Computer Aided Drug Design Introduction
Computers are an essential tool in modern medicinal chemistry and are important in both drug discovery and development. The development of this powerful desktop enabled the chemist to predict the structure and value of properties of known, unknown, stable and unstable molecular species using mathematical equation. Solving this equation gives required data. Graphic package convert the data for the structure of a chemical species into a variety of visual formats.
Table of Contents
Consequently, in medicinal chemistry, it is now possible to visualize the three dimensional shape of both the ligands and their target sites. In addition, sophisticated computational chemistry packages also allow the Medicinal Chemist’s to evaluate the interaction between a compound and its target site before synthesizing that compound.
This means that, medicinal chemists need only to synthesize and test the compounds that considerably increase the potency, that is, it increases the chance of discovering a potent drug. It also significantly reduces the cost of drug development process.
Computer Aided Drug Design Various Models
Molecular Modeling in Computer Aided Drug Design
Molecular modeling is a general term that covers a wide range of molecular graphics and computational chemistry techniques used to build, display, manipulate, simulate and analyze molecular structure and to calculate properties of these structures. Molecular modeling is used in several different researches and therefore the term does not have a rigid definition.
To a chemical physicist, molecular modeling imply performing a high quality quantum mechanical calculation using a super computer on the structure; to a medicinal chemists, molecular modeling mean displaying and modifying a candidate drug molecule on the desktop computer. Molecular modeling techniques can be divided into molecular graphics and computation chemistry.
Molecular Graphics in Computer Aided Drug Design
Molecular graphics is the core of a modeling system, providing the visualization of molecular structure and its properties. In molecular modeling, the data produced are converted into visual image on the computer screen by graphic packages. These images may be displayed in a variety of styles like space fill, CPK (Corey-Pauling-Koltum), stick, ball and stick, mesh and ribbon and colour scheme with visual aids. Ribbon presentation is used for larger molecules like nucleic acid and protein.
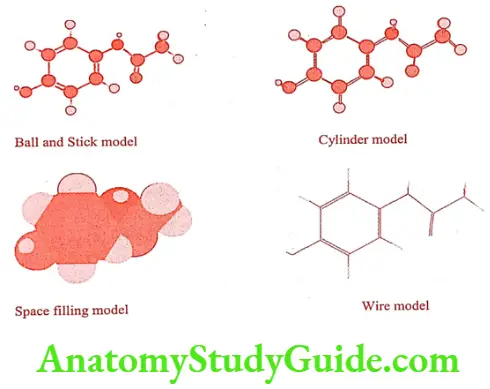
Visualization of molecular properties is an extremely important aspect of molecular modeling. The properties might be calculated using a computational chemistry program and visualized as 3D contours along with associated structure. The most common computational methods are based on either molecular or quantum mechanics. Both these approaches produce equation for the total energy of the structure. In these equation the position of the atom in the structures are represented by either Cartesian or polar co-ordinates.
Once the energy equation is established, the computer computes a set of co-ordinates which corresponds to minimum total energy value for the system. This set of co-ordinate is converted into the required visual display by the graphic package. The program usually indicate that the three dimensional nature of the molecule and it can be viewed from different angles and allow the structure to fit to its target site. In addition, it is also possible by molecular dynamics, to show, how the shape of structure might vary with time by visualizing the natural vibration of the molecule.
Molecular Mechanics in Computer Aided Drug Design
Molecular mechanics is the more popular of the method used to obtain molecular models as it is simple to use and requires considerably less computing time to produce a model. In this technique the energy of structure is calculated. The equation used in molecular mechanics follow the laws of classical physics and applies them to molecular nuclei without consideration of the electrons.
The molecular mechanics method is based on the assumption that, the position of the nuclei of the atom forming the structure is determined by the force of attraction and repulsion operating in that structure. It assumes that the total potential energy [E Total] of a molecule is given by the sum of all the energies of the attractive and repulsive forces between the atoms in the structure.
Molecules are treated as a series of sphere (the atoms) connected by spring (the bond) using this model: E Total is expressed mathematically by the equation known as force fields given by:
E Total = EStretching + ΣΕ bend + ΣΕ Torsion + ΣΕ vdw + ΣΕ Coulombic
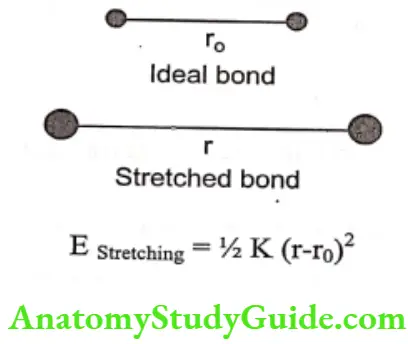
E Stretching: : E stretching is the bond stretching energy. The value of the E stretching bond energy for pair of atoms joined by a single bond can be estimated by considering the bond to be a mechanical spring that obeys Hooke’s law. If r is the stretched length of the bond and ro is the ideal bond length, then
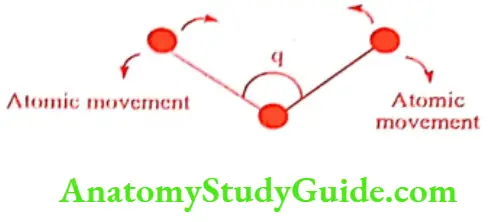
E bend: It is bond energy due to changes in bond angle and estimated as:
Where Oo is ideal bond length that is the minimum energy position of the 3 atoms.
E Torsion: It is bond energy due to changes in the conformation of the bond and given by
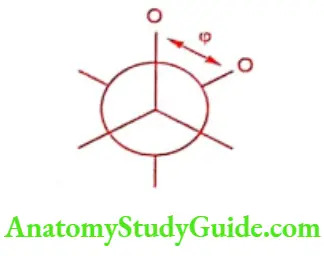
Where k is the energy barrier to the rotation about the torsion angle, m is the periodicity of the rotation and offset is the ideal torsion angle relative to staggered arrangement of two atoms.
Evdw: It is total energy contribution due to Vander Waal’s force and it is calculated from the Lennard Jone potential equation:
The term in this equation represents attractive force, while term represents the short range of repulsive forces between the atoms. The Imin is the distance between two atoms i and j when the energy at a minimum & and r is the actual distance between the atoms.
ECoulomble: It is an electrostatic, attractive and repulsive forces operating in the molecule between the atoms carrying a partial or full charge.
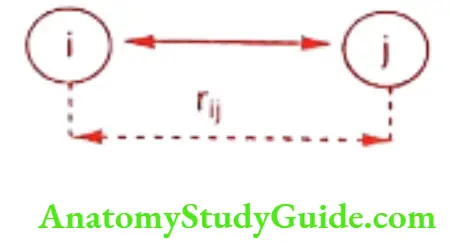
Where
q and q are the point charges on atoms i and j,
rij is the distance between the charges and
D is the dielectric constant of the medium surrounding the charges.
The values of the parameters r, ro, k….. etc., used in the expression for the term in the above equation is either obtained / calculated from experimental observations. The experimental values are derived from variety of spectroscopic techniques; thermodynamic data measurement and crystal structure measurement for inter atomic distances.
The best fit parameters are obtained by looking with known parameter values and stored in the data base of the molecular modeling computer program.
Creating a Molecular Model using Molecular Mechanics
Molecular modeling can be created by any of these methods:
- Commercial force field computer program.
- Assembling model.
Commercial force field Computer Program
Commercial packages usually have several different force fields within the same package and it is necessary to pick the most appropriate one for the structure being modeled. Some more details added here.
Assembling Model
Molecular models are created by assembling a model from structural fragments held in the database of the molecular modeling program. Initially, these fragments are put together in a reasonably sensible manner to give a structure that does not allow for steric hindrance. It is necessary to check that, the computer has selected atoms for the structure whose configuration corresponds to the type of bond required in structure.
For example, if the atom in the structure is double bonded, then the computer has selected a form of atom that is double bonded. These checks are carried out by matching a code for the atoms on the screen against the code given in the manual for the program and replacing atom where necessary.
An outline of the steps involved using INSIGHT II to produce a stick model of the structure of Paracetamol.
INSIGHT II models Step 1
The selection of the structure fragments from the database of the INSIGHT II program. The molecule with the relevant functional group and/or structure is selected.
The INSIGHT II models of these structures.

INSIGHT II models Step 2
The fragments are linked together. Fragments are joined to each other by removing hydrogen atoms at the points at which the fragments are to be linked. The bonding state of each atom is checked and if necessary adjusted.

INSIGHT II models Step 3
The force field of the model is minimized to give the final structure.

A representation of the change in the value of E Total demonstrates how the computation could stop at a local (X) rather than the true (global) minimum value. The use of molecular dynamics gives the structure kinetic energy which allows it to overcome energy barriers, such as Y, to reach the global minimum energy structure of the molecule.
Once the structure is created energy minimization should be carried out. This is because the construction process may have resulted in unfavorable bond lengths, bond angles or torsion angles. The energy minimization process is carried out by a molecular mechanics program, which calculates the energy of the starting molecule, and then varies the bond lengths, bond angle, and torsion angle to create a new structure in whatever software program is used.
The program will interpret the most stable structure and will stop at that stage when the force field reaches the nearest local minimum energy value. This final structure may be moved around the screen and expanded or reduced in size. It can also be rotated about the x and y axis to view different elevations of the model.
The molecular mechanic method requires less computing time than the quantum mechanical approach and may be used for large molecules containing more than a thousand atoms. Energy calculation has a range of applications in molecular modeling.
- They can be used in conformational analysis to evaluate the relative stability of different conformers and to predict the equilibrium geometry of a structure.
- They can also be used to evaluate the energy of two or more interacting molecules, such as when docking a substrate the enzyme active site.
It is not useful for computing properties such as electron density. The accuracy of the structure obtained will depend on the quality and appropriateness of the parameters used in the force field. Moreover, the molecular mechanical calculations are normally based on isolated structures at zero Kelvin and do not normally take into account the effect of the environment on the structure.
Molecular Dynamics
Molecular mechanics calculations are made at zero Kelvin, that is on structures that are frozen in time and so do not show the natural motion in the structure, Molecular dynamics programs allow the modular to show the dynamic nature of the molecule by stimulating the natural motion of the atom in a structure.
Starting with the molecular mechanics energy description of the structure as described above, the force acting as the atom can be evaluated. Since the masses of the atom are known, Newton’s second law of motion (force = mass x acceleration) may be used to compute the acceleration and thus the velocities of the atoms.
The acceleration and velocities may be used to calculate new position for the atom over a short time step thus moving each atom to a new position in the space. The velocities of the atoms are related directly to the temperature at which the stimulation is run. Higher temperature stimulations are used to search conformational shape since more energy is available to climb and cross energy barriers.
These variations are displayed on the monitor as a moving picture. The appearance of this picture will depend on the force field selected for the structure, the time interval and the temperature used for the integration of the Newtonian equation. Molecular dynamics can also be used to find minimal energy structure and conformational analysis.
Conformational Analysis
Using molecular mechanics (MM2), it is possible to generate a variety of different conformations using a molecular dynamics program which ‘heats’ the molecule to 800 to 900°K. Of course, this does not mean that the inside of your computer is about to melt. It means that the program allows the structure to undergo bond stretching and bond rotation as if it were being heated.
As a result, energy barriers between different conformations are overcome, allowing the crossing of energy saddles. In the process, the molecule is ‘heated’ at a high T (900°K) for a certain period (Example: 5 picoseconds), then ‘cooled’ to 300°K for another period (Example: 10 picoseconds) to give a final structure. The process can be repeated automatically as many times as wished to give as many different structures as required.
Each of these structures can then be recovered, energy minimized and its steric energy measured. By carrying out this procedure, it is usually possible to identify distinct conformations, some of which might be more stable than the initial conformation.
For example, 2D drawing of butane shown in Fig. 5.1 was imported into Chem3D and energy minimized. Because of the way the molecule was represented, energy minimization stopped at the first local energy minimum it found, which was the gauche conformation having a steric energy of 3.038 kcal/mol.
The molecular dynamic program was run to generate other conformations and successfully produced the fully staggered trans conformation which, after optimization, had a steric energy of 2.175 kcal/mol, showing that the latter was more stable by about 1 kcal/mol.

This particular problem could be solved more efficiently by stepwise rotation of bonds described below. Molecular dynamic is more useful for creating different conformations of molecule that are not conducive to stepwise bond rotation, (cyclic systems) or which would take too long analyze by that process (large molecular).
For example, the twist boat conformation of cyclohexane remains as twist boat when energy minimization is carried out. ‘Heating’ the molecule by molecular dynamics in Chem3D produces a variety of different conformations, including the more stable chair conformation.

Quantum Mechanics
Unlike molecular mechanics, the quantum mechanic approach to molecular modeling does not require the use of parameters similar to those used in molecular mechanics. It is based on the realization that electrons and all material particles exhibit wave-like properties. This allows the well-defined, parameter-free, mathematics of wave motion to be applied to electrons, atoms,s and molecular structures. The basis of this calculation is the Schrodinger wave equation, which in its simplest form may be stated as:
Ηψ = Εψ
In molecular modeling term Ey represents the total potential and kinetic energy of all the particles in the structure and H is the Hamiltonian operator acting on the wave function y.
The energy of a structure calculated via quantum mechanics can be used in conformational searches, in the same way that, the molecular mechanics energy is used. Quantum mechanics calculations can also be used for energy minimization. However, quantum mechanics calculations typically consume a far greater amount of computer resource than molecular mechanics calculations and are therefore generally limited to small molecules, whereas molecular mechanics can be applied to structures up to the size of large proteins.
Molecular mechanics and quantum mechanics should thus be viewed as complementary techniques. For instance, conformational energy calculations for a peptides are best carried out using molecular mechanics. However, molecular mechanics is generally ineffective for handling conjugated systems, while quantum mechanics, in calculating electronic structure, takes account of conjugation automatically and is therefore recommended for optimizing the structure of a small molecule containing conjugated systems.
The wave function can be used to calculate a range of chemical properties, which can be used in structure-activity studies. These include electrostatic potential, electron density, dipole moment and the energies and positions of frontier orbital. As with the analysis of a molecular dynamics calculation, molecular graphics is essential for visualizing these properties.
Quantum mechanics calculations are also used frequently to derive atom-centered partial charges (although the term charge itself does not have a strict quantum mechanical definition). Charges have a wide range of applications in modeling and are used in the calculation of electrostatic energies in molecular mechanics calculations and in computing electrostatic potentials.
Quantum mechanical methods are suitable for calculating the following
- Molecular orbital energies and coefficients.
- Heat of formation for specific conformations.
- Partial atomic charges are calculated from molecular orbital coefficients.
- Electrostatic potentials.
- Dipole moment.
- Transition state geometries and energies.
- Bond dissociation energies.
Hybrid QM / MM
QM (quantum-mechanical) methods are very powerful, however, they are computationally expensive, while the MM (classical or molecular mechanics) methods are fast but suffer from several limitations (require extensive parameterization; energy estimates obtained are not very accurate; cannot be used to simulate reactions where covalent bonds are broken/ formed; and are limited in their abilities for providing accurate details regarding the chemical environment).
A new class of method has emerged that combines the good points of QM (accuracy) are MM (speed) calculations. These methods are known as mixed or hybrid quantum-mechanical and molecular mechanics methods (hybrid QM/MM). The methodology for such techniques was introduced by Warshel and co-workers.
Leave a Reply