary hypertension which comprises less than 10% of cases have been better understood, whereas the mechanism of essential hypertension, which constitutes about 90% of cases, remains largely obscure.
- In general, normal blood pressure is regulated by 2 haemodynamic forces cardiac output and total peripheral vascular resistance.
- Factors which alter these two factors result in hypertension. The role of the kidney in hypertension, particularly in secondary hypertension, by elaboration of renin and subsequent formation of angiotensin 2, is well established (renin-angiotensin system).
- With this background knowledge, we next turn to the mechanisms involved in the two forms of hypertension.
Essential (Primary) Hypertension
By definition, the cause of essential hypertension is unknown but a number of factors are related to its development. These are as under:
- Genetic factors: The role of heredity in the aetiology of essential hypertension has long been suspected.
- The pieces of evidence support the familial aggregation, the occurrence of hypertension in twins, epidemiologic data, experimental animal studies and identification of the hypertension susceptibility gene (angiotensinogen gene).
- Racial and environmental factors: Surveys in the US have revealed a higher incidence of essential hypertension in African Americans than in whites.
- A number of environmental factors have been implicated in the development of hypertension including salt intake, obesity, skilled occupation, higher living standards and individuals under high stress.
- Risk factors modifying the course of essential hypertension: There is sufficient evidence to show that the course of essential hypertension that begins in middle life is modified by a number of factors. These are as under:
- 1. Age Younger the age at which hypertension is first noted but left untreated, the lower the life expectancy.
- 2. Sex Females with hypertension appear to do better than males.
- 3. Atherosclerosis Accelerated atherosclerosis invariably accompanies essential hypertension.
- This could be due to the contributory role of other independent factors like cigarette smoking, elevated serum cholesterol, glucose intolerance and obesity.
- 4. Other risk factors Other factors which alter the prognosis of hypertension include smoking, excess alcohol intake, diabetes mellitus, persistently high diastolic pressure above normal and evidence of end-organ damage heart, eyes, kidney and nervous system.

Pathogenesis Essential hypertension is explained by many theories:
- High plasma level of catecholamines.
- Increase in blood volume i.e. arterial overfilling (volume hypertension) and arteriolar constriction (vasoconstrictor hypertension).
- Increased cardiac output.
- Low-renin essential hypertension is found in approximately 20% of patients due to altered responsiveness to renin release.
- High-renin essential hypertension is seen in about 15% of cases due to decreased adrenal responsiveness to angiotensin 2.
Secondary Hypertension
Though much less common than essential hypertension, mechanisms underlying secondary hypertension with identifiable causes have been studied more extensively.
Based on the aetiology, these are described under four headings: renal hypertension, endocrine hypertension, hypertension associated with coarctation of the aorta and neurogenic causes.
1. Renal Hypertension: Hypertension produced by renal diseases is called renal hypertension.
Renal hypertension is subdivided into 2 groups:
- Renal vascular hypertension e.g. in occlusion of a major renal artery, pre-eclampsia, eclampsia, polyarteritis nodosa and fibromuscular dysplasia of renal artery.
- Renal parenchymal hypertension example of various types of glomerulonephritis, pyelonephritis, interstitial nephritis, diabetic nephropathy, amyloidosis, polycystic kidney disease and renin-producing tumours.
- In either case, renal hypertension can be produced by one of the following 3 inter-related pathogenetic mechanisms
1. Activation of the renin-angiotensin system: Renin is a proteolytic enzyme produced and stored in the granules of the juxtaglomerular cells surrounding the afferent arterioles of the glomerulus.
- The release of renin is stimulated by renal ischaemia, sympathetic nervous system stimulation, depressed sodium concentration, fluid depletion and decreased potassium intake.
- Released renin is transported through the bloodstream to the liver where it acts upon substrate angiotensinogen, an α2-globulin synthesised in the liver, to form angiotensin I, a decapeptide.
- Angiotensin 1 is converted into angiotensin 2, an octapeptide, by the action of convertase in the lungs.
- Angiotensin 2 is the most potent naturally-occurring vasoconstrictor substance and its pressor action is mainly attributed to peripheral arteriolar vasoconstriction.
- The other main effect of angiotensin II is to stimulate the adrenal cortex to secrete aldosterone via the AT1 receptor and thus promote the reabsorption of sodium and water.
Thus, the renin-angiotensin system is concerned mainly with 3 functions:
-
- Control of blood pressure by altering plasma concentration of angiotensin II and aldosterone.
- Regulation of sodium and water content.
- Regulation of potassium balance.
- The renin-angiotensin mechanism is summarised in.
2. Sodium and water retention: Blood volume and cardiac output, both of which have a bearing on blood pressure, are regulated by the blood level of sodium which is significant for maintaining extracellular fluid volume.
Blood concentration of sodium is regulated by 3 mechanisms:
- Release of aldosterone from activation of the renin-angiotensin system, as already explained.
- Reduction in GFR due to reduced blood flow occurs in reduced renal mass or renal artery stenosis. This results in the proximal tubular reabsorption of sodium.
- Release of atriopeptin hormone from atria of the heart in response to volume expansion. These peptides cause increased GFR and inhibit sodium reabsorption.
3. Release of vasodepressor material: A number of vasodepressor materials and antihypertensives counterbalance the vasopressor effect of angiotensin 2.
These substances include prostaglandins (PGE2, PGF2, PGA or medullin) released from interstitial cells of the medulla, urinary kallikrein-kinin system and platelet-activating factor.
2. Endocrine Hypertension: A number of hormonal secretions may produce secondary hypertension as follows:
- Adrenal gland examples in primary aldosteronism, Cushing’s syndrome, adrenal virilism and pheochromocytoma.
- Thyroid examples are hyper- and hypothyroidism.
- Parathyroid gland for example hypercalcaemia in hyperparathyroidism.
- Oral contraceptives The oestrogen component in oral contraceptives stimulates hepatic synthesis of renin substrate.
3. Coarctation Of the Aorta: Coarctation of the aorta causes systolic hypertension in the upper part of the body due to constriction itself. Diastolic hypertension results from changes in circulation.
4. Neurogenic: Psychogenic, polyneuritis, increased intracranial pressure and section of the spinal cord are all uncommon causes of secondary hypertension.
Consequences Of Hypertension
Systemic hypertension causes major effects on the following five main organs:
- Blood vessels hypertensive arteriolosclerosis
- Heart hypertensive heart disease
- Kidneys nephrosclerosis
- Nervous system stroke
- Eyes hypertensive retinopathy
-
- While the effects of hypertension on other target organs are discussed elsewhere in the textbook, the renal effects in the form of benign and malignant nephrosclerosis are discussed below.
- An important and early clinical marker for renal injury from hypertension and risk factor for cardiovascular disease is macroalbuminuria (albuminuria >150 mg/day or random urine albumin/creatinine ratio of >300 mg/gm creatinine), or microalbuminuria estimated by radioimmunoassay ( microalbumin 30-300 mg/day or random urine microalbumin/creatinine ratio of 30-300 mg/gm creatinine).
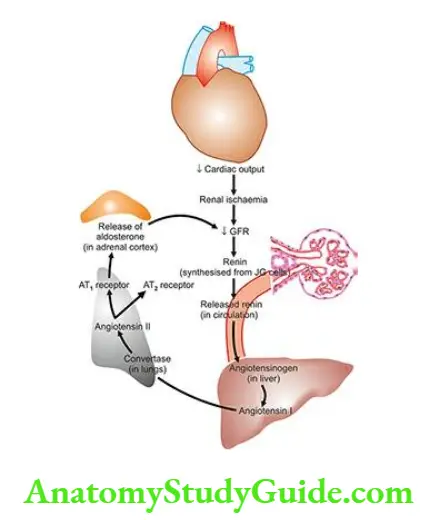
Benign Nephrosclerosis
- Benign nephrosclerosis is the term used to describe the kidney of a benign phase of hypertension.
- Mild benign nephrosclerosis is the most common form of renal disease in persons over 60 years of age but its severity increases in the presence of hypertension and diabetes mellitus.
Morphologic Features Grossly, both the kidneys are affected equally and are reduced in size and weight, often weighing about 100 gm or less.
- The capsule is often adherent to the cortical surface. The surface of the kidney is finely granular and shows V-shaped areas of scarring.
- The cut surface shows a firm kidney and narrowed cortex.
- Microscopically, there are primarily diffuse vascular changes which produce parenchymal changes secondarily as a result of ischaemia.
- The histologic changes are, thus, described as vascular and parenchymal
- Vascular changes: Changes in blood vessels involve arterioles and arteries up to the size of arcuate arteries. There are 2 types of changes in these blood vessels:
- Hyaline arteriolosclerosis results in homogeneous and eosinophilic thickening of the wall of small blood vessels.
- Intimal thickening due to the proliferation of smooth muscle cells in the intima.
- Parenchymal changes: As a consequence of ischaemia, there is a variable degree of atrophy of the parenchyma.
- This includes glomerular shrinkage, deposition of collagen in Bowman’s space, periglomerular fibrosis, tubular atrophy and fine interstitial fibrosis.
- Vascular changes: Changes in blood vessels involve arterioles and arteries up to the size of arcuate arteries. There are 2 types of changes in these blood vessels:
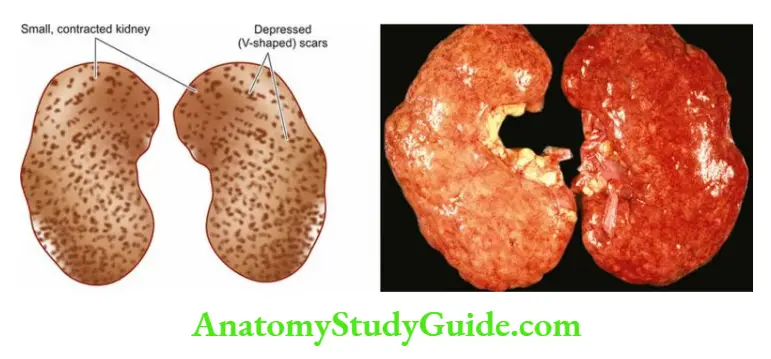
Clinical Features There is variable elevation of the blood pressure with headache, dizziness, palpitation and nervousness. Eye ground changes may be found but papilloedema is absent.
- Renal function tests and urine examinations are normal in the early stage. In long-standing cases, there may be mild proteinuria with some hyaline or granular casts. Rarely, renal failure and uraemia may occur.
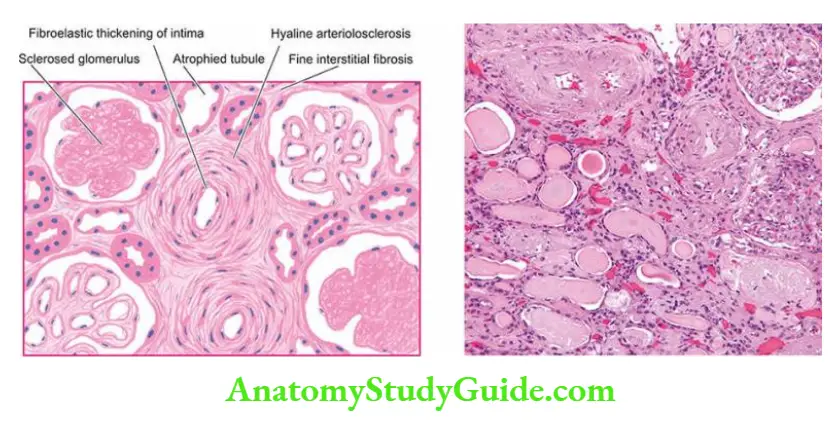
Malignant Nephrosclerosis
- Malignant nephrosclerosis is a form of renal disease that occurs in malignant or accelerated hypertension.
- Malignant nephrosclerosis is uncommon and usually occurs as a superimposed complication in 5% of cases of pre-existing benign essential hypertension or in those having secondary hypertension with identifiable causes such as in chronic renal diseases.
- However, the pure form of the disease also occurs, particularly at younger ages with preponderance in males.
- Morphologic Features Grossly, the appearance of the kidney varies.
- n a case of malignant hypertension superimposed on pre-existing benign nephrosclerosis, the kidneys are small in size, shrunken and reduced in weight and have finely granular surfaces.
- However, the kidneys of a patient who develops malignant hypertension in pure form are enlarged, oedematous and have petechial haemorrhages on the surface producing so-called ‘flea-bitten kidney’.
- The cut surface shows a red and yellow mottled appearance. Microscopically, most commonly the changes are superimposed on benign nephrosclerosis. These changes are under
1. Vascular changes: These are more severe and involve the arterioles. The two characteristic vascular changes seen are as under:
- Necrotising arterioles develop on hyaline arteriolosclerosis. The vessel wall shows fibrinoid necrosis, a few acute inflammatory cells and small haemorrhages.
- Hyperplastic intimal sclerosis or onionskin proliferation is characterised by concentric laminae of proliferated smooth muscle cells, collagen and basement membranes.
2. Ischaemic changes: The effects of the vascular narrowing on the parenchyma include tubular loss, fine interstitial fibrosis and foci of infarction necrosis.
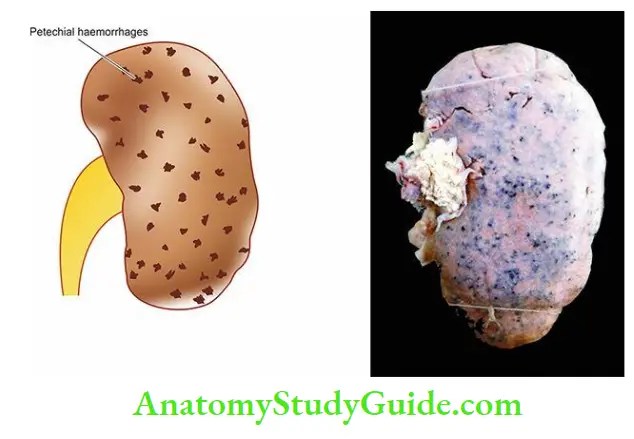
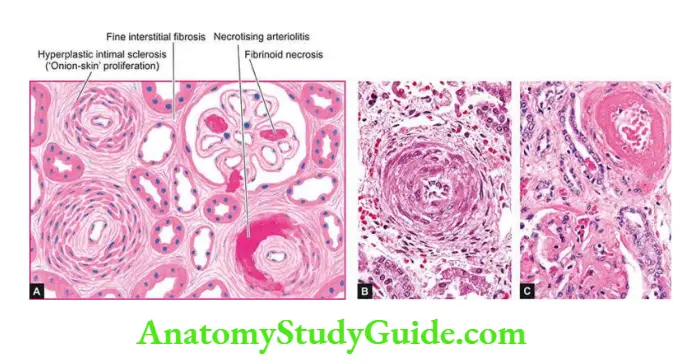
Clinical Features The patients of malignant nephrosclerosis have malignant or accelerated hypertension with SBP of >200 and DBP of ≥140 mmHg. Headache, dizziness and impaired vision are commonly found.
- The presence of papilloedema distinguishes the malignant from the benign phase of hypertension. The urine frequently shows microscopic haematuria and proteinuria.
- Renal function tests show deterioration during the course of the illness. Azotaemia (high BUN and serum creatinine) and uraemia develop soon if malignant hypertension is not treated aggressively.
- Approximately 90% of patients die within one year from causes such as uraemia, congestive heart failure and cerebrovascular accidents.

Thrombotic Microangiopathy
Thrombotic renal disease encompasses a group of diseases having in common the formation of thrombi composed of platelets and fibrin in arterioles and glomeruli of the kidney and culminating clinically in acute renal failure.
- Causes of thrombotic microangiopathy of renal microvasculature are listed in.
- The common clinical manifestations include microangiopathic haemolytic anaemia, thrombocytopenia, DIC, and eventually renal failure.
Pathogenesis In all such cases, endothelial injury appears to be the trigger for vascular changes. The injured endothelial surface causes the following effects:
- Passage of plasma constituents to the subendothelial zone of the microvasculature.
- Promotes thrombosis.
Morphologic Features The lesions closely resemble those of malignant nephrosclerosis. The features are as under:
- Fibrinoid necrosis of arterioles.
- Thrombi in renal microvasculature.
- Oedema of the intima of arterioles.
- Consolidation, necrosis and congestion of glomeruli.
If the renal lesions are massive, the prognosis is generally lethal.
Renal Cortical Necrosis
Renal cortical necrosis is infarction of the renal cortex varying from microscopic foci to a situation where most of the renal cortex is destroyed.
- The medulla, the juxtamedullary cortex and a rim of cortex under the capsule are usually spared.
- The condition develops most commonly as an obstetrical emergency (for example in eclampsia, pre-eclampsia, or premature separation of the placenta).
- Other causes include septic shock, poisoning, severe trauma etc. The lesions may be focal, patchy or diffuse. The gross and microscopic characteristics of infarcts of the cortex are present.
- Patients present with sudden oliguria or anuria and haematuria. If the process has involved the renal cortex extensively, acute renal failure and uraemia develop and the prognosis is grave.
Renal Vascular Diseases
- Hypertension may be primary or essential hypertension in which the cause is unknown (about 90% of cases) or secondary which is caused by diseases of the kidneys, endocrines or some other organs.
- Clinically, hypertension may be benign (moderate and slow elevation of SBP ≥ 130 or DBP of >80 mmHg), or malignant hypertension in which there is a marked and sudden increase of blood pressure SBP≥ 200 and DBP >140 mmHg in a known case of hypertension.
- The main mechanisms of essential hypertension are activation of the renin-angiotensin pathway while secondary hypertension may be due to endocrine, aortic coarctation or neurogenic causes.
- Systemic hypertension causes major effects in five main organs heart (hypertensive heart disease), blood vessels (arteriolosclerosis), nervous system (stroke), eyes (retinopathy), and kidneys (benign and malignant nephrosclerosis).
- Thrombotic renal disease, renal cortical necrosis and infarcts are some other diseases of renal blood vessels.
Tumours Of The Kidney
Both benign and malignant tumours occur in the kidney, the latter being more common.
- These may arise from renal tubules (adenoma, adenocarcinoma), embryonic tissue (mesoblastic nephroma, Wilms’ tumour), mesenchymal tissue (angiomyolipoma, medullary interstitial tumour) and from the epithelium of the renal pelvis (urothelial carcinoma).
- Besides these tumours, the kidney may be the site of the secondary tumours.
- The WHO classification of renal tumours (2016) is given in 20; the important forms are described below.
Benign Tumours
Benign renal tumours are usually small and uncommon, often an incidental finding at autopsy or nephrectomy.
Papillary Adenoma
- Cortical tubular adenomas are more common than other benign renal neoplasms. They are frequently multiple and associated with chronic pyelonephritis or benign nephrosclerosis.
- Grossly, these tumours may form tiny nodules up to 3 cm in diameter. They are encapsulated and white or yellow.
Microscopically, they are composed of tubular cords or papillary structures projecting into cystic space. The cells of the adenoma are usually uniform, cuboidal with no atypicality or mitosis.
- However, the size of the tumour rather than histologic criteria is considered a more significant parameter to predict the behaviour of the tumour those larger than 3 cm in diameter are potentially malignant and metastasising.
Oncocytoma
- Oncocytoma is a relatively more common benign epithelial renal tumour arising from collecting ducts.
- Grossly, the tumour is encapsulated and has variable size. The Cut section is homogenous and has a characteristic mahogany-brown or tan colour.
- Microscopically, the tumour cells are plump with abundant, finely granular, acidophilic cytoplasm and round nuclei. Electron microscopy demonstrates numerous mitochondria in the cytoplasm.
- The tumour has an excellent prognosis and rarely ever undergoes malignant transformation.
Other Benign Tumours
- Angiomyolipoma is a hamartoma of the kidney that contains differentiated tissue elements derived from blood vessels, smooth muscle and fat. Patients of tuberous sclerosis, a multisystem disease characterised by skin lesions, CNS and renal involvement, frequently have bilateral angiomyolipomas.
- Mesoblastic nephroma is a congenital benign tumour.
- Grossly, the tumour resembles a uterine leiomyoma in having a whorled appearance. Microscopically, it shows the cellular growth of spindle cells derived from secondary mesenchyme.
- Multicystic nephroma is another uncommon tumour of early infancy.
- Grossly, it is a solitary, unilateral well-demarcated tumour of varying size. The cut surface shows a characteristic multilocular appearance.
- Microscopically, the cysts are lined by tubular epithelium while the stroma between the cysts contains mesenchymal tissue with some immature blastemal or abortive tubules.
- Some authors consider this entity as a fully-differentiated variant of Wilms’ tumour. However, clinically multicystic nephroma is always benign compared to Wilms’ tumour.
- A medullary interstitial cell tumour is a tiny nodule in the medulla composed of fibroblast-like cells in the hyalinised stroma.
- These tumours used to be called renal fibromas but electron microscopy has revealed that the tumour cells are not fibrocytes but are medullary interstitial cells.
- Juxtaglomerular tumour or reninoma is a rare tumour of the renal cortex consisting of sheets of epithelioid cells with many small blood vessels.
- The tumour secretes excessive quantities of renin and, thus, the patients are likely to have hypertension.
Malignant Tumours
The two most common primary malignant tumours of the kidney are renal cell carcinoma and Wilms’ tumour.
- A third malignant renal tumour is a urothelial tumour occurring in the renal pelvis and the rest of the lower urinary tract; it is described in the next section along with other tumours of the lower urinary tract.
Renal Cell Carcinoma
- Renal cell carcinoma (RCC) is an adenocarcinoma arising from proximal renal tubular epithelium.
- RCC comprises 70 to 80% of all renal cancers and occurs most commonly in 50 to 70 years of age with male preponderance (2:1).
Etiology And Pathogenesis Various etiologic factors implicated in the aetiology of RCC are as follows:
- Tobacco Tobacco is the major risk factor for RCC, whether chewed or smoked and accounts for 20-30% of cases of RCC. Cigarette smokers have a two-fold higher risk of developing RCC.
- Genetic factors Heredity and first-degree relatives of RCC are associated with higher risk. Although the majority of cases of RCC are sporadic but about 5% of cases are inherited. These cases have the following associations:
-
- von Hippel-Lindau (VHL) disease It is an autosomal dominant cancer syndrome that includes: haemangioblastoma of the cerebellum, retinal angiomas, multiple RCC (clear cell type), pheochromocytoma and cysts in different organs.
- Patients of VHL disease have germline mutations of the tumour suppressor VHL gene located on chromosome 3p, commonly as
homozygous loss of the VHL gene. About 35% of cases of VHL develop RCC.
- Hereditary clear cell RCC These are cases of clear cell type RCC confined to the kidney without other manifestations of VHL but having autosomal dominant inheritance.
- Papillary RCC This form of RCC is characterised by bilateral and multifocal cancer with a papillary growth pattern. The genetic abnormality in these cases lies in the MET gene located on chromosome 7.
- Hereditary leiomyomatosis and RCC These cases having non-renal leiomyomatosis associated with RCC have germline FH (fumarate hydratase) mutations on chromosome 1q42.
-

-
- Chromophobe RCC These cases have genetic defects in the form of multiple losses of whole chromosomes i.e. they have an extreme degree of hypodiploidy.
- Succinate dehydrogenase (SDH)-deficient RCC These cases of RCC have a loss of expression of SDH due to germline mutation in the SDH gene.
- Cystic diseases of the kidneys: Both hereditary and acquired cystic diseases of the kidney have an increased risk of the development of RCC.
- Patients on long-term dialysis develop acquired cystic disease which may evolve into RCC and adenomas. Adult polycystic kidney disease and multicystic nephroma is associated with a higher occurrence of papillary RCC.
- Other risk factors: Besides the above, the following other factors are associated with a higher incidence of RCC:
-
- Exposure to asbestos, heavy metals and petrochemical products-trichloroethylene
- In women, on oestrogen therapy
- Obesity is a risk factor in both males and females
- Analgesic nephropathy
- Tuberous sclerosis
- Hypertension, even on treatment, is a risk factor
Classification Based on the cytogenetics of sporadic and familial tumours, RCC has been reclassified into several types.
- However, major histologic types are clear cell, papillary, and chromophobe types; their contrasting features are summarised in.
Morphologic Features Grossly, RCC commonly arises from the poles of the kidney as a solitary and unilateral tumour, more often in the upper pole.
- The tumour is generally large, golden yellow and circumscribed. Papillary tumours have grossly visible papillae and may be multifocal. About 1% of RCCs are bilateral.
- The cut section of the tumour commonly shows large areas of ischaemic necrosis, cystic change and foci of haemorrhages.
- Another significant characteristic is the frequent presence of tumour thrombus in the renal vein which may extend into the vena cava.
Histologically, there are three major types of RCC while other types are uncommon.
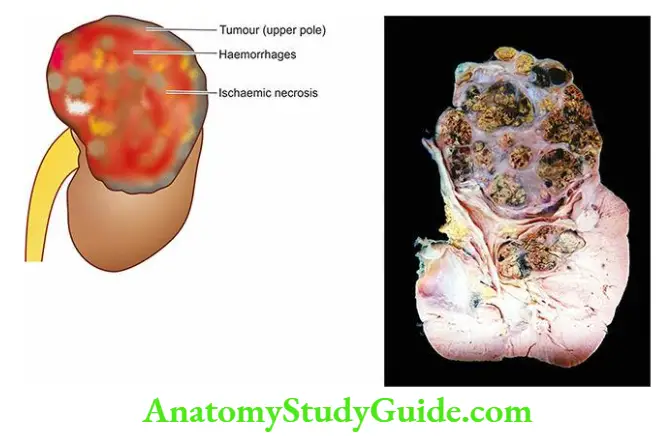
- Clear cell type RCC (75%) This is the most common pattern. The clear cytoplasm of tumour cells is due to the removal of glycogen and lipid from the cytoplasm during the processing of tissues.
- The tumour cells have a variety of patterns: solid, trabecular, acinar and tubular, separated by delicate vasculature. The majority of clear cell tumours are well differentiated.
- Papillary type RCC (15%) The tumour cells are arranged in a papillary pattern over the fibrovascular stalks. The tumour cells are cuboidal with small round nuclei.
- Psammoma bodies may be seen. Based on different molecular pathways and inheritance patterns, though with overlapping morphology, papillary RCC has been divided into type 1 and type 2:
- Type 1 papillary RCC These cases have a germline mutation in the MET gene on chromosome 7. Their morphology shows papillae lined by a single layer of tumour cells having scanty basophilic cytoplasm and low nuclear grade. These cases have a better prognosis.
- Type 2 papillary RCC They have germline mutation of the FH gene in the Kreb cycle and other multiple mutations; this group is also called non-type 1 RCC.
- Morphologically, papillae in these cases are lined by pseudostratified layers of tumour cells having more abundant eosinophilic cytoplasm and higher nuclear grade. These cases have a worse prognosis.
- Chromophobe type RCC (5%) This type shows an admixture of pale clear cells with perinuclear halo and acidophilic granular cells. The cytoplasm of these tumour cells contains many vesicles.
- 4. Other types (5%) Various other histologic types are uncommon. These include: collecting duct RCC, multilocular cystic RCC of low malignant potential, hereditary leiomyomatosis-associated RCC, medullary RCC, tubulocystic RCC, clear cell papillary RCC, and mucinous tubular and spindle carcinoma.
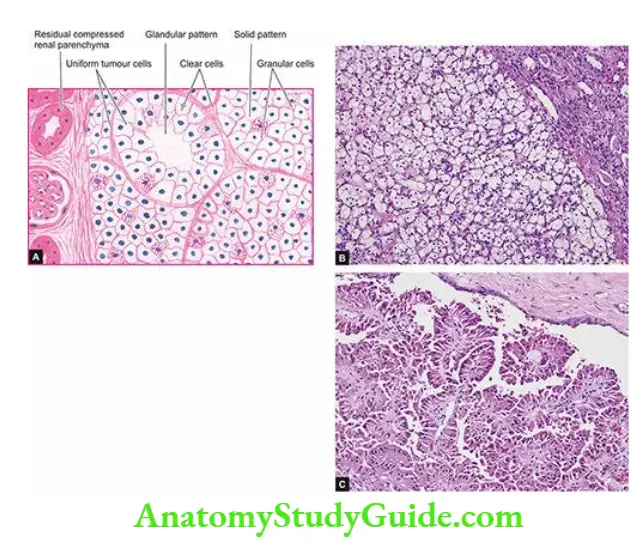
Clinical Features Renal cell carcinoma is generally a slow-growing tumour and the tumour may have been present for years before it is detected.
- The classical clinical evidence for a diagnosis of renal cell carcinoma is the triad of gross haematuria, flank plain and palpable abdominal mass.
- The most common presenting abnormality is haematuria which occurs in about 60% of cases. By the time the tumour is detected, it has spread to distant sites via a haematogenous route to the lungs, brain and bone, and locally to the liver and perirenal lymph nodes.
- Systemic symptoms of fatiguability, weight loss, cachexia and intermittent fever unassociated with evidence of infection are found in many cases at presentation.
- A number of paraneoplastic syndromes due to ectopic hormone production by renal cell carcinoma have been described.
- These include polycythaemia (by erythropoietin), hypercalcaemia (by parathyroid hormone and prostaglandins), hypertension (by renin), effects of feminisation or masculinisation (by gonadotropins) and Cushing’s syndrome (by glucocorticoids).
- The prognosis in renal cell carcinoma depends upon the extent of tumour involvement at the time of diagnosis.
- The overall 5-year survival rate is about 70%. The presence of metastases, renal vein invasion and higher nuclear grade of the tumour are some of the predictors of poor prognosis.
Nephroblastoma (Wilms’ Tumour)
Nephroblastoma or Wilms’ tumour is an embryonic tumour derived from primitive renal epithelial and mesenchymal components.
- It is the most common abdominal malignant tumour of young children, seen most commonly between 1 to 6 years of age with equal sex incidence.
Etiology And Pathogenesis Wilms’ tumour has the following etiologic associations:
- A higher incidence has been seen in monozygotic twins and cases with family histories.
- Association of Wilms’ tumour with some other congenital anomalies has been observed, especially of the genitourinary tract.
- A few other malignancies are known to have a higher incidence of Wilms’ tumour. These include osteosarcoma, botryoid sarcoma, retinoblastoma, neuroblastoma etc.
The genetic pathogenesis of Wilms’ tumour is explained as under:
- Wilms’ tumour-associated gene, WT1 gene, is located on chromosome 11p13. The normal function of the WT1 gene is a synthesis of protein required for the development of kidneys and gonads (ovaries/testes).
- A mutated form of the WT1 gene is often by a deletion in people with Wilms’ tumour, aniridia (i.e. absence of the iris), genitourinary anomalies, and mental retardation syndrome, known by the acronym WAGR syndrome.
- As a result of this deletion, affected individuals are missing one copy of the WT1 gene in each cell and such individuals are prone to develop Wilms’ tumour and several other forms of cancers.
- Due to a common genetic mutation, individuals with gonadal dysgenesis are at higher risk of developing Wilms’ tumour.
- Another molecular mechanism for development of sporadic cases of Wilms’ tumour is synergism due to a common mutation between the β-catenin pathway and WNT (wingless) signalling pathway.
Morphologic Features Grossly, the tumour is usually quite large, spheroidal, replacing most of the kidney. It is generally solitary and unilateral but 5-10% of cases may have bilateral tumours.
- On the cut section, the tumour shows a characteristic variegated appearance soft, fish flesh-like grey-white to cream-yellow tumour with foci of necrosis and haemorrhages and grossly identifiable myxomatous or cartilaginous elements.
- Invasion into the renal vein is grossly evident in half the cases.
Microscopically, nephroblastoma shows a mixture of primitive epithelial and mesenchymal elements. Most of the tumour consists of small, round to spindled, anaplastic, sarcomatoid tumour cells.
- In these areas, abortive tubules and poorly-formed glomerular structures are present. Mesenchymal elements such as smooth and skeletal muscle, cartilage and bone, fat cells and fibrous tissue, may be seen.
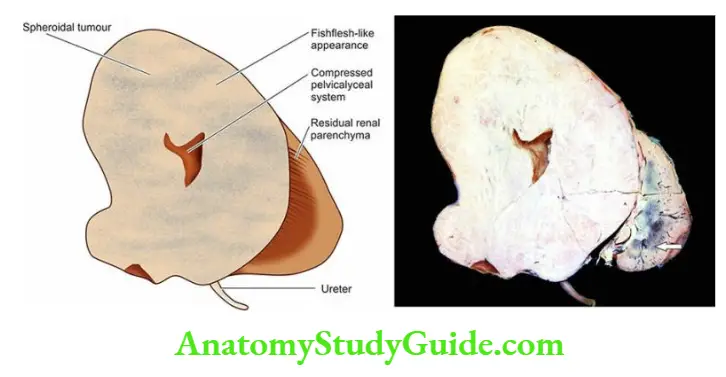
Clinical Features The most common presenting feature is a palpable abdominal mass in a child. Other common abnormalities are haematuria, pain, fever and hypertension. The tumour rapidly spreads via blood, especially to the lungs.
- The prognosis of the tumour with combination therapy of nephrectomy, post-operative irradiation and chemotherapy, has improved considerably and the 5-year survival now is 80- 90%.
Metastatic Tumours
Leukaemic infiltration of the kidneys is a common finding, particularly in chronic myeloid leukaemia. The kidney is a common site for blood-borne metastases from different primary sites, chiefly from cancers of the lungs, breast and stomach.
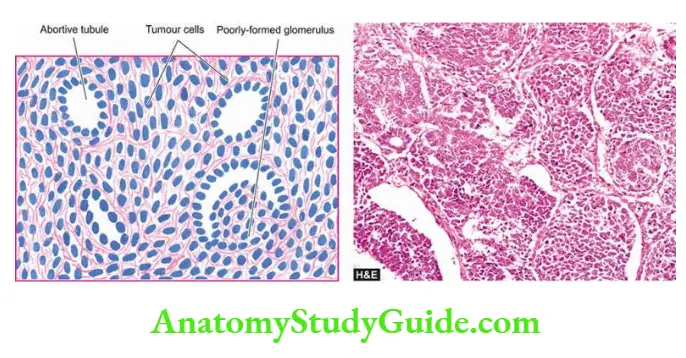
Tumours of Kidneys
- Benign renal tumours are usually small and are often an incidental finding at autopsy or nephrectomy. These include cortical adenoma, oncocytoma, angiomyolipoma etc.
- Renal cell carcinoma (RCC) is an adenocarcinoma arising from the renal tubular epithelium and occurs in adults. It has three major histologic types: clear cell, papillary, and chromophobe type. Extension of the tumour into a renal vein is common.
- Nephroblastoma or Wilms’ tumour is an embryonic tumour derived from primitive renal epithelial and mesenchymal components and occurs in children.
- Urothelial carcinoma is seen in the renal pelvis along with the rest of the lower urinary tract.
- The kidney is a frequent site for blood-borne metastases from different primary sites.
Lower Urinary Tract
Normal Structure
The lower urinary tract consists of ureters, urinary bladder and urethra.
Ureters These are tubular structures, 30 cm in length and half a centimetre in diameter, and extend from the renal pelvis (pelvis-ureteric junction) to the urinary bladder (vesicoureteric junction).
- Normally they enter obliquely into the bladder so that the ureter is compressed during micturition, thus preventing vesicoureteric reflux. Ureters lie retroperitoneally throughout their course.
Histologically, the ureter has an outer fibrous investing layer which overlies a thick muscular layer and is lined internally by transitional epithelium or urothelium similar to the lining of the renal pelvis above and bladder below.
Urinary Bladder It lies extraperitoneally and the peritoneum is reflected on its superior surface. Besides the superior surface (or dome), the bladder has a posterior surface (or base) and two lateral surfaces.
- The trigone is at the base of the bladder and continues as the bladder neck. Normally, the capacity of the bladder is about 400 to 500 ml without over-distension.
- Micturition is partly a reflex and partly a voluntary act under the control of sympathetic and parasympathetic innervation.
Histologically, the greater part of the bladder wall is made up of a muscular layer (detrusor muscle) having 3 coats internal, middle and external.
- The trigone muscle is derived from the prolongation of the longitudinal muscle layer of each ureter. The inner layer of the bladder consists of urothelium 6-7 layers in thickness.
- The superficial epithelial layer is made of larger cells in the form of a row and has abundant eosinophilic cytoplasm; these cells are called umbrella cells.
Urethra It runs from the bladder up to the external meatus. The male urethra consists of 3 parts—prostatic, membranous and penile.
- It is lined in the prostatic part by urothelium but elsewhere by stratified columnar epithelium except near its orifice where the epithelium is stratified squamous.
- The urethral mucosa rests on the highly vascular submucosa and the outer layer of striated muscle. There are numerous small mucous glands in the urethral mucosa.
- The female urethra is shorter and runs from the bladder parallel to the anterior wall of the vagina. The mucous membrane in the female urethra is lined throughout by columnar epithelium except near the bladder where the epithelium is transitional.
- The other layers and mucous glands are similar to those in the male urethra.
- Conditions of the lower urinary tract discussed here are congenital anomalies, inflammation, and tumours.
Congenital Anomalies
Vesicoureteric reflux is the most common anomaly described already. A few others are considered below.
Double Ureter This is a condition in which the entire ureter or only the upper part is duplicated.
- A double ureter is invariably associated with a double renal pelvis, one in the upper part and the other in the lower part of the kidney.
- If a double ureter affects the entire length, then there are two separate openings into the bladder on one side but more commonly they are joined in the intravesical portion and open by a single ureteric orifice.
Ureterocele is a cystic dilatation of the terminal part of the ureter which lies within the bladder wall. The cystic dilatation lies beneath the bladder mucosa and can be visualised by cystoscopy.
Ectopia Vesicae (Exstrophy) This is a rare condition owing to congenital developmental deficiency of the anterior wall of the bladder and is associated with the splitting of the overlying anterior abdominal wall.
- This results in the exposed interior of the bladder. There may be prolapse of the posterior wall of the bladder through the defect in the anterior bladder and abdominal wall.
- The condition in males is often associated with epispadias in which the urethra opens on the dorsal aspect of the penis.
- If the defect is not properly repaired, the exposed bladder mucosa gets infected repeatedly and may undergo squamous metaplasia with a subsequently increased tendency to develop carcinoma of the bladder.
Urachal Abnormalities Rarely, there may be a persistence of the urachus in which urine passes from the bladder to the umbilicus. More often, part of the urachus remains patent which may be the umbilical end, bladder end, or central portion.
-
- Persistence of the central portion gives rise to a urachal cyst lined by transitional or squamous epithelium. Adenocarcinoma may develop in the urachal cysts.
- Other abnormalities due to urachal remnants are patent urachus, urachal-umbilical sinus and vesico-urachal diverticulum.
Inflammation
Urinary tract infection (UTI) is common, especially in females and has been described already along with its morphologic consequences. Inflammation of the tissues of the lower urinary tract (ureteritis, cystitis and urethritis) is considered here.
Ureteritis
Infection of the ureter is almost always secondary to pyelitis above, or cystitis below. Ureteritis is usually mild but repeated and long-standing infection may give rise to chronic ureteritis.
Cystitis
- Inflammation of the urinary bladder is called cystitis. Primary cystitis is rare since the normal bladder epithelium is quite resistant to infection.
- Cystitis may occur by the spread of infection from the upper urinary tract as seen following renal tuberculosis, or may spread from the urethra such as in instrumentation.
- Cystitis is caused by a variety of bacterial and fungal infections as discussed in the etiology of pyelonephritis. The most common pathogenic organism in UTI is E. coli, followed in decreasing frequency by Enterobacter, Klebsiella, Pseudomonas and Proteus.
- Infection with Candida albicans may occur in the bladder in immunosuppressed patients. Besides bacterial and fungal organisms, parasitic infestations such as Schistosoma haematobium are common in Middle-East countries, particularly in Egypt.
- Chlamydia and Mycoplasma may occasionally cause cystitis. In addition, radiation, direct exposure to chemical irritants, foreign bodies and local trauma may all initiate cystitis.
- Cystitis, like UTI, is more common in females than in males because of the shortness of the urethra which is liable to faecal contamination and due to mechanical trauma during sexual intercourse.
- In males, prostatic obstruction is a frequent cause of cystitis. All forms of cystitis are clinically characterised by a triad of symptoms—frequency (repeated urination), dysuria (painful or burning micturition) and low abdominal pain.
- There may, however, be systemic manifestations of bacteraemia such as fever, chills and malaise.
Morphologic Features Cystitis may be acute or chronic.
Acute Cystitis Grossly, the bladder mucosa is red, swollen and haemorrhagic. There may be suppurative exudate or ulcers on the bladder mucosa.
- Microscopically, this form of cystitis is characterised by intense neutrophilic exudate admixed with lymphocytes and macrophages. There is oedema and congestion of mucosa.
Chronic Cystitis Repeated attacks of acute cystitis lead to chronic cystitis.
-
- Grossly, the mucosal epithelium is thickened, red and granular with the formation of polypoid masses. Long-standing cases result in a thickened bladder wall and shrunken cavity.
Microscopically, there is patchy ulceration of the mucosa with the formation of granulation tissue in the regions of polypoid masses.
- Submucosa and muscular coat show fibrosis and infiltration by chronic inflammatory cells. A form of chronic cystitis characterised by the formation of lymphoid follicles in the bladder mucosa is termed cystitis follicular.
- A few other special forms of cystitis having distinct clinical and morphological appearance are described below.
Interstitial Cystitis (Hunner’S Ulcer) This variant of cystitis occurs in middle-aged women. The patients get repeated attacks of severe and excruciating pain on distension of the bladder, frequency of micturition and a great decrease in bladder capacity.
- Cystoscopy often reveals a localised ulcer. The etiology of the condition is unknown but it is thought to be neurogenic in origin.
Microscopically, the submucosa and muscle coat show increased fibrosis and chronic inflammatory infiltrate, chiefly lymphocytes, plasma cells and eosinophils.
Cystitis is cystica As a result of long-standing chronic inflammation, there occurs a downward projection of epithelial nests known as Brunn’s nests from the deeper layer of the bladder mucosa.
- These epithelial cells may appear as small cystic inclusions in the bladder wall, or may actually develop columnar metaplasia with secretions in the lumen of cysts.
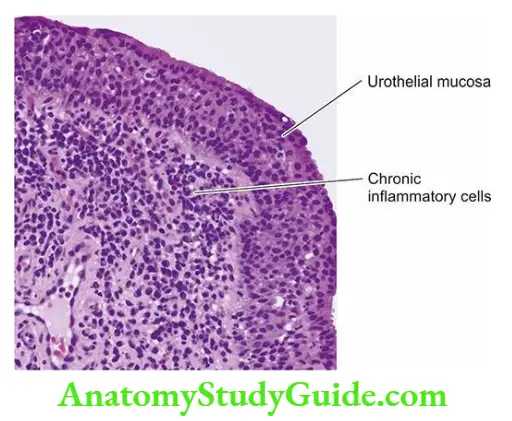
Malakoplakia: This is a rare condition most frequently found in the urinary bladder but can occur in the ureters, kidney, testis and prostate, and occasionally in the gut.
- The etiology of the condition is unknown but it probably results from the persistence of chronic inflammation with the defective phagocytic process by the macrophages.
- Malakoplakia occurs more frequently in immunosuppressed patients and in recipients of transplants.
Grossly, the lesions appear as soft, flat, yellowish, slightly raised plaques on the bladder mucosa. They may vary from 0.5 to 5 cm in diameter.
Microscopically, the plaques are composed of a massive accumulation of foamy macrophages with occasional multinucleate giant cells and some lymphocytes.
- These macrophages have granular PAS-positive cytoplasm and some of them contain cytoplasmic laminated concretions of calcium phosphate called Michaelis-Gutmann bodies.
- These bodies ultrastructurally represent lysosomes filled with partly digested debris of bacteria phagocytosed by macrophages which have not been digested fully by them due to defective phagocytosis.
Polypoid Cystitis: Polypoid cystitis is characterised by papillary projections on the bladder mucosa due to submucosal oedema and can be confused with transitional cell carcinoma. The condition occurs due to indwelling catheters and infection.
Urethritis
Urethritis may be gonococcal or non-gonococcal.
- Gonococcal (gonorrhoeal) urethritis is an acute suppurative condition caused by gonococci (Neisseria gonorrhoeae). The mucosa and submucosa are eventually converted into granulation tissue which becomes fibrotic and scarred resulting in urethral stricture.
- Non-gonococcal urethritis is more common and is most frequently caused by E. coli.
- The infection of the urethra often accompanies cystitis in females and prostatitis in males. Urethritis is one of the components in the triad of Reiter’s syndrome which comprises arthritis, conjunctivitis and urethritis.
- The pathologic changes are similar to inflammation of the lower urinary tract elsewhere but strictures are less common than following gonococcal infection of the urethra.
Tumours Of the Lower Urinary Tract
The majority of lower urinary tract tumours are epithelial. Both benign and malignant tumours occur; the latter being more common.
- About 90% of malignant tumours of the lower urinary tract occur in the urinary bladder, 8% in the renal pelvis and the remaining 2% are seen in the urethra or ureters.
Tumours Of The Bladder:
The tumours of the urinary bladder are divided into epithelial and non-epithelial, the latter being uncommon. Thus, epithelial tumours are the main tumours, the vast majority of which are transitional cell type (urothelial) tumours.
Urothelial (Transitional Cell) Tumours:
More than 90% of bladder tumours arise from the transitional epithelial (urothelium) lining of the bladder in continuity with the epithelial lining of the renal pelvis, ureters, and the major part of the urethra.

Bladder cancer comprises about 3% of all cancers. Most of the cases appear beyond 5th decade of life with 3 times higher preponderance in males than females.
Etiopathogenesis Urothelial tumours in the urinary tract are typically multifocal and the pattern of disease becomes apparent over a period of years.
A number of environmental and host factors are associated with an increased risk of bladder cancer. These are as under:
- Smoking Tobacco smoking is associated with a 2 to 4-fold increased risk of developing bladder cancer, probably due to increased urinary excretion of carcinogenic substances.
- Industrial occupations Workers in industries that produce aniline dyes, rubber, plastic, textiles, and cable have high incidence of bladder cancer.
- Bladder cancer may occur in workers in these factories after a prolonged exposure of about 20 years. The carcinogenic substances responsible for bladder cancer in these cases are the metabolites of β-naphthylamine and benzene.
- Schistosomiasis There is an increased risk of bladder cancer, particularly squamous cell carcinoma, in patients having bilharzial infestation (Schistosoma haematobium) of the bladder.
- Schistosomiasis is common in Egypt and Sudan and accounts for the high incidence of bladder cancer in these countries. It is thought to induce a local irritant effect and initiate squamous metaplasia followed by squamous cell carcinoma.
- Dietary factors Certain carcinogenic metabolites of tryptophan are excreted in the urine of patients with bladder cancer. These metabolites have been shown to induce bladder cancer in experimental animals.
- The role of artificial sweeteners like saccharin, coffee or caffeine and chronic alcoholism in the aetiology of bladder cancer in men is controversial.
- Local lesions A number of local lesions in the bladder predispose to the development of bladder cancer.
- These include ectopia vesicae (atrophied bladder), vesical diverticulum, leukoplakia of the bladder mucosa and urinary diversion in the de-functionalised bladder.
- All these conditions are associated with squamous metaplasia and a high incidence of bladder cancer.
- Drugs Immunosuppressive therapy with cyclophosphamide and patients having analgesic abuse (phenacetin-) nephropathy have a high risk of developing bladder cancer.
- Prior irradiation Patients who have received prior irradiation for some other pelvic cancers have a higher risk of developing bladder cancer.
- The multicentric nature of urothelial cancer and its high rate of recurrence has led to the hypothesis that a field effect in the urothelium is responsible for this form of cancer.
- This is responsible for polychronotropism in bladder cancer i.e. the tumour tends to recur with time and develops at new locations within the urinary tract.
- Several cytogenetic abnormalities have been seen in bladder cancer. These include several mutations of fibroblast growth factor receptor 3, p53, RB gene and p21 gene.
- These mutations are associated with a higher rate of recurrences and metastasis.
Morphologic Features Grossly, urothelial tumours may be single or multiple. About 90% of the tumours are papillary (non-invasive or invasive), whereas the remaining 10% are flat indurated (non-invasive or invasive).
- The most common location in the bladder is the lateral walls, followed by the posterior wall and region of the trigone. The papillary tumours have a free-floating fern-like arrangement with a broad or narrow pedicle.
- The non-papillary tumours are bulkier with ulcerated surfaces. More common locations for either of the two types are the trigone, the region of ureteral orifices and the lateral walls.
- Histologically, the most common epithelial tumours of the bladder are urothelial (90%); others are squamous cells, glandular, small cells and mixed.
Urothelial (Transitional Cell) Tumours The WHO and ISUP (International Society of Urologic Pathology), in 1998 have proposed consensus histologic criteria to categorise urothelial tumours into papillomas (exophytic, inverted), carcinoma in situ (CIS), papillary urothelial neoplasms of low malignant potential (PUNLMP), and urothelial carcinoma (low grade and high grade). The salient histologic features of this categorisation are summed up and briefly given below
- Urothelial papilloma Papillomas may occur singly or may be multiple. These may be exophytic or inverted.
- Exophytic papillomas are generally small, less than 2 cm in diameter, having delicate papillae.
- Each papilla is composed of a fibrovascular stromal core covered by normal-looking transitional cells having a normal number of layers (up to 6-7) in thickness.
- The individual cells resemble the normal transitional cells and do not vary in size and shape. Mitoses are absent and basal polarity is retained. Patients with exophytic papillomas may sometimes develop recurrences and require long-term follow-up.
- Inverted papillomas have an endophytic growth pattern and are benign tumours.
- 2. Carcinoma in situ (Flat urothelial carcinoma) Carcinoma in situ (CIS) is characterised by anaplastic malignant cells confined to layers superficial to the basement membrane of the bladder mucosa.
- These flat lesions have cytologic features similar to high-grade urothelial carcinoma such as lack of cohesiveness, nuclear pleomorphism, and mitoses.
- If left untreated, these cases progress to invasive bladder cancer in the vast majority of cases (50-75%).
- Papillary urothelial neoplasms of low malignant potential (PUNLMP) These cases have many features similar to papillomas with additional features such as the increase in the number of epithelial layers, having round to oval nuclei and diffuse nuclear enlargement.
- PUNLMP is unassociated with invasion and may have recurrences.
- Papillary urothelial (Transitional cell) carcinoma Histologic criteria for categorising these tumours are based on architecture, cytologic features and invasiveness:
-
- Architecture takes into consideration the type of papillae and the relationship of cell layers with the basement membrane as regards polarity.
- Cytologic criteria of neoplasm are the extent of resemblance with normal cells, crowding, variation in nuclear size, shape, chromatin, nucleoli (their presence and type), and mitotic figures.
- Criteria for invasion in papillary as well as non-papillary tumours are penetration of the basement membrane of the bladder mucosa and the presence or absence of invasion by neoplastic cells in the muscle. Based on these salient features, the characteristics of low and high grades of the tumour are as under
- Non-invasive papillary urothelial carcinoma, low grade These tumours show fused and branching papillary patterns but overall there is an orderly arrangement of layers of cells.
- These cells are cohesive and show mild variation in polarity, nuclear size, chromatin and shape (round to oval), and inconspicuous small and regular nucleoli. Mitotic figures are infrequent and are seen in the lower half.
- Non-invasive papillary urothelial carcinoma, high-grade High-grade tumours have increased thickness and have fused and branching papillae which show quite a disorderly arrangement.
- The tumour cells show nuclear enlargement, moderate to marked variation in nuclear size, shape, hyperchromatism, and multiple prominent nucleoli. Mitoses are frequent and atypical and seen at any level in the epithelial layers.
- Invasive urothelial carcinoma Any grade of papillary urothelial carcinoma may show invasion into lamina propria or further into muscularis propria (detrusor).
- Several variations in patterns and cellular morphology of invasive urothelial carcinoma are seen: nested, microcystic, micropapillary, lymphoepithelioma-like, plasmacytoid, sarcomatoid, giant cell, clear cell and poorly differentiated.
- Foci of squamous or glandular metaplasia may be seen in any grade of the tumour.
- Other Variants Although urothelial carcinoma is the most common, a few less common histologic types are seen in the bladder as under:
- Squamous cell carcinoma comprises about 5% of bladder carcinomas. Unlike TCC which is mostly papillary and non-ulcerating, most squamous carcinomas of the bladder are sessile, nodular, infiltrating and ulcerating.
- The association between squamous carcinoma and schistosomiasis has already been highlighted. The carcinoma may be
well-differentiated with keratin pearl formation, or maybe anaplastic. - Adenocarcinoma of the bladder is rare. Adenocarcinoma has an association with exstrophy of the bladder with glandular metaplasia or may arise from urachal rests, periurethral and periprostatic glands, or from cystitis cystica.
- The tumour is characterised by glandular and tubular patterns with or without mucus production.
- Urachal carcinoma This is a rare type of carcinoma occurring in the dome of the bladder and arises from remnants of the urachus.
- Morphologically, it is a variant of adenocarcinoma.
- Neuroendocrine carcinoma (Small cell carcinoma) This variant has morphologic resemblance with small cell carcinoma of the lung or other neuroendocrine carcinomas and has a worse outcome.
-
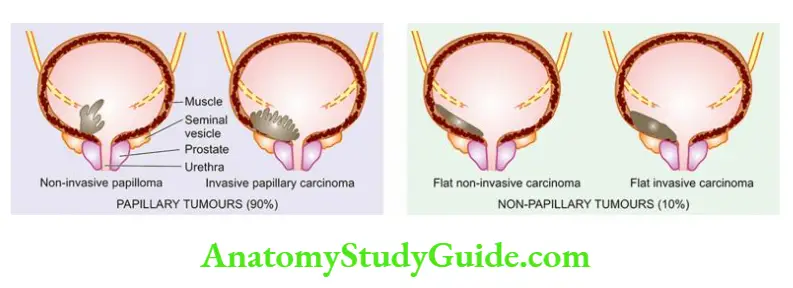
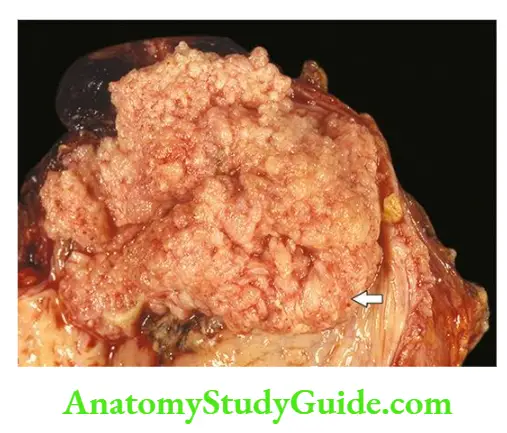
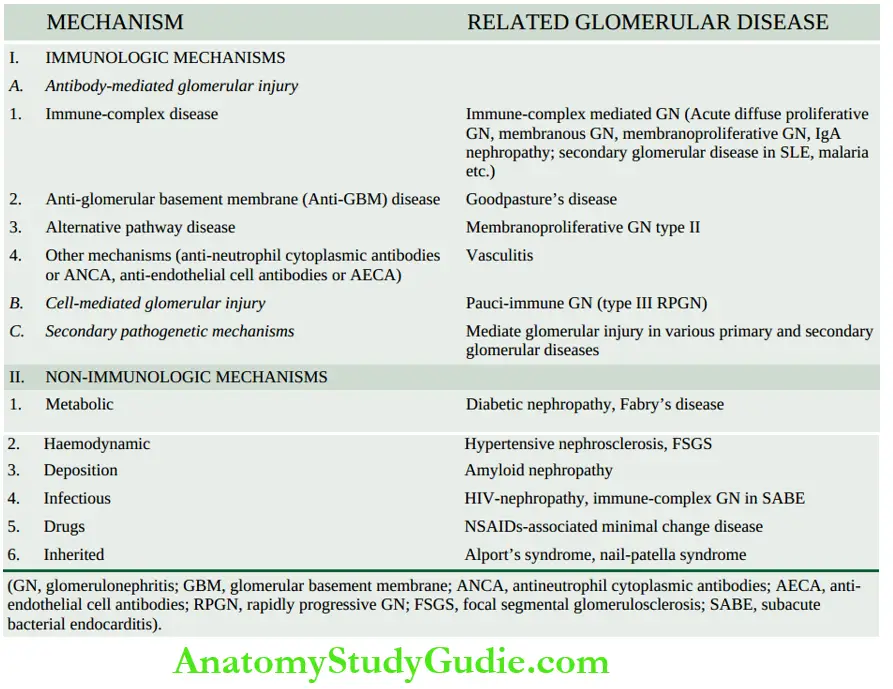
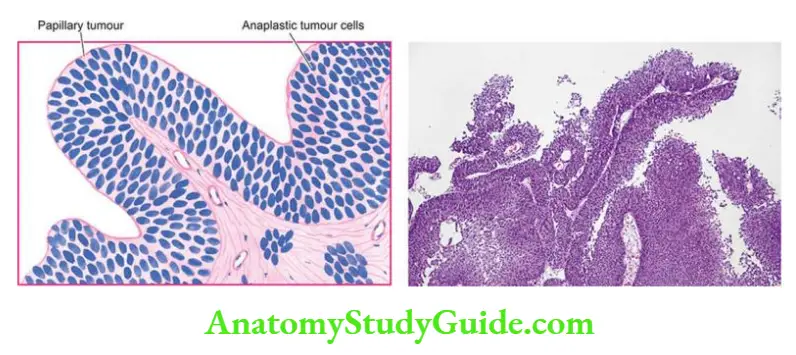
Staging Of Bladder Cancer The clinical behaviour and prognosis of bladder cancer can be assessed by the following simple staging system:
Stage 0: Carcinoma confined to the mucosa.
Stage A: Carcinoma invades the lamina propria but not the muscular.
Stage B1: Carcinoma invades the superficial muscle layer.
Stage B2: Carcinoma invades the deep muscle layer.
Stage C: Carcinoma invades the perivesical tissues.
Stage D1: Carcinoma shows regional metastases.
Stage D2: Carcinoma shows distant metastases.
Non-epithelial Bladder Tumours:
Mesenchymal tumours of the bladder are less common and may be benign or malignant.
Benign mesenchymal tumours of the bladder are uncommon but the most common is leiomyoma. Other less common examples are neurofibroma, haemangioma and granular cell myoblastoma.
Malignant Rhabdomyosarcoma is the most frequent malignant mesenchymal tumour. It exists in 2 forms:
- The adult form occurs in adults over 40 years of age and resembles the rhabdomyosarcoma of skeletal muscle.
- Childhood form occurs in infancy and childhood and appears as a large polypoid, soft, fleshy, grapelike mass and is also called sarcoma botryoides or embryonal rhabdomyosarcoma.
- It is morphologically characterised by masses of embryonic mesenchyme consisting of masses of highly pleomorphic stellate cells in the myxomatous background. Similar tumours occur in the female genital tract.
Tumours Of The Renal Pelvis And Ureters
Almost all the tumours of the renal pelvis and ureters are of epithelial origin. They are of the same type as are seen in the urinary bladder. However, tumours in the ureters are quite rare.
Tumours Of The Urethra
Tumours of the urethra are rare except for the urethral caruncle which is a tumour-like lesion.
Urethral Caruncle Urethral caruncle is not uncommon. It is an inflammatory lesion present on the external urethral meatus in elderly females.
Grossly, the caruncle appears as a solitary, 1 to 2 cm in diameter, pink or red mass, protruding from the urethral meatus. It is quite friable and ulcerated.
Microscopically, the mass may be covered by squamous or transitional epithelium or there may be an ulcerated surface.
The underlying tissues show proliferating blood vessels, fibroblastic connective tissue and intense acute and chronic inflammatory infiltrate. Thus, the histologic appearance closely resembles a pyogenic granuloma.
Urethral Carcinoma Carcinoma of the urethra is uncommon. In most cases, it occurs in the distal urethra near the external meatus and thus is commonly squamous cell carcinoma.
Less often, there may be transitional cell carcinoma or adenocarcinoma arising from periurethral glands.
Diseases of the Lower Urinary Tract
- Vesicoureteric reflux, ectopia vesicae and urachal abnormalities are some of the congenital anomalies of the lower urinary tract.
- Ureteritis and cystitis may accompany pyelitis.
- Urethritis may be gonococcal or non-gonococcal.
- Epithelial tumours are the main tumours, the vast majority of which are transitional cell type (urothelial) tumours.
- Urothelial tumours include papillomas (exophytic, inverted), carcinoma in situ, papillary urothelial neoplasms of low malignant potential (PUNLMP), and urothelial carcinoma (low grade and high grade).
- Based on cellular features (architecture, atypia, invasion), papillary urothelial carcinoma may be low-grade or high-grade.
- The urethral caruncle is an inflammatory lesion on the external urethral meatus in elderly females.
Leave a Reply