Differential Diagnosis Of Common Abnormal Physical Signs
Failure To Thrive
Table of Contents
Most parents are worried about the growth of their children. Failure to thrive (FTT) is a common symptom for which children are brought for evaluation.
It should not be confused with the food fussiness of children of well-to-do parents when the “child is not eating or growing to the satisfaction of his parents.’
The diagnosis of FTT cannot be made on the basis of a single observation. It is characterized by failure to gain weight or weight loss observed over a period of time.
The diagnosis can be made reliably if the weight gain pattern of the child is maintained on a Road-to-Health card.
Read and Learn More Pediatric Clinical Methods Notes
In preterm babies, corrected age or post-conceptional age should be used for recording anthropometric measurements on a Road-to-Health card during the first year of life.
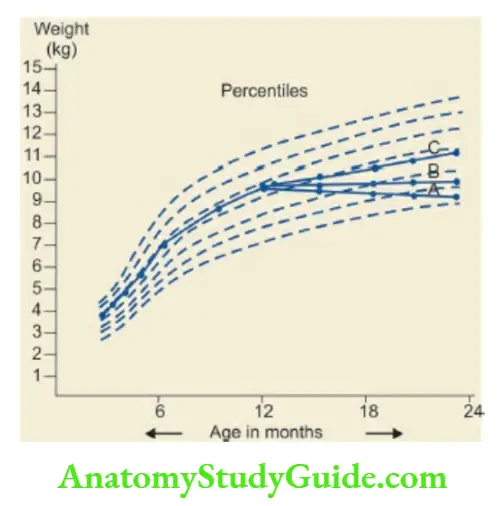
When the growth curve runs along the 25th percentile line without any downward trend, it indicates low genetic potential and not any disease process.
The weight curve of the child with FTT shows a plateau or a downward trend so that it drops below two major percentile lines.
The diagnosis of FTT can also be suspected if the weight-for-height of the child is less than the 10th percentile on the WHO or National Center for Health Statistics (NCHS) weight-for-length/height charts.
The child is diagnosed to be underweight when his weight is less than 2SD of the reference mean. Wasting is diagnosed when weight-for-height is less than 2SD of the reference mean value.
A slowing in the rate of linear growth may take 6 months to manifest while slowing of weight gain or weight loss can be demonstrated during a short period of observation.
The size of the baby at birth depends upon the maternal health and adequacy of the intrauterine environment rather than the genetic or constitutional factors.
Children with poor genetic growth potential may grow normally during the first 1–2 years of life.
After the physiological adjustments, the child “finds his genetic growth curve” and then grows along a percentile line at a slow growth velocity.
Some LBW infants with fetal malnutrition (especially babies with symmetric IUGR) continue to follow the trend of intrauterine growth velocity after birth and grow as constitutionally light children.
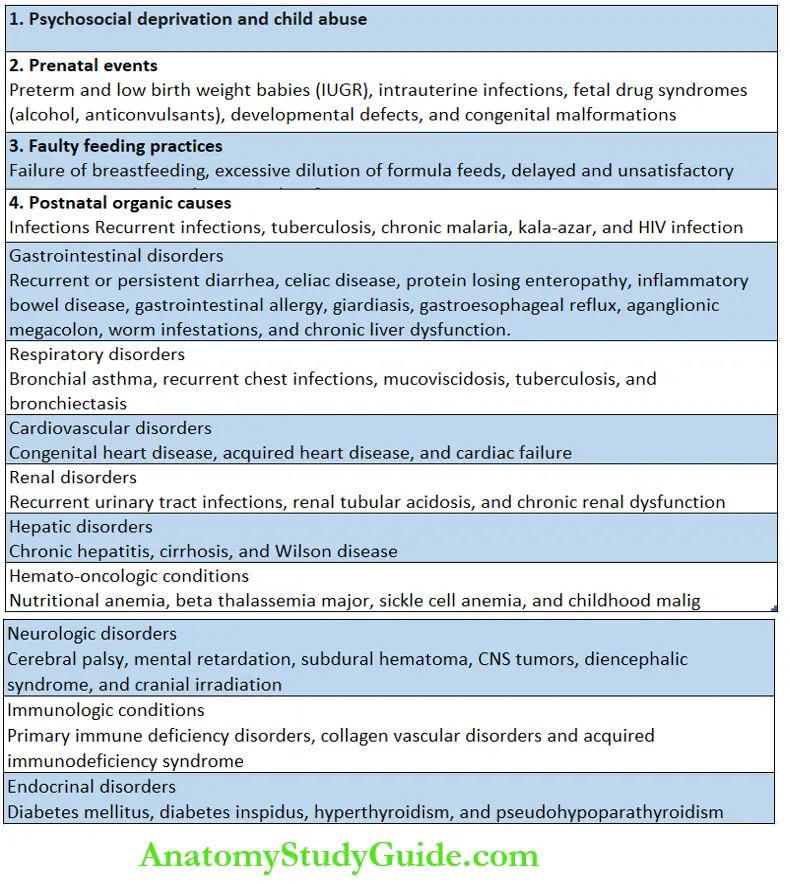
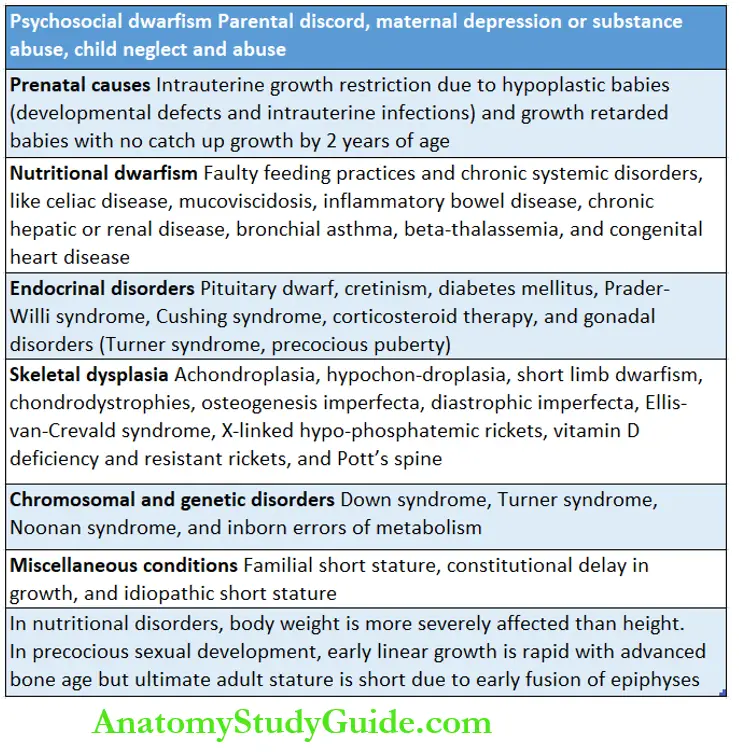
In these children, there is global retardation in all the growth parameters including body weight, head size, and linear growth. The causes of FTT may be psychosocial or organic.
The majority of children with FTT in our country have nutritional or organic causes. The important causes of FTT are given in.
The common psychosocial correlates of FTT include parental discord, lack of an emotional support system, financial problems, and substance abuse.
There is a history of irritability, sleep disturbances, excessive crying, and temper tantrums.
Infants with sensory deprivation are characterized by decreased vocalization, minimal smiling, abnormal posture, lack of cuddliness, head banging, rocking movements, and rumination.
In apparently healthy children, suboptimal physical growth and stunting may occur due to sub-clinical deficiencies of micronutrients (vitamins and trace minerals).
Nutritional supplements containing micronutrients in these children can augment their growth by optimal expression of their genetic potential.
Stunting Or Short Stature
Stunting or short stature is diagnosed when height is less than -2SD or below the 3rd percentile of mean height-for-age as per the WHO growth reference standard.
According to National Family Health Survey 2005–2006 (NFHS-3), about 48% of under-5 children are stunted in India.
The prevalence of stunting should also be taken as a poor social indicator for any nation just like the high infant mortality rate.
The height achieved at the age of 3 years is crucial because it is a good predictor of ultimate adult height.
Dwarfism is diagnosed when height is less than -3SD below the mean height-for-age. Height velocity should be calculated over an observation period of 6 months.
In a school-going child, when height velocity is less than 5 cm per year, it indicates slow linear growth and a cause for concern.
A large number of conditions can adversely affect physical growth. Most conditions are associated with poor weight gain as well as subnormal linear growth.
Nutritional disorders and systemic diseases are likely to have greater adverse effects on body weight than on linear growth and they are leading causes of failure to thrive.
Stunting or dwarfism occurs more commonly due to endocrinal disorders, skeletal dysplasias, and dysmorphic syndromes because of chromosomal or genetic disorders.
A detailed history should be taken to identify any psychosocial factors, faulty feeding practices, and chronic systemic disorders.
Anthropometric parameters should be recorded on a periodic basis on a Road-to-Health chart to identify the age of onset and severity of growth retardation.
Height-for-age and height age (age calculated on the basis of actual height) should be recorded.
The Target Height of the child should be recorded on the basis of mid-parental height as follows:
Boys: Father’s height in cm + mother’s height in cm divided by 2 + 6.5 cm
Girls: Father’s height in cm + mother’s height in cm divided by 2–6.5 cm
The child should be examined for any facial dysmorphism and skeletal dysplasia.
Body proportions should be assessed to identify whether stunting is mainly affecting the trunk (hypogonadism, Turner syndrome, eunuchoidism, chondrodystrophic, spinal deformities) or limbs (achondroplasia, short-limbed dwarfism, cretinism, sexual precocity, bowed legs).
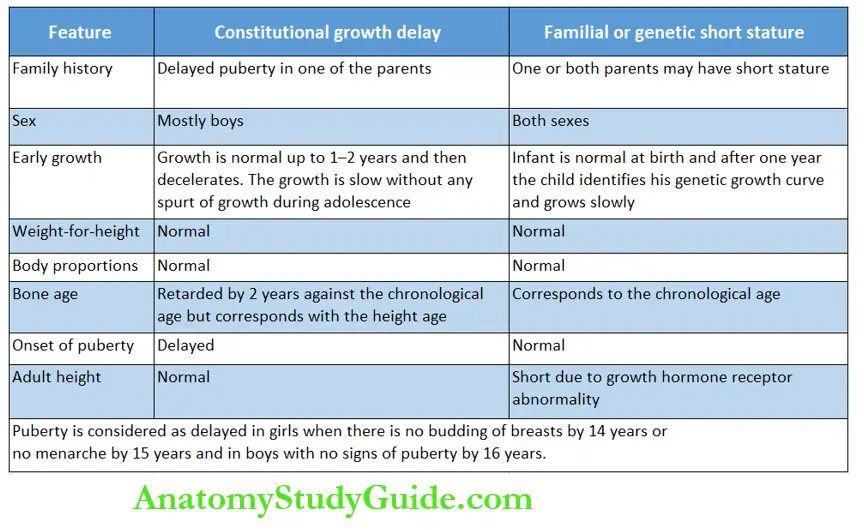
The distinctive clinical differences between constitutional growth delay (CGD) and familial short stature (FSS) should be identified.
Bone age should be checked by taking a skiagram of the left wrist and elbow in all children with stunting.
Bone age may be retarded in children with hypopituitarism, hypothyroidism, Cushing syndrome, malnutrition, constitutional dwarfism with delayed adolescence, male hypogonadism, and chronic systemic disease.
When bone age is less than chronological age, it is possible to have catch-up linear growth In constitutional growth delay, the onset of pubertal growth spurt is delayed but the ultimate adult height is normal.
Sexual maturity rating should be assessed to diagnose sexual infantilism and precocious puberty because both can adversely affect the ultimate adult height.
Precocious puberty is diagnosed when secondary sexual characteristics appear before 8 years of age in girls and 10 years in boys while delayed puberty or sexual infantilism is suspected when there is no budding of breasts by 14 years or no menarche by 15 years in girls and absence of signs of puberty (facial hair and enlargement of testes) by 16 years in boys.
The term idiopathic short stature (ISS) is used to describe a child or adolescent with a height below -2SD of the mean height forage on the reference standard and in whom no cause has been identified on the basis of current diagnostic tools.
Before assigning a label of ISS, it is important to exclude important causes of stunting, like celiac disease, Turner syndrome, hypopituitarism, cretinism, chronic inflammatory or systemic disease, and renal tubular acidosis.
Excessively Tall Child
As opposed to stunting, excessively tall stature is rare. It is suspected when height is more than +2SD (>97th percentile) of the mean height-for-age and the predicted adult height is >3SD above the mean, about 78 inches in males and 71 inches in females.
Bone age should be checked and if it is less than the chronological age, there is a greater likelihood of abnormally tall stature.
The common conditions which are associated with excessively tall stature include familial or constitutional tall stature, exogenous obesity, Klinefelter syndrome (XXY), Marfan syndrome, fragile-X syndrome, excess GH secretion, hyperthyroidism, precocious puberty (ultimate adult height is likely to be short) and testicular feminization.
Cyanosis
Cyanosis is characterized by bluish discoloration of the skin and mucous membranes due to desaturation of arterial blood. It should be looked for in bright natural light.
Cyanosis manifests clinically when the level of deoxygenated hemoglobin exceeds 5 g/dl. Cyanosis is, therefore, unlikely to manifest in children with severe anemia.
In contrast, infants with polycythemia become cyanosed at higher arterial oxygen tensions.
Pulse oximetry is more reliable to assess oxygen saturation and cyanosis becomes evident only when arterial oxygen saturation (SaO2) falls below 85 percent.
The clinical manifestations of hypoxia include bradycardia, irritability, restlessness, and excessive crying which is followed by lethargy and somnolence. Cyanosis may be peripheral or central.
Peripheral cyanosis occurs due to sluggish peripheral circulation as a consequence of exposure to cold, hypothermia, hypoglycemia, polycythemia, sepsis, shock, and vasomotor changes.
In peripheral cyanosis, bluish discoloration is limited to the tip of the nose, nails, and ear lobe.
In newborn babies, peripheral or acrocyanosis usually manifests as circumoral grayness due to the presence of prominent superficial venous plexuses in the region.
In peripheral cyanosis, extremities are cold, and arterial oxygen tension is normal but arterial oxygen saturation (SpO2) as recorded by pulse oximetry is reduced.
Central cyanosis is characterized by the blueness of mucous membranes, like lips, mucosa, and tongue.
When hypoxia or cyanosis is chronic, it is associated with elevation of hemoglobin and clubbing of nails of fingers and toes.
In certain cardiac malformations, differential cyanosis may be seen. In coarctation of the aorta with transposition of great arteries, hands, and face are blue while feet are pink.
This can be reliably assessed by simultaneously recording SpO2 on the right hand and left foot.
On the other hand, in a patient with ductus arteriosus with reversal of shunt due to pulmonary hypertension or preductal coarctation of the aorta, the face and hands are pink while the feet are blue.
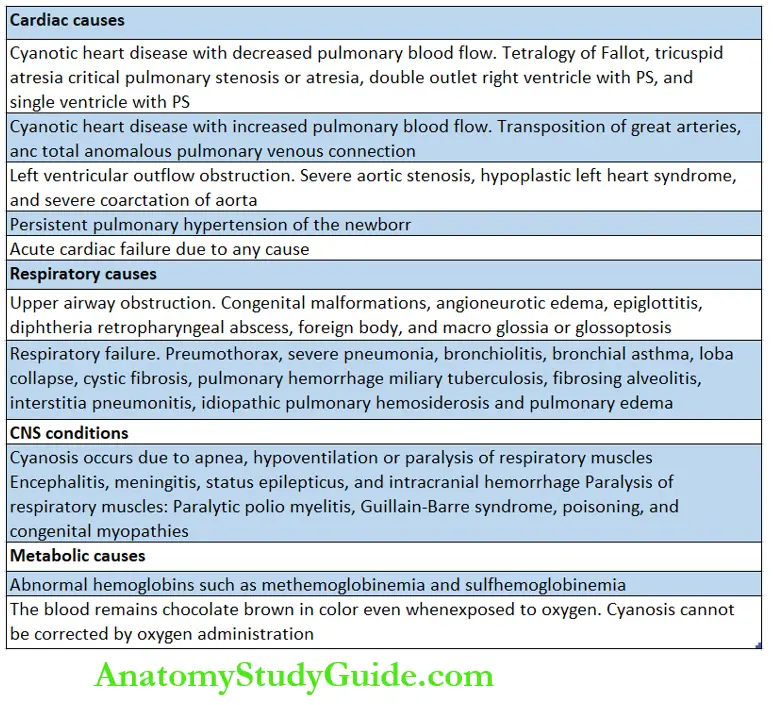
Cyanosis is most commonly seen in association with cardiac, pulmonary, CNS, and metabolic conditions.
The presence of cyanosis in the absence of respiratory difficulty is highly suggestive of cyanotic heart disease, methemoglobinemia, and polycythemia.
The cyanosis due to the right-to-left shunt becomes worse when the baby cries.
The presence of severe respiratory difficulty with marked cyanosis is more common due to pulmonary rather than cardiac disorders. Respiratory cyanosis usually responds to oxygen administration or assisted ventilation.
When administration of 100% oxygen fails to raise PaO2 above 50 mm Hg, it is highly suggestive of cyanotic heart disease.
Anemia
Anemia or low hemoglobin is suspected on the basis of pallor of the skin (face, palms, and soles), lower palpebral conjunctiva, dorsum of the tongue, lips, oral mucosa, and nail bed.
When anemia is severe (hemoglobin <5 g/dl), the pallor is marked and palmar creases become pale.
You can compare the color of a child’s palm with yours if you are not anemic! The skin of the face may appear pale in fair-complexioned children and it should not be confused with pallor.
Iron deficiency anemia may be associated with flat or spooning of nails (koilonychia) with longitudinal ridges but it is less common in children compared to adults.
It is difficult to assess pallor in a severely jaundiced child. The extremities may become pale and cold in the absence of anemia in critically sick children with sepsis, shock, cardiac failure, and generalized edema.
The normal hemoglobin level varies widely in children with the highest mean hemoglobin level of 16.5 g/dl at birth and the lowest level of 11.0 g/dl at 2 months of age due to physiological anemia.
According to WHO, hemoglobin level below 11.0 g/dl in children between 6 months and 6 years and below 12 g/dl in older children is suggestive of anemia.
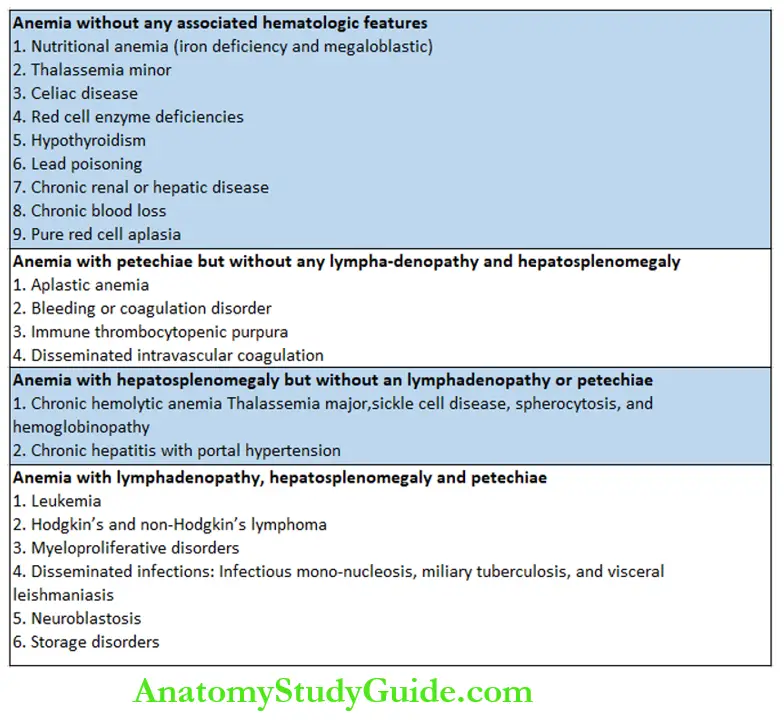
The classification of anemia on the basis of associated clinical findings is shown in.
Jaundice
Jaundice manifests as yellowish discoloration of the skin and mucous membranes due to the accumulation of bilirubin in the blood.
Jaundice should be looked for in natural daylight over the bulbar conjunctiva (yellow sclera), under the surface of the tongue, soft palate, and skin, especially over the face, palms, and soles.
Urine may be highly colored and deep yellow when direct reacting or conjugated bilirubin is elevated. Anorexia is common when hepatic cellular dysfunction is present.
The characteristic clinical features of jaundice due to various causes are listed.
Edema
Edema or swelling of subcutaneous tissues occurs due to the accumulation of fluid in the interstitial spaces.
Edema may occur due to a fall in the oncotic or osmotic pressure of plasma (fall in plasma protein, especially albumin), a rise in capillary hydrostatic pressure at the arteriolar end, or passive venous congestion and capillary damage.
Edema may be localized to certain parts of the body or generalized as manifested by puffiness, and swelling of feet, legs, and sacrum.
Edema occurs more commonly over the periorbital region, scrotum, vulva, and dorsum of hands and feet because of loose areolar tissue with reduced tissue tension.
When edema is associated with the collection of fluid in one or more serosal cavities, it is called generalized anasarca. Localized edema with signs of inflammation is a characteristic feature of infection or cellulitis.
At times, edema may be limited to certain body organs, like airways (angioneurotic edema), the brain, lungs, or an isolated serosal cavity.
In newborn babies and infants, edema manifests as excessive weight gain, puffiness of the face, and swelling over the sacrum because they mostly lie in a supine position.
Edema is assessed by looking for pitting after applying sustained pressure. It is best looked for over the medial malleolus, dorsum of feet, and shins.
Sustained pressure is applied with the thumb or index finger for 5 seconds and the site is watched for visible or palpable depression.
Edema due to accumulation of interstitial fluid pits on pressure and skin returns back to normal after some time.
Abdominal wall edema is tested by pinching the skin and subcutaneous tissues with the thumb and index finger or after sustained pressure with a chest piece of the stethoscope.
Edema due to lymphatic obstruction feels firm and does not pit on pressure. It is associated with rough or coarse skin. The common causes of edema in children are listed.
Lymphadenopathy
Lymph nodes serve as protective sentinels to eliminate antigens or pathogens that are transported to them through the lymphatics.
They are widely distributed throughout the body but are mostly concentrated over the neck, around the joints, axillae, groins, elbows (epitrochlear), knees, (popliteal), hilum of lungs, mesenteric and retroperitoneal sites.
Lymph nodes (including tonsils and adenoids) are overreactive and readily enlarge in size in children to ward off infections through cell-mediated immune responses because young children lack specific humoral antibodies because of a lack of previous exposure to pathogenic organisms.
Newborn babies
Localized edema
- Edema on the presenting part
- Inflammatory edema
- Turner syndrome
- Milroy disease
Generalized
- Edema of prematurity
- Hydrops fetalis
- Rh-isoimmunization
- Non-immunologic causes
- Severe anemia (homozygous alpha-thalassemia, twin-to-twin or fetomaternal
- hemorrhage, osteopetrosis)
- Congenital malformations
- Intrauterine infections
- Miscellaneous conditions, like chorioangioma hemangioendothelioma of the placenta, arteriovenous anastomosis of the fetus or placenta, venous thromboses, fetal neuroblasts, skeletal abnormalities
Older children
Hypoalbuminemia (decreased oncotic pressure)
- Protein-energy malnutrition, kwashiorkor
- Nephrotic syndrome
- Malabsorption syndrome
- Protein-losing enteropathy
- Chronic hepatic and renal disease increased hydrostatic pressure and Congestive heart failure
- Constrictive pericarditis
- Budd-Chiari syndrome
- Veno-occlusive disease
- Thrombophlebitis
- Superior vena cava obstruction
- Extrinsic pressure by a tumor massSodium and water retention Congestive heart failure
- Acute glomerulonephritis
- Cirrhosis
- Chronic anemia
- Fluid and salt overload
- Corticosteroid therapy
- Premenstrual syndrome
- Increased capillary permeability
- Allergic reaction. Urticaria, and angioneurotic edema
- Inflammatory reaction. Dengue fever, Rocky Mountain spotted fever
- Lymphatic obstruction
- Turner syndrome
- Milroy disease
- Chylous ascites
Lymph nodes should be examined for their site, size, number, and consistency, whether discrete or matted, free or attached to the underlying structures or overlying skin, softening with abscess or sinus formation, and any evidence of acute inflammation, like pain, tenderness, redness, and warmth.
When lymph node enlargement is more than 1.0 cm (>1.5 cm in the case of inguinal nodes) in size, it is considered as significant.
In localized infections, regional groups of lymph nodes are enlarged. When two or more lymph node groups are enlarged, it is designated as generalized lymphadenopathy. The common causes of lymphadenopathy are listed in.
Delayed Closure Of Anterior Fontanel
Anterior fontanel at birth varies in size between 2.0 ±1.0 cm and is slightly depressed relative to the frontal and parietal bones.
The anterior fontanel normally closes between 10 and 18 months of age. Early closure of anterior fontanel should not be a cause for concern if head growth is proceeding normally and there is no ridging of sutures.
The presence of excessively large anterior fontanel and its delayed closure is a recognized clinical feature in several conditions listed.
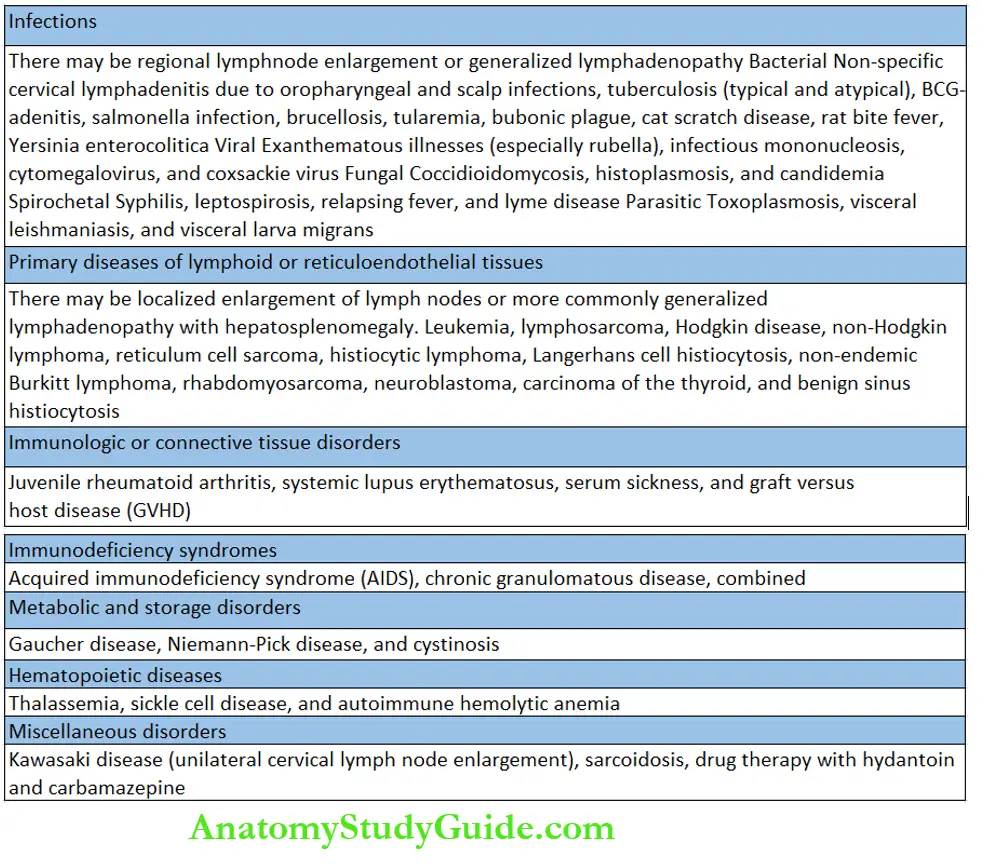
Causes of delayed closure of anterior fontanel
- Malnutrition
- Hydrocephalus
- Achondroplasia
- Pituitary dwarf
- Rickets
- Alpert syndrome
- Cretinism
- Trisomy 13 and 18
- Down syndrome (trisomy-21)
- Russell-Silver syndrome
- Gorgoylism (mucopolysaccharidoses)
- Hypophosphatasia
- Congenital syphilis
- Progeria
- Thalassemia major
- Hallermann-Streiff syndrome
- Osteogenesis imperfecta
- Pyknodysostosis
- Cleidocranial dysostosis
- Zellweger syndrome
- Congenital rubella syndrome
Bulging Of Anterior Fontanel
The fontanel should be examined in a quiet child held in an upright position. The anterior fontanel is normally flat or slightly depressed relative to the frontal and parietal bones and is pulsatile.
Bulging fontanel is a reliable sign of raised intracranial tension during infancy. The pulsations may disappear when the fontanel becomes tense because of marked elevation of intracranial tension.
The causes of bulging anterior fontanel are given in.
Causes of bulging of the anterior fontanel
- Crying infant
- Raised intracranial tension: Hydrocephalus, meningitis, intracranial bleeding, shaken baby syndrome, tumor, sinus thrombosis, and pseudotumor cerebri.
- Hyperparathyroidism
- Congenital hypophosphatasia
- Maple syrup urine disease
- Urea cycle enzyme defects
- Galactosemia
- Tetracycline and fluoroquinolone therapy
- Vitamin D-dependent rickets
- Vitamin A poisoning
- Nalidixic acid overdose
- Corticosteroid therapy (following cessation of therapy)
- Lead poisoning
Craniotabes
Craniotabes refer to softened and parchmentlike skull bones that can be indented like a ping pong ball. The sign should be elicited away from the suture line.
It is normally eligible in preterm babies. The common causes are listed in.
Causes of craniotabes
- Physiological
- Rickets
- Osteogenesis imperfecta
- Marasmus
- Lacunar skull and craniofenestria
- Congenital syphilis
- Hypervitaminosis A
- Hydrocephalus
- Mandibulofacial dysostosis (Treacher Collins syndrome)
Bossing Of Skull
The prominence of skull bones is called bossing. It may affect frontal, parietal, or occipital bones though frontal bossing is most common. The important causes of the bossing of the skull are listed in.
Causes of bossing of the skull
- Rickets
- Ectodermal dysplasia
- Thalassemia major
- Ehlers-Danlos syndrome
- Congenital syphilis
- Lowe’s syndrome
- Achondroplasia
- Hallermann-Streiff syndrome
- Hurler’s syndrome (mucopolysaccharidoses)
- Generalized gangliosidosis type 1
- Cleidocranial dysostosis
- 10 p deletion syndrome
- Pyknodysostosis
Head Nodding Or Banging
Nodding of the head up and down or side to side and head banging or head rolling may be seen in some normal children. Nodding is greater in the sitting than in the supine position.
When head nodding is persistent or excessive, the conditions listed should be considered.
Causes of head nodding
- Spasmus mutants (with nystagmus)
- Emotional deprivation
- Mental retardation
- Pelizaeus-Merzbacher disease (with poor head control and cog-wheel nystagmus)
- Ocular albinism
- Autism spectrum disorder
- Boredom and stress spectrum disorder
- Bobble-head doll syndrome (hydrocephalus due to lesion in the region of the third ventricle)
Macrocephaly
It is diagnosed when the head circumference exceeds 2.5 cm of the mean-for-age or is above two standard deviations or above the 97th centile of the mean-for-age, sex, height, and weight of the child.
The salient causes of large heads are listed in Box 8.6. It may occur due to an increase in the thickness of the scalp, skull bones, and contents of the cranium (megalencephaly and hydrocephalus)
Causes of macrocephaly
- Familial macrocephaly
- Hydrocephalus. It is characterized by dilatation of the ventricular system. The salient clinical features include large bulging anterior fontanel, separated sutures, bossing of frontal bones, engorged veins over the scalp, and sun-setting signs. Serial head circumference should be taken to identify whether it is progressive (active) or arrested hydrocephalus.
- Thick skull bones (achondroplasia, osteopetrosis, pyknodysostosis, craniometry dysplasia, oro-digital-facial dysostosis, rickets, leontiasis pose, etc.)
- Cerebral gigantism (Sotos syndrome)
- Mucopolysaccharidoses
- Cerebral lipodosis (gangliosidosis)
- Metachromatic leukodystrophy
- Fragile X syndrome
- Porencephaly
- Subdural hematoma
- Hydranencephaly
- Subdural effusion
- Intracranial tumor
- Neurofibromatosis type 1
- Tuberous sclerosis
- Weaver syndrome
- Glutaryl-coenzyme a dehydrogenase deficiency
- Autism spectrum disorder
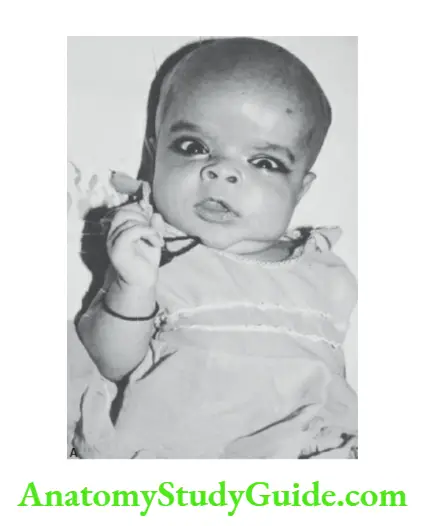

Microcephaly
It is defined as a head circumference below two standard deviations of the mean for age, sex, height, and weight. It may be primarily due to impaired growth of the brain or secondary because of premature fusion of sutures.
It may be associated with mental subnormality and neuromotor disabilities. The common causes of microcephaly are listed.
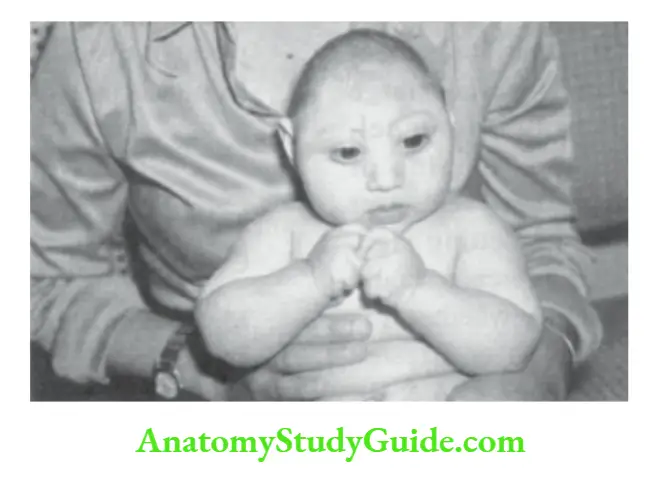
Salient causes of microcephaly
- Familial microcephaly
- Perinatal hypoxia
- Low birth weight babies (premature and small-for-dates babies)
- Cri-du-Chat syndrome
- Craniosynostosis (odd-shaped skull, prominence of sutures)
- Trisomy-13 and trisomy-21
- Intrauterine infections (CMV, rubella, toxoplasmosis, HIV)
- Cerebral dysgenesis
- Cockayne’s syndrome
- Fetal alcohol, hydantoin, or cocaine syndrome
- Smith-Lemli-Opitz syndrome
- Rothmund-Thomson syndrome
- Lissencephaly
- Wolf-Hirschhorn syndrome (p-syndrome)
- Rett syndrome
- Incontinentia pigmenti
- 18 short-arm deletion (18p-) and long-arm deletion (18 q-) syndromes
Blue Sclerae
The sclerae of infants and young children are usually grayish-blue in color and they become white as a child grows.
In infants, the sclera is thin so that underlying dark-colored structures are seen through the white of the eye. The conditions with distinctly blue sclerae are listed.
Causes of blue sclerae
- Physiological during early infancy
- Osteogenesis imperfecta
- Ehlers-Danlos syndrome
- Marfan syndrome
- Glaucoma
- Roberts syndrome
- Russell-Silver syndrome
- Marshall-Smith syndrome
- Hallermann-Streiff syndrome
- Pyknodysostoses
- Pseudoxanthoma elasticum
- Alkaptonuria
- High myopia
Setting-Sun Sign
The eyes are rolled downwards so that the iris is completely covered by the lower eyelids and the sclera is uncovered by the upper eyelids.
It occurs due to the involvement of the center of the upward gaze which is located in the pretectal area of the brainstem.
The sign is easily visible when the infant is quickly lowered from sitting to a supine position. The common causes of the setting-sun sign are listed.
Causes of setting sun sign
- Physiological. Transient and episodic ‘sun-setting’ signs are common in preterm and some term infants
- Hydrocephalus (compression of the brainstem due to dilatation of the third ventricle)
- Kernicterus
- Perinaud’s syndrome (vertical gaze palsy)
- Laron dwarfism
Hypertelorism
Increased interpupillary distance between the two eyes is called hypertelorism.
In Waardenburg syndrome, the interpupillary distance is not increased but eyes look widely placed because both the inner canthi are displaced laterally.
It occurs due to hypertrophy of the lesser wing of the sphenoid. The important causes of hypertelorism are listed in.
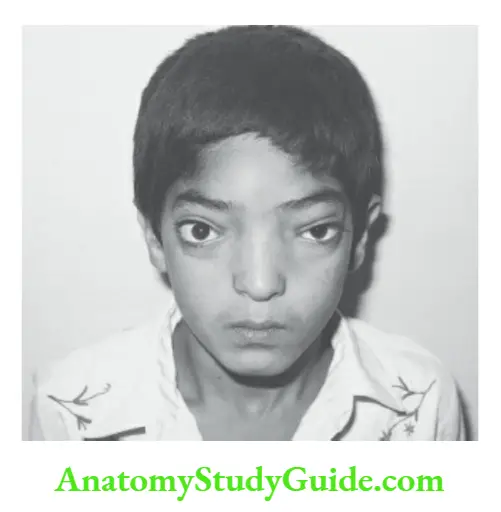
Causes of hypertelorism
- Racial
- Cerebral gigantism (Sotos syndrome)
- Down syndrome
- Nevoid basal cell carcinoma
- Cretinism
- Di George syndrome
- Chondrodystrophies
- Larsen syndrome
- Craniofacial dysostosis
- Multiple lentigines syndrome
- Thalassemia major
- Orofaciodigital dysostosis
- Ehlers-Danlos syndrome
- Apert syndrome
- Turner syndrome
- Coffin-Lowry syndrome
- Waardenburg syndrome
- Crouzon disease
- Cat cry syndrome
- Fetal hydantoin syndrome
- Aarskog syndrome
- Whistling face syndrome
- Optiz syndrome
- Williams syndrome
- Noonan syndrome
- Carpenter syndrome
- Rubinstein-Taybi syndrome
- 10q deletion syndrome
Hypotelorism
The decreased distance between orbits is less common and is seen in the conditions listed.
Causes of hypotelorism
- Cyclops
- Fetal alcohol syndrome
- Patau syndrome (trisomy-13)
- Fragile-X syndrome
- Holoprosencephaly
- Ethmocephaly
- Cebocephaly
- Arrhinencephaly
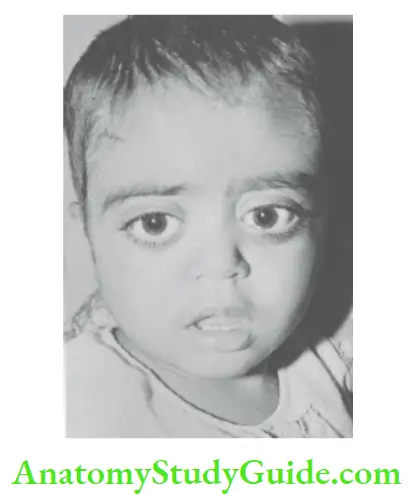
Proptosis
Proptosis or abnormal protrusion of the eyeball(s) occurs due to a space-occupying lesion in the orbit. It may be unilateral or bilateral.
The term exophthalmos is used when proptosis occurs in association with Graves’ disease. The sclera is visible above and below the cornea and lid lag is often present.
Prominent eyes should not be confused with proptosis wherein the eyes bulge forwards. The severity of proptosis can be assessed with an exophthalmometer and CT scans of the orbits. The common causes of proptosis are listed in.
Causes of proptosis
- Orbital cellulitis and abscess
- Retinoblastoma
- Thyrotoxicosis
- Anterior meningocele
- Hand-Schüller-Christian disease
- Rhabdomyosarcoma
- Optic glioma
- Orbital cellulitis and abscess
- Chloroma (acute myeloid leukemia)
- Crouzon disease
- Langerhans cell histiocytosis
- Polyostotic fibrous dysplasia
- Cavernous hemangioma or lymphangioma
- AV aneurysm
- Dermoid cyst
- Apert syndrome
- Cavernous sinus thrombosis
- LEOPARD syndrome
- Retro orbital hemorrhage
- Basal skull fracture
- Pyknodysostosis
- Neuroblastoma
- Sickle cell disease
- Neurofibromatosis
- Visceral larva migrans
- Wegener’s granulomatosis
Ptosis
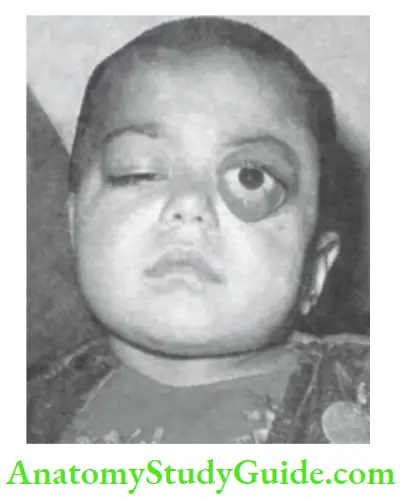
Ptosis or drooping of the upper eyelid may be congenital or acquired, unilateral or bilateral drooping of one or both eyelids.
It may be present at birth or occur later in life. If congenital ptosis is not treated, it can lead to amblyopia (lazy eye).
Rarely neurogenic ptosis may be associated with a jaw-winking phenomenon, the droopy lid ‘flicks” open when the child sucks on a bottle or chews food. The common conditions which are associated with ptosis are listed in.
Causes of ptosis
- Congenital unilateral/bilateral ptosis
- Aarskog syndrome
- Turner syndrome
- Oculomotor palsy (ptosis with mydriasis)
- Moebius syndrome
- Horner syndrome (ptosis, enophthalmos, miosis, and lack of sweating)
- Intracranial tumor or hemorrhage
- Noonan syndrome
- Fetal alcohol syndrome
- Myasthenia gravis
- Whistling face syndrome
- Botulism
- Sticky eyes
- Myotonic dystrophy
Cataract
The opacities in the lens are best seen through the +10 diopter lens of an ophthalmoscope at a distance of 10 inches from the patient’s eyes.
The red reflex of the eye is replaced by white opacity. Cataracts may be congenital or acquired, unilateral or bilateral, central or complete.
If treatment is delayed beyond 2 months, the child develops a “lazy” eye or amblyopia which leads to nystagmus, strabismus, and inability to fix the gaze.
A number of developmental disorders, genetic and chromosomal diseases, and infections may be associated with cataracts.
Causes of cataract
- Familial or idiopathic (developmental)
- Homocystinuria
- Rubella syndrome
- Rothmund-Thomson syndrome
- Galactosemia
- Cortisone therapy
- Marfan syndrome
- Osteopetrosis
- Iridocyclitis
- Zellweger syndrome
- Post-traumatic
- Conradi’s disease
- Lowe’s syndrome
- Ectodermal dysplasia syndrome
- Fabry disease
- Hallermann-Streiff syndrome
- Hypoparathyroidism
- Incontinentia pigmenti
- Progeria
- Refsum syndrome
- Alport syndrome
- Mannosidosis
- Intrauterine infections (toxoplasmosis, CMV, varicella, syphilis, herpes simplex)
- Mandibulofacial dysostoses
- Cockayne syndrome
- Trisomy 13, 18, and 21
- Smith-Lemli-Opitz syndrome
- Diabetes mellitus
- Turner syndrome
- Wilson disease
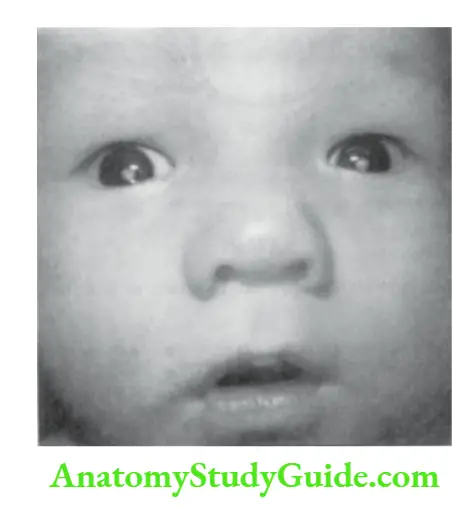
White Reflex In The Eye (CAT’S EYE)
When you shine a light through the pupil or look at the pupil through an ophthalmic scope, normally a red flare is seen. The absence of a red reflex is a recognized feature of several conditions that demand urgent attention.
The common causes of “white pupil” or leukocoria are shown in.
Causes of white reflex
- Cataract
- Retinoblastoma
- Retrolental fibroplasia or retinopathy of prematurity
- Pupillary membrane or persistent central hyaloid artery
- Vitreous opacity
- Eosinophilic granuloma (visceral larva migrans or toxocariasis)
- Retinal detachment
- Coats disease
- Choroidoretinal coloboma
Depressed Nasal Bridge (SADDLE NOSE)
During vaginal delivery, most babies are born with a flat or misshapen nose which gets corrected within a few days. A flat nose is common in certain ethnic and racial groups.
A depressed or flat nose may be associated with hypertelorism. The salient causes of the flat nasal bridge are listed.
Causes of the depressed nasal bridge
- Racial and ethnic feature
- Osteopetrosis
- Down syndrome
- Cleidocranial dysostosis
- Cretinism
- Ectodermal dysplasia
- Thalassemia major
- Conradi syndrome
- Congenital syphilis
- Williams syndrome
- Hurler syndrome
- Smith-Lemli-Opitz syndrome
- Chondrodystrophies
- Fetal alcohol or hydantoin syndrome
- Larson syndrome
Slanting Of Eyes
The face is normally symmetrical and both the eyes and palpebral fissures are aligned horizontally.
A number of developmental defects and chromosomal disorders are associated with upward (mongoloid) or downward (antimongoloid) slanting of both the eyes
Upward slanting of eyes (mongoloid slant)
- Racial
- Down syndrome
- Prader-Willi syndrome
- Leri’s pleonosteosis
- Ectodermal dysplasia
- Aarskog syndrome
- Zellweger syndrome
Downward slanting of eyes (antimongoloid slant)
- Mandibulofacial dysostosis
- Whistling face syndrome
- Turner syndrome
- Alagille-Watson syndrome
- Trisomy 17–18
- Alport syndrome
- Cri-du-Chat syndrome
- Smith-Lemli-Opitz syndrome
- Apert syndrome
- Noonan syndrome
- Treacher Collins syndrome
- ιop depletion syndrome
Puffiness Of Eyelids And Periorbital Tissues
Mild puffiness with dark circles under the eyes may be seen in children with recurrent respiratory infections, nasal allergies, chronic sinusitis, or as a family trait. The important causes of puffiness are listed in.
Causes of facial puffiness
- Familial
- Excessive crying
- Allergic rhinitis
- Chronic sinusitis
- Orbital trauma and cellulitis
- Conjunctivitis
- Mediastinal obstruction
- Insect bites
- Hypothyroidism (myxedema)
- Spasmodic cough
- Hypoproteinemia (nephritis, nephrotic syndrome, anemia, kwashiorkor, etc.)
- Ethmoid sinusitis
- Cavernous sinus thrombosis
- Angioneurotic edema or drug reaction
- Chronic cor pulmonale
- Congestive heart failure
- Constrictive pericarditis
- Dermatomyositis (heliotrope color)
- Sickle cell veno-occlusive crisis
Causes of the long philtrum
- Hurler syndrome
- Robinow syndrome
- Fetal alcohol syndrome
- Femoral hypoplasia
- Fetal hydantoin and valproic acid syndrome
- Aarskog syndrome
- Gangliosidosis
- Smith-Lemli-Opitz syndrome
- Wagler-Stickler syndrome
- Williams syndrome
- Weaver syndrome
- Ectodermal dysplasia
- Arthrogryposes multiplex congenital
- Coffin-Siris syndrome
- Cornelia de Lange syndrome

Abnormalities Of Philtrum
The midline depressed area or vertical groove between the columella nasi (lower margin of nasal septum) and the upper lip is called the philtrum. Philtrum may be abnormally long in several developmental disorders listed.
The philtrum may be abnormally short in children with DiGeorge syndrome, (CATCH-22 syndrome, 18q deletion syndrome), Marsolf syndrome, and orofacial-digital syndrome.
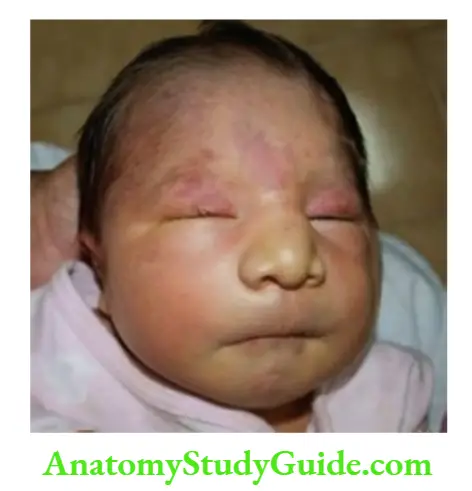
The philtrum may be flat or smooth in children with fetal alcohol syndrome and Prader-Willi syndrome. The vertical groove or philtrum is rather broad in children with autism spectrum disorder.
Low Set Ears
The upper and lower limits of the pinna normally correspond to the level of eyebrows or palpebral fissures and the base of the alae nasi, respectively.
The horizontal interpalpebral line, when projected posteriorly, should bisect the ears into upper one-third and lower two-thirds portions.
If the line passes above the ears, or helix, it is suggestive of low-set ears. The common causes of low-set ears are listed.
All newborns with external ear anomalies should be assessed for renal malformations and hearing tests.
Causes of low-set ears
- Down syndrome
- Smith-Lemli-Opitz syndrome
- Renal agenesis (Potter facies)
- Treacher Collins syndrome
- Gorgoylism
- Cri-du-Chat syndrome
- Turner syndrome
- DiGeorge syndrome
- Noonan syndrome
- Edwards syndrome
- Patau syndrome
- Carpenter syndrome
- Trisomy 17–18, 13–15
- Apert syndrome
- Idiopathic hypercalcemia
Micrognathia (Hypoplasia Of Mandible)
A small chin giving an appearance of “bird facies” is a recognized feature of a number of developmental conditions and may be associated with posterior displacement of the jaw or retrognathia.
It may be associated with feeding and breathing difficulties. The salient causes of micrognathia are listed in.
Causes of micrognathia
- Pierre-Robin syndrome (micrognathia, retrognathia, cleft palate, and glossoptosis)
- TreacherCollins syndrome
- Cerebro-costo-mandibular syndrome (micrognathia, cleft palate, glossoptosis, multiple posterior rib gaps, and cerebral maldevelopment)
- Pyknodysostosis
- Hallermann-Streiff syndrome
- Cri-du-Chat syndrome
- Marfan syndrome
- Seckel syndrome
- Progeria
- Turner syndrome
- Arteriohepatic dysplasia
- Fetal alcohol syndrome
- DiGeorge syndrome
- Russell-Silver syndrome
- Rubinstein-Taybi syndrome
- Schwartz-Jampel syndrome
- Smith-Lemli-Optiz syndrome
- Wagner-Stickler syndrome
- Trisomy 13 and 18
- Weaver syndrome
- Warkany syndrome
- Juvenile rheumatoid arthritis
High Arched Palate
The hard palate is usually flat or slightly arched. The palate becomes dome-shaped or higharched in children with mouth breathing, prolonged ventilation following orotracheal intubation, and repair of cleft palate.
Other causes of high-arched palate include Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, fragile-X syndrome, Friedrich ataxia, craniosynostosis, and crowding of molars.
It is usually asymptomatic but may cause difficulty in feeding, clarity of speech, and obstructive sleep apnea.
Causes of macroglossia
- Criticism
- Beckwith-Wiedemann syndrome
- Down syndrome (tongue is normal but oral cavity is small)
- Primary amyloidosis
- Glycogen storage disease type 2 (Pompe disease)
- Puppet-like syndrome of Angelman
- New growth of tongue (lymphangioma, hemangioma, neurofibromatosis, rhabdomyoma)
- Klippel-Trenaunay-Weber syndrome
- Lingual thyroid
- Duchenne muscular dystrophy
- Generalized gangliosidosis
- Sandhoff disease
- Hurler syndrome
Big Tongue (Macroglossia)
The tongue may be large and protrude out of the mouth in several conditions. The child with a long tongue may be able to protrude his tongue up to the chin or tip of the nose.
The condition may be congenital or acquired. Rhythmic protrusion of the tongue (which is normal in size) is a feature of intracranial hemorrhage and cerebral edema in newborn babies, athetosis, and Sydenham chorea. The common causes of macroglossia are listed.
It may be associated with difficulties in feeding, swallowing, speaking, and sleeping (obstructive sleep apnea).
In contrast, the tongue may be small and atrophic in children with hypoglossal nerve palsy, multiple cranial nerve palsies (Moebius syndrome), and Werdnig-Hoffman disease.
Gum Hyperplasia
Hyperplasia of gums is most commonly due to poor or dental hygiene, mouth breathing, and phenytoin therapy. The common causes are listed in.
Causes of hyperplasia of gums
- Poor oral hygiene (gingivitis)
- Drug-induced: Phenytoin, cyclosporine, and calcium channel antagonists
- Xanthomatosis
- Epulis
- Scurvy
- Diffuse fibromatosis
- Acute monocytic leukemia
- Histiocytosis X
- Hurler syndrome
- Granulomatous diseases
Thrush
Oral thrush is caused by Candida albicans. It is characterized by white raised membranous patches that resemble milk curds and are located on the tongue, buccal mucosa, gums, lips, and pharynx.
The patches cannot be removed easily and when scraped, they leave behind mucosal lesions with oozing blood. Except during the newborn period, thrush does not occur in healthy individuals.
Neonates may develop thrush, if the mother is having vaginal candidiasis or candidal infection of the breast nipples or when the infant is bottle fed.
Prolonged use of broad-spectrum antibiotics may be associated with oral thrush.
Recurrent or chronic oral and pharyngeal candidiasis is a recognized feature of acquired immunodeficiency syndrome (AIDS), severe combined immunodeficiency, T cell disorder, DiGeorge syndrome, biotinidase deficiency, and autoimmune polyendocrine syndrome.
Causes of delayed dentition
- Constitutional or hereditary delay
- Protein-energy malnutrition
- Chronic systemic disorder
- Rickets
- Down syndrome
- Endocrinal disorders like congenital hypothyroidism, hypopituitarism, and hypoparathyroidism
- Amelogenesis imperfecta
- Apert syndrome
- Chondroectodermal dysplasia
- Osteogenesis imperfecta type 1
- Cleidocranial dysostosis
- Mucopolysaccharidosis
- Incontinentia pigmenti
- Progeria
Delayed Dentition
Eruption of primary dentition usually starts around 6 to 8 months of age. The dentition may be delayed up to one year due to hereditary or constitutional factors.
Dentition is considered delayed if there is no eruption of teeth by the first birthday.
Apart from constitutional delay (history of delayed dentition in siblings and parents), common causes of delayed dentition include nutritional or systemic disorders, and chromosomal and genetic disorders associated with orofacial dysmorphism.
The common causes of delayed dentition are listed in. The child should be evaluated by a dentist, if there is no primary dentition by 18 months or when secondary dentition is delayed beyond 8 years.
Causes of delayed dentition
- Constitutional or hereditary delay
- Protein-energy malnutrition
- Chronic systemic disorder
- Rickets
- Down syndrome
- Endocrinal disorders like congenital hypothyroidism, hypopituitarism, and hypoparathyroidism
- Amelogenesis imperfecta
- Apert syndrome
- Chondroectodermal dysplasia
- Osteogenesis imperfecta type 1
- Cleidocranial dysostosis
- Mucopolysaccharidosis
- Incontinentia pigmenti
- Progeria
Brownish Discoloration Of Teeth
Brownish discoloration of teeth may occur due to amelogenesis because of malfunctioning of ameloblasts with the poor formation of enamel or due to staining of teeth by drugs and endogenous metabolites.
Greenish-yellow staining of teeth may occur due to the elevation of direct or conjugated bilirubin which is water-soluble.
Costochondral Beading
The common causes of costochondral beading include rickets, scurvy, and chondrodystrophy.
The beading is broad and dome-shaped in rickets while it is sharp like a bayonet because of posterior subluxation of the sternum in cases of scurvy.
Clubbing
The exact mechanism of clubbing is unclear and is probably caused by hypervascularity and the opening up of anastomotic channels in the nail bed.
It appears to be due to the production of humoral substances which cause dilatation of blood vessels of the fingertips and endothelial proliferation.
The clubbing can appear after about 6 weeks of the appearance of a predisposing factor. There are four clinical stages of clubbing.
Grade 1: Softening of the nail bed with increased fluctuations.
Grade 2: Obliteration of the angle between the nail bed and skin fold.
Grade 3: Increased curvature and thickening of the nail producing a parrot-beak appearance.
Grade 4: Drumstick appearance because of thickening of the whole distal phalanx.
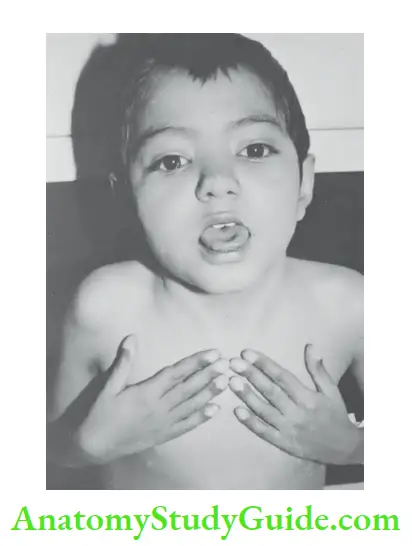
Schamroth sign When dorsal surfaces of terminal phalanges of corresponding fingers are
opposed, there is a diamond-shaped space at the base of the nail bed. In clubbing, the diamond-shaped space is lost.
Examine the profile view of the distal phalanges. When clubbing is present, the vertical height at the base of the nail will be greater than the height of the distal interphalangeal joint.
Clubbing occurs in association with a number of cardiopulmonary and abdominal conditions. The important causes of clubbing are listed in.
Clubbing in association with pain over the wrists and ankles because of subperiosteal bone formation over distal diaphyses of the radius, ulna, tibia, and fibula is called hypertrophic osteoarthropathy.
It may occur due to mesothelioma, bronchiectasis, and cirrhosis of the liver.
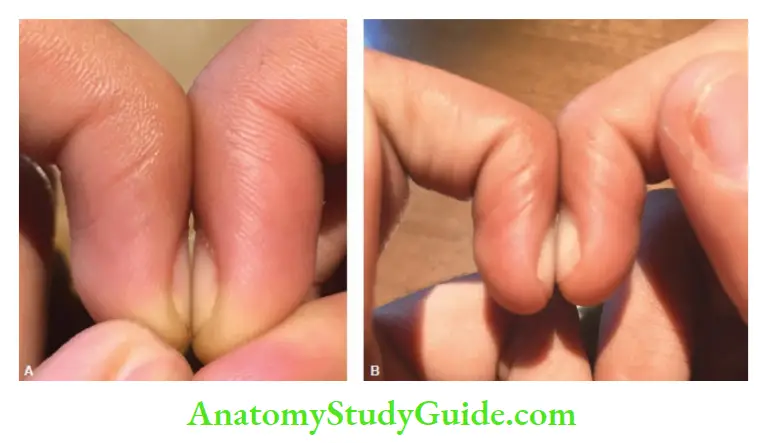
Causes of clubbing
- Familial or congenital
- Cyanotic heart disease (drumstick clubbing)
- Pulmonary suppuration (bronchiectasis, lung abscess, empyema, cystic fibrosis)
- Malabsorption syndrome
- Ulcerative colitis
- Polyposis of the colon
- Fibro-caseous pulmonary tuberculosis (parrot-beak type clubbing)
- Regional enteritis or Crohn’s disease
- Subacute bacterial endocarditis
- Cirrhosis of life
- Clubbing should be looked for in fingernails because toenails may become curved due to tight shoes.
Causes of lockjaw
- Tetanus
- Syndrome of brainstem dysfunction
- Tumor of the jaw (rhabdomyosarcoma)
- Arthritis of the temporomandibular joint
- Encephalitis
- Primary hypoparathyroidism (DiGeorge syndrome)
- Nasopharyngeal carcinoma
- Brain tumor
- Acute post-streptococcal polymyalgia
- Teeth grinding
- Injury to the Jaw
- Strychnine poisoning
- Anesthetic-induced malignant hyperthermia
- Phenothiazine and neuroleptic toxicity
- Infantile Gaucher’s disease
- Maple syrup urine disease
Conditions associated with a short neck
- Chondrodystrophy (Morquio’s disease)
- Klippel-Feil deformity
- Down syndrome
- Platybasia
- Cretinism
- Turner syndrome
- Hurler syndrome
- Bilateral Sprengel’s deformity
- Goldenhar syndrome
- Spondyloepiphyseal dysplasia congenital
- Noonan syndrome
Causes of neck stiffness
- Meningitis
- Kernicterus
- Meningismus
- Acute poliomyelitis
- Tetanus
- Phenothiazine toxicity
- Vertebral anomalies
- Leukemic infiltrates in CNS
- Vertebral trauma
- Hypernatremia
- Caries cervical spine
- Toxic shock syndrome
- Retropharyngeal abscess
- Posterior fossa brain tumor with herniation
- Juvenile chronic arthritis
- Lyme disease
- Subarachnoid hemorrhage
- Calcification of cervical intervertebral disks
- Arnold-Chiari malformation
- Infantile Gaucher’s disease
- Leptospirosis
- Behcet syndrome
- Decerebrate rigidity
- Lesch-Nyhan syndrome
Signs of meningeal irritation without any abnormalities in the CSF may be seen in children with pneumonia, pyelonephritis, salmonellosis, bacillary dysentery, leukemic infiltrates, etc.
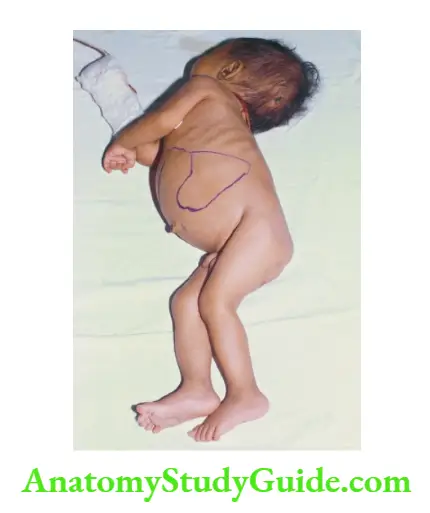
Trismus (Lockjaw)
There is the inability to open the mouth. The common conditions causing lockjaw are listed.
Short Neck
Webbing of the neck (pterygium colli) gives the appearance of a short neck due to the presence of a thick web of skin that extends from behind the ears to the outer end of the clavicles and the acromial process.
A number of conditions are associated with a short neck.
Neck Stiffness And Opisthotonos
Neck stiffness is diagnosed when the chin cannot be touched to the front of the chest by flexion of the neck.
In a struggling infant, the head should be suspended beyond the edge of the examination table to relax the neck.
In infants below 3 months, severely malnourished and immunocompromised children, neck rigidity may not develop despite the presence of meningitis.
Opisthotonos is characterized by marked neck stiffness with extensor arching of the whole body. The causes of neck stiffness are listed in.
Torticollis (Wry Neck)
Torticollis is an asymmetric deformity of the neck and head characterized by lateral flexion or tilt of the head towards the involved side and rotation of the chin towards the opposite shoulder.
In normal infants, the head can be turned so that the chin can touch each shoulder and the ear can be made to touch the ipsilateral shoulder.
Infants and children with double vision may tilt their heads to avoid double vision. Sudden exposure to cold may lead to torticollis.
Causes of torticollis
- Sternocleidomastoid ‘tumorâ€
- Strabismus and diplopia
- Klippel-Feil syndrome
- Acute oral infections and retropharyngeal abscess
- Injury to the cervical spine
- Acute cervical adenitis
- Pott’s disease of the cervical spine
- Torsion dystonia
- Acute dystonic drug reaction
- Athetoid cerebral palsy
- Hiatus hernia
- Tumor of the cervical spine (eosinophilic granuloma, osteoid osteoma)
- Tonsillar herniation (astrocytoma of the cerebellum)
- Subluxation of the atlantoaxial joint
- Down syndrome
- Skeletal dysplasias
- Juvenile chronic arthritis
- Morquio’s syndrome
- Occipitalization and basilar impression of a skull
- Larsen syndrome
- Pseudoachondroplasia
- Congenital anomalies of the odontoid process
- Benign paroxysmal torticollis due to wrong posture or exposure to cold
- Sandifer syndrome
Erythema Nodosum
They are characterized by tender, painful, reddish-blue nodules which are typically located on the shins and rarely may spread to thighs and arms.
They fade slowly over several weeks leaving behind bruised patches but they never ulcerate. It is a manifestation of hypersensitivity panniculitis due to a variety of inflammatory conditions.
The common causes of erythema nodosum are listed.
Conditions associated with erythema nodosum
- Viral upper respiratory tract infections
- Streptococcal infection
- Tuberculosis
- Sarcoidosis
- Behcet’s disease
- Leprosy
- Systemic mycosis and histoplasmosis
- Toxoplasmosis
- Inflammatory bowel disease
- Systemic lupus erythematosus
- Cat-scratch disease
- Yersinia enterocolitic infection
- Sulphonamide and penicillin therapy
Micropenis
It refers to a small penis. There is minimal growth of genitals during childhood followed by a sudden spurt of growth during adolescence.
The normal stretched dorsal length of the penis in infants is 3.9 ±0.8 cm. Micropenis is diagnosed when the length of the penis is less than 2.0 cm.
The penis may be buried in the pubic fat in obese children. The length of the stretched penis should be recorded from the pubic ramus to the tip of the glans penis (after retracting the foreskin) over the dorsal side.
The suprapubic pad of fat should be pressed inward as much as possible. A disposable 10 ml plastic syringe can be used to measure penile length.
The needle-bearing end of the syringe is cut and the piston is introduced through the cut end. The smooth flanged end of the syringe is firmly pressed against the pubis after enclosing the penis into the barrel of the syringe.
Gentle suction is applied to stretch the penis. A piston is aligned to the glans and the length of the penis is read with a scale. Micropenis is a recognized clinical feature of several syndromes.
Sydromes associated with micropenis
- Hypogonadotropic hypogonadism (Kallmann syndrome, Prader-Willi syndrome, Ruds’ syndrome, Alstrom syndrome, septo-optic dysplasia)
- Klinefelter syndrome
- Fanconi’s anemia
- Carpenters syndrome
- Robinows syndrome
- Williams syndrome
- Hallermann-Streiff syndrome
- Deletion of the long arm of 18 chromosome
- Noonan syndrome
- X-linked hypogammaglobulinemia
- Down syndrome
- Hypopituitarism
- Cornelia de Lange syndrome
- CHARGE association
- Maternal intake of fertility drugs like diethyl-stilbestrol (DES)
Abnormalities Of Testicular Size
The volume of testes is best evaluated with the help of a Prader orchidometer which has testis-shaped plastic balls of different sizes on a string.
During preadolescence, the size of testes varies between 1.5 and 2.0 cm. The adult size of testes varies between 3.5 and 5.0 cm or an average volume of 20 ml.
The right testis is slightly larger in size and hangs lower than the left. Failure of testicular enlargement by the age of 14 years is suggestive of delayed sexual maturation or hypogonadism.
Micro-orchid. If testes are less than 1.0 cm in length or 10 ml in volume, it is suggestive of micro-orchid. It is a recognized feature of the following conditions:
Rudimentary testes syndrome
Klinefelter syndrome
Laurence-Moon-Biedl syndrome
Hypopituitarism
Hypothalamic disorders
Testicular atrophy due to mumps
May be associated with various syndromes having micropenis.
Macro-orchidism. The testicular length that exceeds 2 cm or a volume of greater than 20 ml in preadolescent children is suggestive of macro-orchid-ism. It may occur in the following conditions:
Fragile-X syndrome (macro-orchid-ism occurs after puberty)
Neurogenic or idiopathic sexual precocity
Hypothyroidism
Testicular tumors (teratoma, interstitial cell tumor, and rhabdomyosarcoma)
Sexual Infantilism And Delayed Puberty
The age of onset of puberty is variable but usually occurs between 10 and 12 years in girls and 12 and 14 years in boys.
Puberty is considered delayed if there is no budding of breasts by 12 years in girls or a lack of increase in the volume of testes by the age of 14 years in boys.
The upper age limit for onset of menstruation is 15 years. Delayed puberty is more common in boys than girls.
The most common cause of delayed puberty is a constitutional delay in boys and Tinner syndrome in girls. The common causes of delayed sexual maturation are listed in Table 8.9.
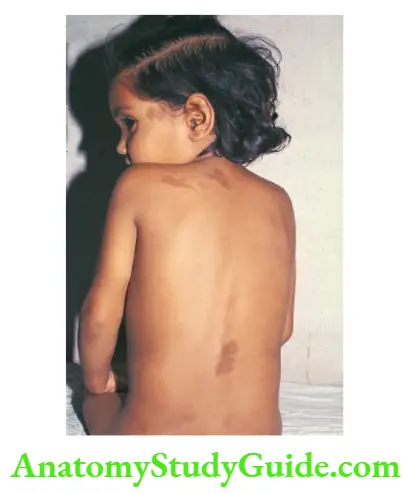
Café-Au-Lait Spots
These are flat, sharply demarcated brown-colored (coffee with milk) macules on the skin, hi children, up to 5 spots of less than 1.0 cm diameter are considered normal.
When the spots are large in size (>5 mm in preadolescents or >15 mm in adolescents) or excessive in number (>6), they serve as useful markers of diseases that are associated with cafe-au-lait spots.

Causes of cafe-au-lait spots
- Neurofibromatosis type 1
- McCune-Albright syndrome
- Gaucher disease
- Russell-Silver syndrome
- Ataxia-telangiectasia
- Bloom syndrome
- Tuberous sclerosis
- Chediak-Higashi syndrome
- Legins syndrome
- Hunter syndrome
- Noonan syndrome
- Watson syndrome
- Wiskott-Aldrich syndrome
- Fanconi’s anemia
- Epidermal nevus syndrome
- Chronic myeloid leukemia
- Multiple lentigines
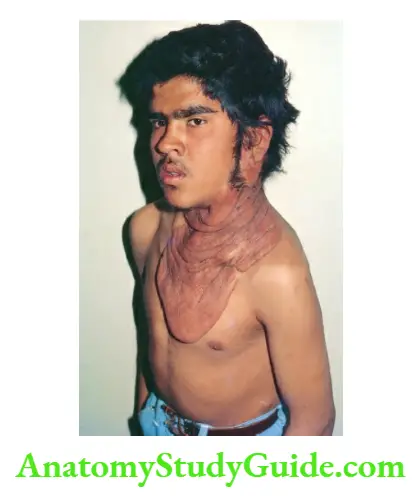
Hemihypertrophy
It is characterized by enlargement of one-half of the body or hypertrophy may be limited to the face or one of the extremities. The common causes and their correlates are listed.
Palmar Erythema
There is marked erythema of the palms, especially at the thenar and hypothenar eminences. Soles may or may not be affected.
Erythema may occur due to elevation of estrogen level, inflammation, increased circulation, polycythemia, telangiectasia, or increased vascularity due to the release of nitric oxide.
Palmar (and plantar) erythema may occur due to various physiological and pathological conditions.
Causes of palmar erythema
- Pregnancy
- Steroid therapy
- Chronic liver disease with or without portal hypertension
- Rheumatoid arthritis
- Kawasaki disease
- Infective disorders like scarlet fever, hand-foot-mouth disease, secondary syphilis and
- Rocky Mountain spotted fever
- Chronic mercury poisoning (acrodynia)
- Thyrotoxicosis
- Polycythemia
- Deep telangiectasias
- Eczema and cirrhosis
- Idiopathic
Epistaxis
Bleeding from the nose or epistaxis is common is children. It is usually benign and occurs during the summer months because of excessive drying of nasal secretions and nose picking.
The bleeding most commonly arises from Tittle’s area (Kiesselbach plexus) which is an anastomotic site for terminal arterioles.
It is located over the nasal septum about 0.5 cm within the nostril and above the nasal floor. The common causes of epistaxis are listed in.
Causes of epistaxis
- Bleeding from Little’s area by picking or blowing the nose
- Chronic allergic or viral rhinitis
- Acute febrile illness: Diphtheria, rheumatic fever, infectious mononucleosis, dengue fever, and viral hemorrhagic fevers
- Bleeding diathesis: Thrombocytopenia, hemophilia, von Willebrand disease
- Prolonged instillation of nasal drops, insufflations, and sprays
- Nasal polyps
- Foreign body and nasal catheter
- Nasal trauma and head injury with fracture of the base of the skull
- Hemangioma and hereditary telangiectasia (Osler-Weber-Rendu syndrome)
- Tumors like juvenile angiofibroma, lymph-epithelioma, and rhabdomyosarcoma of the nasopharynx. Capillary hemangioma and hamartoma may present as epistaxis
- Chronic liver disease
- Hypertension
Drooling Of Saliva
Saliva is produced to keep the oral mucosa wet and contains anti-infective agents and the digestive enzyme ptyalin. It aids in swallowing, digestion, and maintenance of oral hygiene.
Excessive drooling and choking during feeding in a newborn baby are suggestive of esophageal atresia. When atresia is suspected, an attempt should be made to pass a Fr. 8 or 10 stiff rubber catheter into the esophagus.
In esophageal atresia, it is not possible to pass the catheter into the stomach.
Drooling with the putting of fingers in the mouth is a physiological oral phase (mouthing) of development and is often considered a useful correlate of teething in infants.
Upper respiratory tract infection with nasal congestion is usually associated with drooling.
Drooling occurs either due to excessive production of saliva (sialorrhea) or inability to swallow the saliva as a result of oropharyngeal obstruction or neuromotor incoordination.
Persistent drooling is common in children with mental retardation, cerebral palsy, familial dysautonomia (Riley-Day syndrome), Rett syndrome, and pseudobulbar palsy (Wilson disease).
Drooling of acute onset and short duration is seen in children with stomatitis, tonsillitis, acute epiglottitis, diphtheria, peritonsillar or retropharyngeal abscess, foreign body, caustic ingestion, scorpion sting, organophosphate poisoning.
Halitosis
Halitosis or bad breath is uncommon in children compared to adults. It is a reliable index of dental hygiene.
Mouth breathing due to persistent nasal blockage is a leading cause of foul breath. The salient causes of halitosis are listed in.
Excessive Sweating
Excessive sweating or hyperhidrosis is a common complaint in children and is usually localized over the head and palms.
It is most commonly due to constitutional abnormality and is of no clinical significance although most parents are worried that it may cause weakness.
Increased perspiration is commonly seen in children with congestive heart failure, acute respiratory failure, and narcotic withdrawal syndrome.
Excessive sweating of palms along with nail biting is suggestive of anxiety. The common causes of excessive sweating are listed in.
Common causes of halitosis
- Poor oriental hygiene
- Caries teeth
- Adenoidal hypertrophy
- Sinusitis
- Nasal foreign body
- Atrophic rhinitis
- Vincent stomatitis or ulcerative gingivostomatitis
- Prolonged febrile illness
- Bronchiectasis and lung abscess
Causes of excessive sweating
- Constitutional or idiopathic
- The hot and humid environment
- Lysis of fever
- Physical activity
- Hyperthyroidism
- Autonomic dysfunction
- Anxiety and stress
- Pheochromocytoma
- Serotonin-secreting tumor of the gut (ganglioneuroma or ganglioneuroblastoma)
- Pulmonary tuberculosis (night sweating)
- Night terror
- Hypoglycemia
- Cystic fibrosis
- Fabry disease
- Shock (cold and clammy extremities)
Abnormal Body Odor
Body odor depends upon a large number of factors including personal hygiene, odor of breath and smell of sweat, urine, feces, or pheromones elaborated by the skin, and use of medicated soaps and various cosmetics.
Abnormal and specific odors may be observed in certain systemic disorders and inborn errors of metabolism.
However, among various special senses which are used for the evaluation of a patient, the sense of smell and taste are the least important.

Xerostomia (Dry Oral Mucosa)
Reduced secretion of salivary glands with dryness of mouth and tongue may cause foul breath, tooth decay, and poor digestion.
The common causes include mouth breathing (nasal obstruction), severe dehydration, parotitis, calculus in one of the major salivary glands, and psychological disorders like anxiety, stress, and depression.
Xerostomia is a recognized feature of Parkinson’s disease, human immunodeficiency virus (HIV) infection, and Sjogren syndrome, and because of side effects of several medications namely atropine, antihistamines, antidepressants, antipsychotics, sedatives, methyldopa, diuretics, chemotherapy agents and radiotherapy of head and neck.
Leave a Reply