The Dysmorphic Child
Most developmental defects and chromosomal disorders are seen during childhood. A large number of childhood diseases can be diagnosed by the identification of typical facies.
Table of Contents
By virtue of constant effort and practice, the pediatrician should sharpen his observational faculties to identify subtle abnormalities of the head and face.
Front and profile views of the face should be examined to identify facial dysmorphism.
Read and Learn More Pediatric Clinical Methods Notes
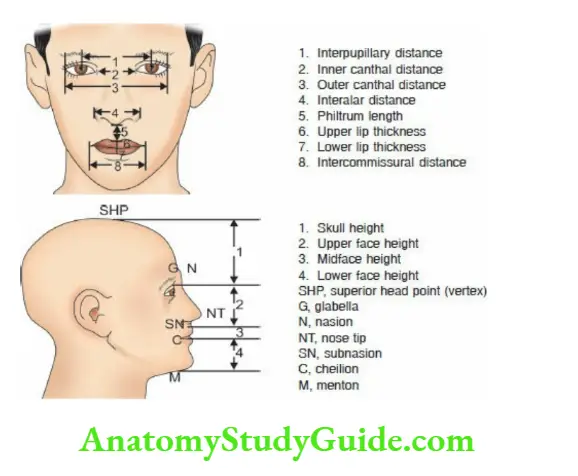
Look for the size and alignment of eyes, the distance between two eyes, the shape of the nasal tip and bridge, the size of the philtrum, the size, and position of the ears, and the size of the chin and forehead.
Data is available to make an objective assessment of dysmorphism in different ethnic groups and populations.
Abnormal Shape Of Head
The abnormalities in the shape and size of the head are common and easily visible. They may be isolated or associated with other anomalies as a part of a syndrome.
Abnormalities in the shape of the scalp due to edema (caput succedaneum) or cephalhematoma are common at birth in vaginal-born babies especially following prolonged labor or vacuum extraction.
These abnormalities are transient and gradually resolve over a few hours or days.
A number of physiological or pathological conditions may be associated with abnormalities in skull bones with odd-shaped skulls and are listed in.

Typical Facies
In certain systemic disorders in children, the diagnosis can be suspected on the basis of characteristic facies. The typical facies are usually evident during the course of the disease or when the disease is well advanced.
In genetic or chromosomal disorders, dysmorphism is evident at birth or soon after.
Chronic hemolytic facies In beta thalassemia major, the increased erythropoiesis in the bone marrow results in the expansion of the medullary cavity of various bones leading to the development of typical facies.
There is bossing of the skull with a flattened vertex giving the appearance of the hot-cross bun.
The facial bones or malar eminences become prominent due to hypertrophy of the maxilla.
There is hypertelorism (expansion of the lesser wing of the sphenoid), epicanthic folds, and a depressed bridge of the nose.
There is crowding of teeth with malocclusion and protrusion of teeth giving a “rodent look”.
Cretinism The facial features appear coarse with bloated cheeks, depressed nasal bridge, large protruding tongue, and drooling of saliva.
The anterior fontanel is large in size and cranial sutures are widely separated. The skin is usually dry, coarse, cold, and mottled. Hair is coarse and brittle.
Mucopolysaccharidosis It is characterized by coarse facial features, a depressed or flat nose, thick lips, a large tongue, clouded cornea, and widely separated peg-like teeth.
Additional features include a short neck, pigeon-shaped chest, long extremities with stiff joints, and hepatosplenomegaly.
Moon facies Moon-shaped facies are seen in children with cushingoid features due to prolonged corticosteroid therapy or Cushing syndrome.
The face is rounded with prominent flushed or plethoric cheeks, double chin, hirsutism, and acne. There is an accumulation of fat over the upper back and supraclavicular region producing a typical buffalo hump.
In nephrotic syndrome, moon facies occur due to marked swelling of the face, puffiness, and lid edema. During the course of corticosteroid therapy, cushingoid appearances supervene.
Hepatic facies In chronic liver disease and/or cirrhosis, facial features are characterized by icteric tinge, sallow or muddy complexion, pinched nose, sunken eyes, hollow temporal regions, and cheeks.
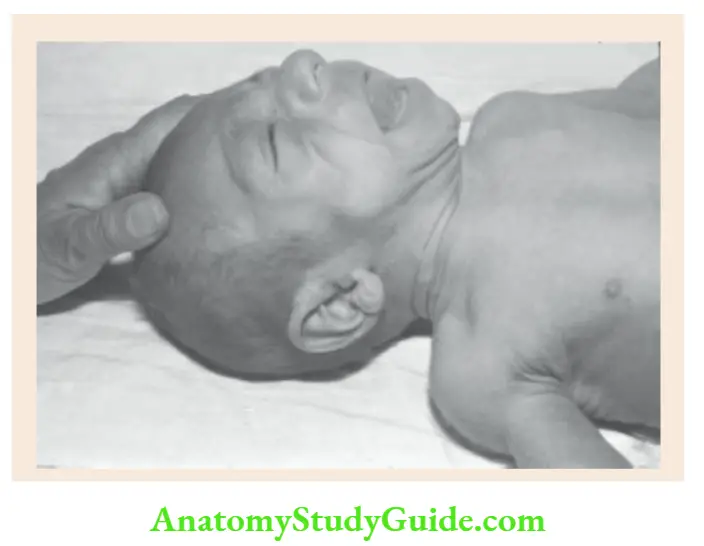
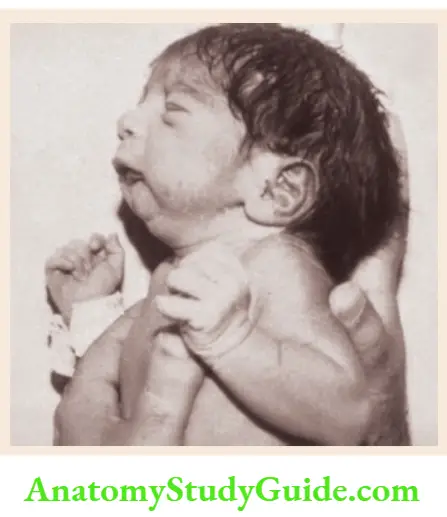
Potter facies In infants with renal agenesis, there is oligohydramnios and the fetus is squashed in utero.
Potter facies is characterized by a small receding chin, low-set ears with folded helices, compressed nose, epicanthic folds, prominent skin folds below the eyes, and an antimongoloid slant of the eyes.
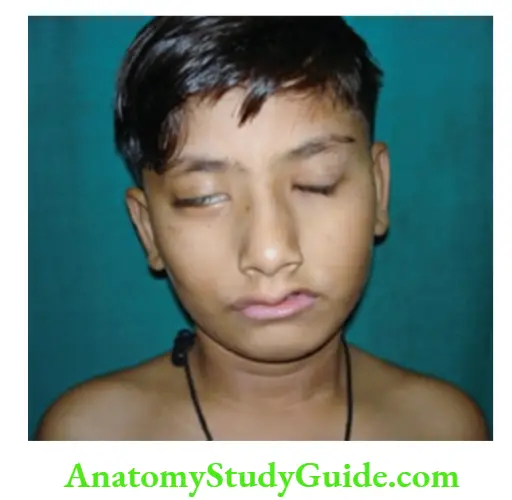
Facial palsy In unilateral facial palsy, there is asymmetry of the face with the absence of a nasolabial fold and the inability to close the eye on the affected side.
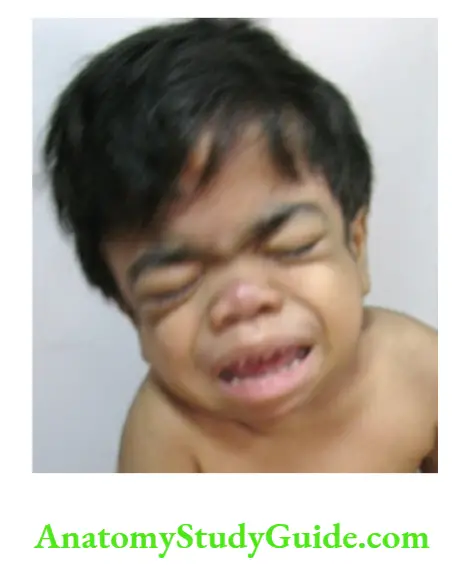
Expressionless face Bilateral facial palsy either at the nuclear (bulbar palsy) or internuclear level (bilateral facial nerve palsy) is characterized by expressionless or masklike facies.
The various conditions causing mask-like face include Mobius syndrome, Guillain-Barre syndrome, infantile botulism, Wilson disease, myotonic dystrophy, facioscapulohumeral muscular
dystrophy and mental depression.
Risus sardonicus The infant with tetanus looks alert, the eyebrows are raised and angles of the mouth are drawn out due to spasms of facial muscles.
The mouth may be kept slightly open due to downward pull because of spasms of the neck muscles. Trismus or reflex spasm of masseters is invoked when an attempt is made to open the mouth.
The Developmental Defects
A child with developmental defects or congenital structural anomalies is called a dysmorphic child.
Dysmorphism is manifest during childhood and every pediatrician should be conversant with the clinical approach to such a child. The congenital structural anomalies may be classified as follows
1. Structural or normal variants These are normal developmental or anatomical variations without any clinical or therapeutic implications.
Examples include simian crease, clinodactyly (medial or lateral deviation of a finger), camptodactyly (clawlike fingers), abnormal dermatoglyphics, wide anterior fontanel, wide forehead, sacral dimple, beaked or bulbous nose, etc.
These variants may or may not be of any clinical relevance.
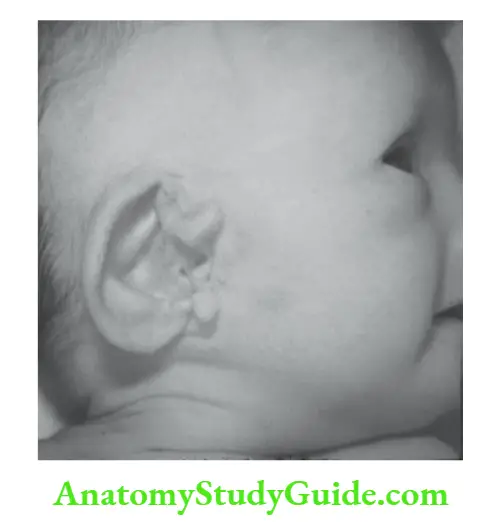
2. Minor anomalies They are true anomalies and are primarily of cosmetic concern. Isolated minor anomalies may be present in over 10 percent of normal newborn babies.
Examples include preauricular skin tags, supernumerary nipples, and various nevi and pigmentary disorders. Children with three or more minor anomalies are likely to have dysmorphic syndrome.
3. Major anomalies The major developmental defects produce functional disability and may compromise normal life expectancy.
Around 2 to 3 percent of newborn babies may have major anomalies. The anomaly may be detectable at birth or manifest at any time during childhood.
4. Isolated versus multiple anomalies The majority of birth defects (almost two-thirds) are isolated involving a single organ or system.
The common examples are cleft lip, cleft palate, congenital heart disease, etc.
The cause of isolated major anomalies is usually multifactorial or polygenic. Less commonly, congenital anomalies may affect several organs or body systems producing multiple congenital defects.
The common patterns of multiple structural anomalies include associations, sequences, field defects, and syndromes.
“Association” defects refer to the non-random combination of anomalies wherein the individual components occur together more frequently than would be expected by chance.
The clinical picture may vary from case to case but is consistent enough for recognition as a syndrome.
For example, the VACTERL association comprises vertebral anomalies, anal atresia, cardiac defects, tracheoesophageal atresia, renal and radial anomalies, and limb defects.
The recognition of two or three cardinal features should prompt a search for other occult anomalies.
CHARGE association is characterized by coloboma of the eye, heart anomalies (tetralogy of Fallot, PDA, VSD, ASD), atresia of choanae, retardation (mental and physical), genital hypoplasia (cryptorchidism and micropenis), ear anomalies and deafness.
VATER association is characterized by vertebral (hemivertebrae or sacral deformity) or vascular anomalies, anal malformations, tracheoesophageal fistula, and radial or renal defects.
The recurrence risk of association anomalies is low and this information is useful for genetic counseling.
The “sequence anomalies†is a pattern of multiple congenital malformations that cannot be explained on a developmental and embryologic basis and occur as a result of a cascade of seemingly unrelated consequences.
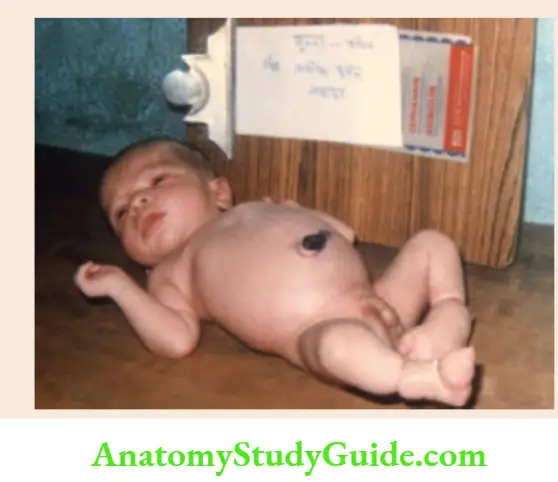
The typical example of sequence anomaly is “Potter oligohydramnios’ due to renal agenesis.
Oligohydramnios due to renal agenesis leads to intrauterine compression of a fetus with limb deformities, flattened or compressed facial appearance, and pulmonary hypoplasia which is usually the cause of death.
The “field defects†refers to the constellation of anomalies of different body organs which differentiate together during embryogenesis due to anatomical proximity.
Some adverse uterine factors may interfere with the normal development of the structures differentiating during the critical phase of embryogenesis.
They are often caused by an adverse vascular event and the recurrence risk is low. Examples include Poland anomaly and Moebius syndrome.
The “syndrome†is defined as a unique constellation of multiple anomalies that repeatedly occur in a consistent pattern.
The presence of multiple dysmorphic features in a child is suggestive of a chromosomal defect.
When there are two or more major anomalies affecting different body systems or one major and two minor congenital malformations, chromosomal disorders should be ruled out.
In patients with multiple malformations, there are no confirmatory laboratory tests, and diagnosis is based on the identification of a typical pattern of anomalies.
Based on the major anomalies present, these dysmorphic syndromes can be clinically divided into ten broad groups. The major diagnostic features of common dysmorphic syndromes are listed in
At times, the constellation of anomalies in a patient may not conform to any recognized syndrome.
A number of computerized dysmorphology databases are available to assist the clinician to make a reliable diagnosis of a child with multiple malformations.
Popular computerized databases or software include LDDB (London dysmorphology database), LNDB
(London neurogenetic database), POSSUM (Pictures of standard syndromes and unidentified malformations) and SYNDROC (syndrome congenital malformation database).
These databases can be screened on a personal computer. If a database search does not suggest a definitive diagnosis, the combination of anomalies may be a new, hitherto, unreported syndrome.
It is recommended to perform a prometaphase chromosomal analysis to rule out a rare chromosomal anomaly (usually interstitial deletions or translocations) as a cause of the syndrome.
Chromosomal Syndromes
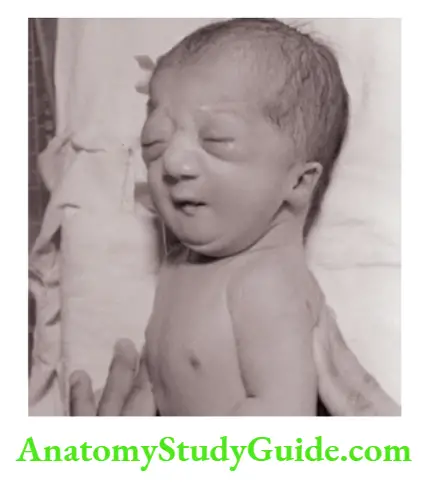
- Down syndrome (Trisomy-21)
Brachycephaly, flat facies, hypertelorism with mongoloid slant (upwards and outwards) of eyes with epicanthic folds, excess skin over the nape of the neck, open mouth with protruding tongue, low set small ears, Brushfield spots in the iris, single transverse palmar crease or simian crease, small incurved little finger, clinodactyly, sandal gap in the toes, hypotonia, or hyper flexibility, and mental subnormality.

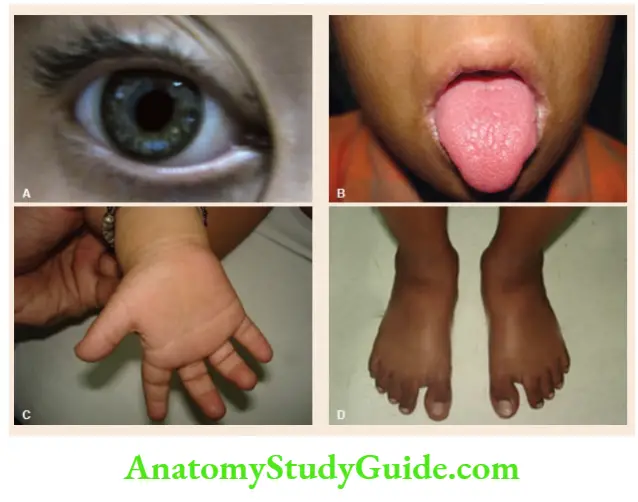
- Edward syndrome (Trisomy-18)
Intrauterine growth retardation, microcephaly with prominent occiput, low-set malformed ears, microphthalmia, clenched hands with index finger overlapping third finger and fifth finger overlapping fourth finger, short dorsiflexed first toe, short sternum, rocker bottom feet, inguinal or umbilical hernia, low-arch dermal ridge pattern, congenital heart defects (VSD, PDA, ASD), and severe neuromotor retardation.
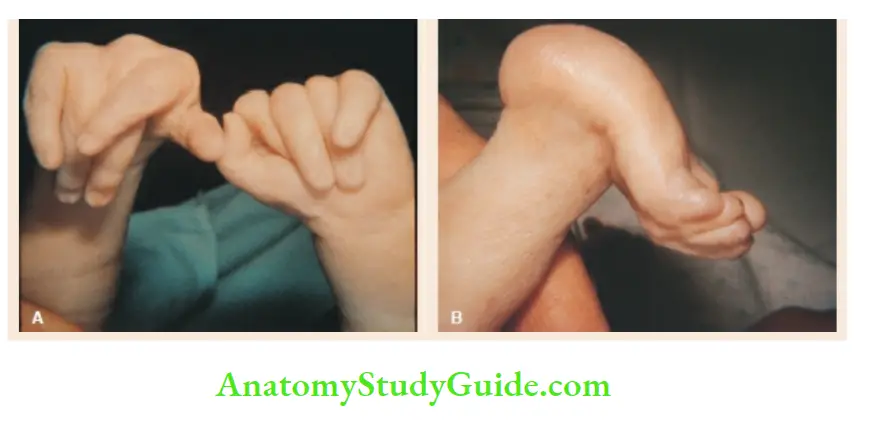
- Patau syndrome (Trisomy-13)
Microcephaly with localized scalp defects, holoprosencephaly, cleft lip/palate or both, broad flat nose, hypotelorism, low-set ears, microphthalmia, coloboma of the iris, postaxial polydactyly of hands and feet, inguinal hernia, omphalocele, congenital heart defects(VSD, ASD, PDA), neuromotor retardation.
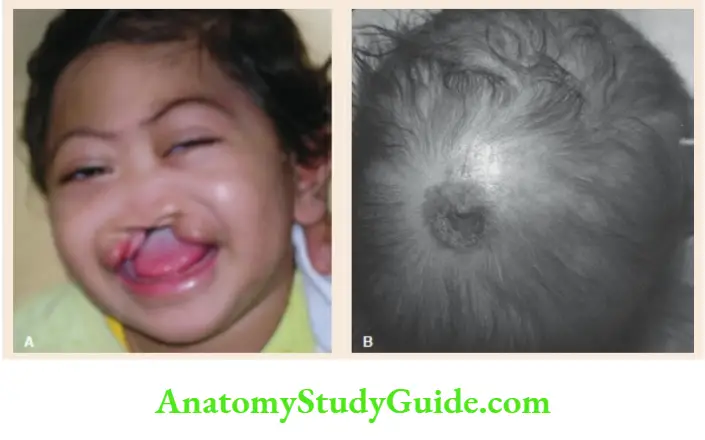
- Cri-du-chat syndrome or Lejeune syndrome
Cat-like cry in infancy, round facies, microcephaly, hypertelorism, epicanthic folds, hypertrichosis, failure to thrive, and antimongoloid slant of eyes. - Turner syndrome (Monosomy X)
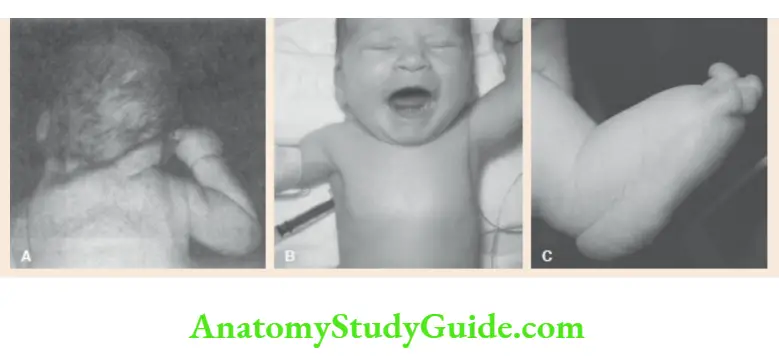
Short webbed neck, low posterior hairline, lymphedema of dorsa of hands and feet, cubitus valgus, shield-like chest with widely spaced nipples, short stature, primary amenorrhea. The associated findings include gonadal dysgenesis, renal anomalies, coarctation of the aorta, and deafness.

Syndromes with extremely Short Stature
- Cornelia de Lange syndrome
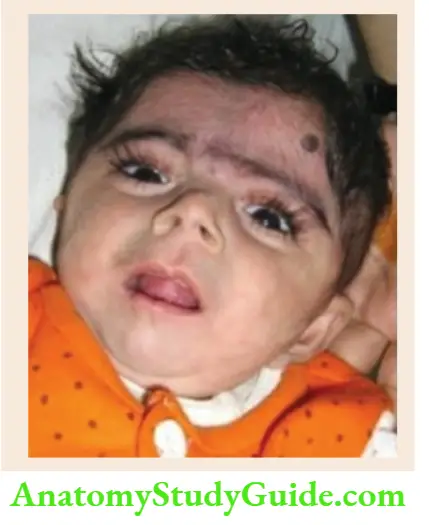
Microbrachycephaly, hirsutism, long curly eyelashes, thin downturned upper lip, long philtrum, bushy eyebrows with synophrys (eyebrows merge in the midline), micromelia, and mental retardation. - Rubinstein-Taybi syndrome
Microcephaly, hypoplastic maxilla with a narrow palate, low set ears, the antimongoloid slant of palpebral fissures, beaked nose with nasal septum extending below the level of nares, hypertrichosis, broad thumbs and toes, mental retardation, the tendency to form keloids. - Russell-Silver syndrome
Triangular facies with down turning of corners of the mouth, skeletal asymmetry (facial or limbs), clinodactyly, cafe-au-lait spots. - Seckel syndrome (Bird-headed dwarf syndrome)
Microcephaly, facial hypoplasia with a prominent nose, prominent eyes, low-set malformed ears, and mental retardation.
- Hallermann-Streiff syndrome
Brachycephaly with frontal and parietal bossing, bird-like facies, hypotrichosis of scalp, eyebrows, and eyelashes, bilateral microphthalmos with cataracts, small pinched nose, and micrognathia with a double chin. - Laron syndrome
Marked dwarfism (resistance or insensitivity to hGH), frontal bossing, saddle nose, ‘setting sun sign, obesity, small genitalia, and high-pitched voice.
Syndromes with Moderate Short Stature, Facial and/or Genital Defects
Rothmund-Thomson syndrome Photosensitivity, alopecia, cataracts, hypogonadism, and short stature.
- Smith-Lemli-Opitz syndrome
Ptosis, epical folds, anteverted nostrils, lowset ears, micrognathia, syndactyly of second or third toes, hypospadias and cryptorchidism in males, failure to thrive, and mental retardation. - Laurence-Moon-Biedl syndrome
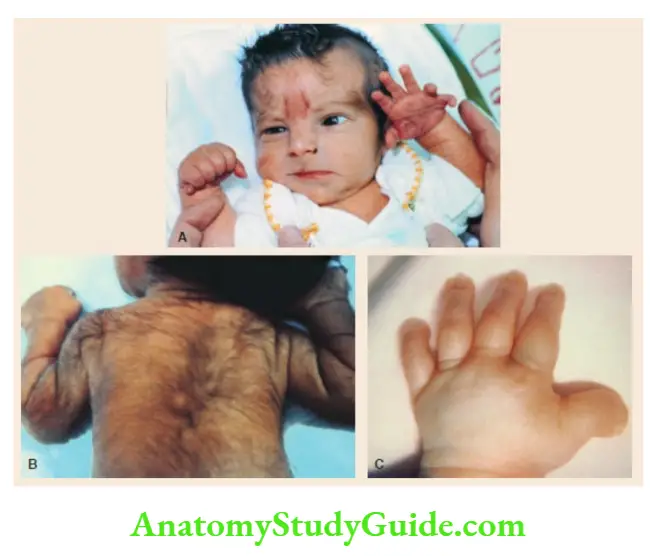
Obesity, retinitis pigmentosa (night blindness), mental retardation, hypogenitalism, and polydactyly. - Williams syndrome
Elfin facies, long philtrum, anteverted or upturned nostrils, depressed nasal bridge, thick patulous upper lip with drooping lower lip, wide smile, hypotelorism, periorbital fullness, strabismus, blue eyes, hoarse voice, supra valvular aortic stenosis, peripheral pulmonary stenosis, friendly ‘cocktail party’ mischievous personality, hypercalcemia during infancy, mental retardation with relative sparing of language, senile look due to wrinkling of skin and graying of hair. - Noonan syndrome
Turner-like phenotype, webbed neck, hypertelorism with the antimongoloid slant of eyes, broad forehead, ptosis, low-set posteriorly rotated or folded ears, rectum excavate carinate or both, pulmonary valve stenosis or ASD, and cryptorchidism. - Aarskog syndrome
Round face, small broad nose with anteverted nostrils, long philtrum with clinodactyly of 5th finger, “shawl scrotum” (scrotal skin fold encircles the base of the phallus), and short stature.
Syndromes with Physical Over Growth and Associated Defects
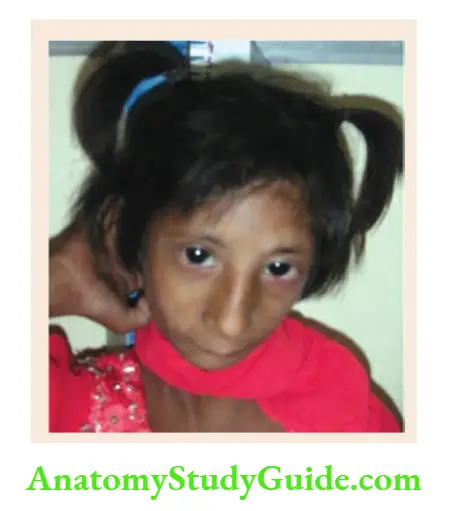
- Fragile X syndrome
Long face, macrocephaly, prominent ears, large chin or prognathism, hyperextensible distal interphalangeal joints, postpubertal macro-orchid-ism, tall stature, mental subnormality, cluttered speech, hyperkinetic, autistic or aggressive behavior. It is the most common genetic cause of intellectual disability. - Sotos syndrome
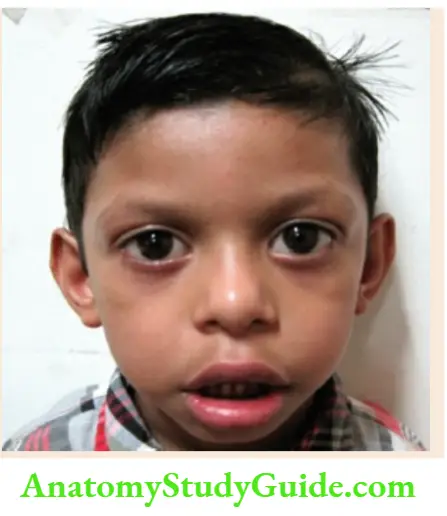
Large dolichocephalic head, coarse-looking facies, hypertelorism, prominent chin (macrognathia), large hands and feet, overgrowth of all physical parameters, hypotonia, laxity of joints, advanced bone age, seizures, autistic features, mental retardation, cardiac anomalies. - Beckwith-Wiedemann syndrome
Exomphalos, macroglossia, nevus flammeus over the forehead, prominent eyes, typical creases or clefts in ear lobes, visceromegaly, hypoglycemia, and hemihypertrophy.
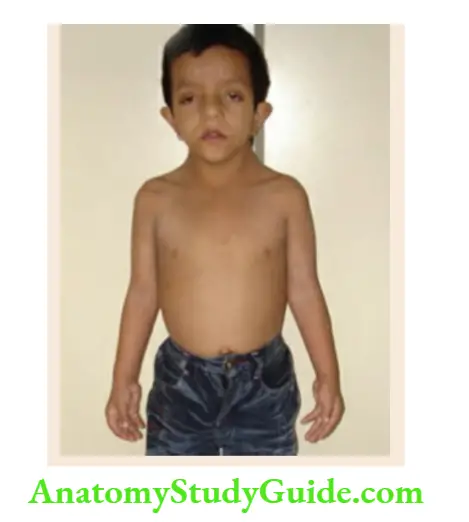
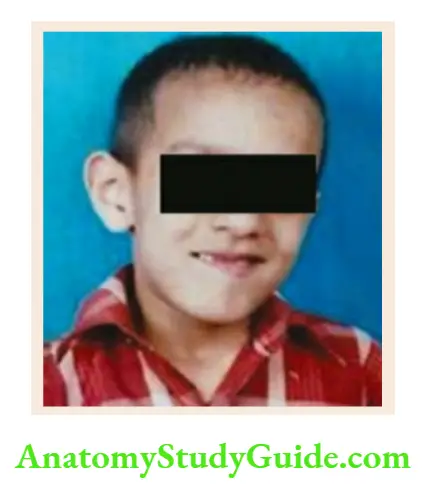
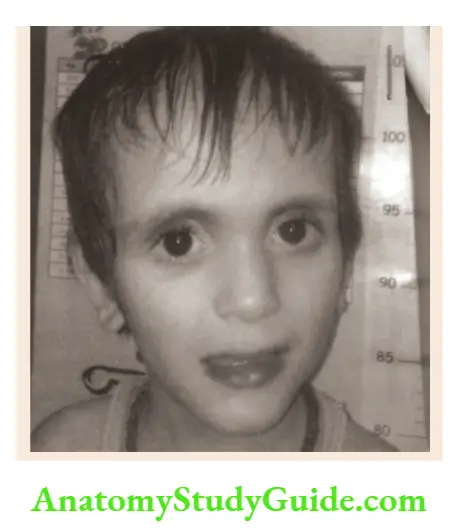
- Berardinelli lipodystrophy syndrome
Muscular body habitus without subcutaneous fat, large hands and feet, acanthosis nigricans, splenomegaly, hepatomegaly, and large phallus. - Marshall-Smith syndrome
Large-for-dates, prominent forehead, upturned nose, blue sclerae, micrognathia, advanced physical and skeletal growth, and mental retardation. - Weaver-Smith syndrome
Large baby at birth, macrocephaly, wide forehead, hypertelorism, the antimongoloid slant of eyes, elongated philtrum, large ears, small chin, camptodactyly, broad thumbs, hypertonia with contractures.
Syndromes with Unusual CNS or Neuromuscular Findings and Associated Defects
- Arthrogryposis multiplex congenital
Multiple contractures of major joints, bilateral club feet, the policeman-tip position of arms and hands, and decreased muscle mass. - Meckel-Gruber syndrome
Occipital encephalocele, microcephaly, cleft lip/palate, abnormal genitalia, postaxial polydactyly, enlarged palpable kidneys due to multicystic dysplasia. - Sjögren-Larsson syndrome
Ichthyosis, spastic diplegia, retinal degeneration, and mental retardation. - Ataxia-telangiectasia
Telangiectasia of the bulbar conjunctiva, auricles, and nasal bridge, progressive cerebellar ataxia, recurrent respiratory infections, and sinusitis. - Prader-Willi syndrome
Obesity, fish-like mouth, hypotonia, micromelia, almond-shaped palpebral fissures, hypogonadism in males, and mental retardation. - Zellweger syndrome
Long flat facies, high forehead, epicanthic folds, large fontanel, micrognathia, abnormal ears, hepatomegaly, hypotonia, hepatic and renal cysts. - Myotonic dystrophy syndrome
Myopathic facies with atrophy of temporalis muscle, ptosis, cataracts (usually on slit-lamp examination), hypotonia during infancy and myotonia during childhood, and testicular atrophy in pubertal boys.

Syndromes with Facial Dysmorphism as a Major Feature
- Moebius sequence
Ptosis, bilateral 6th nerve palsy, bilateral facial nerve palsy with mask-like facies, and micrognathia. - Pierre Robin sequence
Micrognathia and retrognathia, glossoptosis with choking episodes, and U-shaped cleft of the soft palate. - Frontonasal dysplasia sequence
Broad-notched nasal tip or bifid nostrils with nasal tags, ocular hypertelorism or telecanthus, midline deficit of frontal bone with encephalocele. - Waardenburg syndrome
Telecanthus or widely placed eyes, with nasal tags, outer displacement of inferior lacrimal puncta (dystopia cantharus), partial albinism or piebaldism (white forelock of hair), heterochromia of iris, and sensorineural deafness. - Treacher Collins syndrome
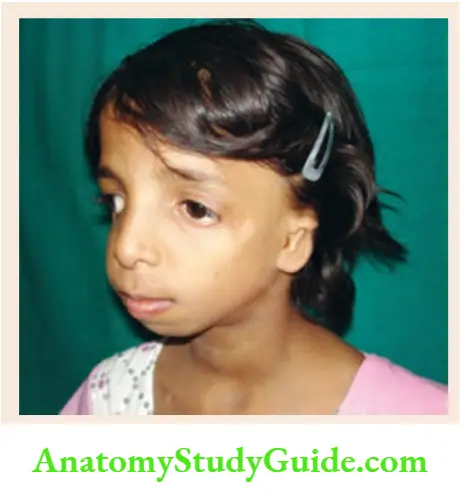
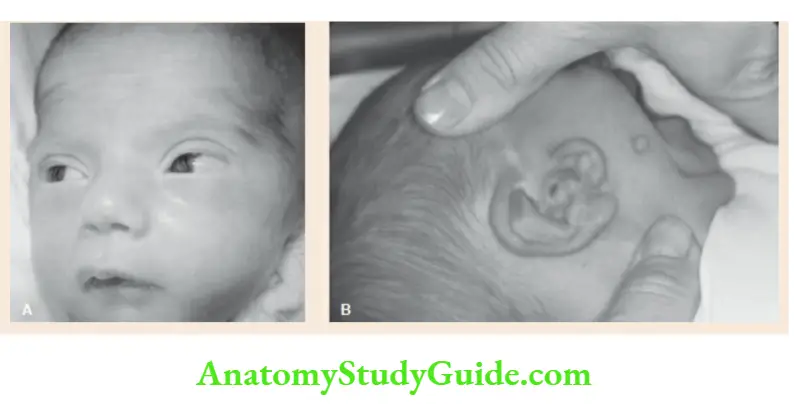
Malar hypoplasia, the antimongoloid slant of palpebral fissures, coloboma of lower eyelids, malformation of external ears, micrognathia, and retrognathia. - Goldenhar syndrome
Facial asymmetry, microtia (small, crumpled, or complete absence of ears), preauricular skin tags, dermoid, a lateral cleft-like extension of the corner of the mouth, and cervical vertebral anomaly.
Syndromes with Facial and Limb Defects
- Langer-Giedion syndrome
Prominent laterally protruding ears, large bulbous pear-shaped nose, sparse scalp hair, multiple bony exostoses, and cone-shaped epiphyses of phalanges. - Whistling face syndrome
“Whistling face”, grooves over the chin, contractures of small joints, and ulnar deviation of fingers. - Coffin-Lowry syndrome
Coarse facies, the antimongoloid slant of palpebral fissures, bulbous nose, thick tapering fingers, drumstick appearance of distal phalanges, and severe mental retardation. - Coffin-Siris syndrome
Coarse facies, sparse scalp hair, hypoplastic or absent fifth finger and toenails, hypotonia, and mental retardation. - Pyknodysostosis
Bossing of the skull, large anterior fontanel, wide sutures, small mandible, parrot-like nose, blue sclerae, short-limbed dwarfism, osteopetrosis, dystrophic nails, widened drumstick appearance of fingers and toes, and tendency to fractures.
Syndromes with Limb Defects as a Major Feature
- Poland anomaly
Unilateral hypoplasia or absence of pectoralis major muscle with ipsilateral brachydactyly of hand. - Escobar syndrome
Ptosis, the antimongoloid slant of palpebral fissures, micrognathia, multiple pterygia of the neck, axillae, antecubital, popliteal, and intercrural areas, and rocker-bottom feet. - Holt-Oram syndrome
Atrial septal defect with a finger-like triphalangeal or absent thumb, complete or partial absence of radius, and a number of associated anomalies. - Thrombocytopenia with absent radius syndrome (TAR syndrome)Thrombocytopenia with absence or hypoplasia of megakaryocytes in early infancy, bilateral absence or hypoplasia of radii (thumbs are usually present), and cardiac anomalies.
- Amniotic band disruption sequenceConstriction rings or transverse amputation of digits or limbs, pseudo syndactyly of digits, unusual facial clefts not conforming to the usual anatomical embryonic planes of fusion.
Craniosynostoses Syndromes
- Carpenter syndrome
Brachycephaly, obesity, flat nasal bridge, lateral displacement of inner canthi, preaxial polydactyly of feet, partial syndactyly of hands and feet. - Apert syndrome
Brachycephaly with high forehead (acrocephaly), midfacial hypoplasia, flat facies, the antimongoloid slant of palpebral fissures, proptosis, syndactyly usually with complete fusion of 2nd, 3rd, and 4th fingers (mitten hands), broad distal phalanx, thumb, and hallux.
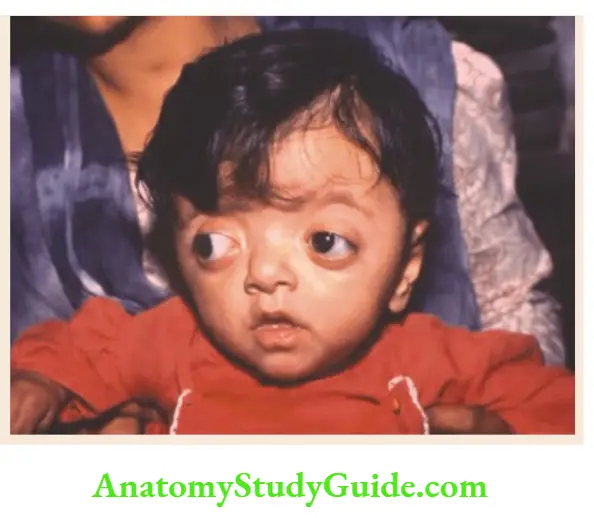
Crouzon syndrome
Oxicephaly, maxillary hypoplasia, shallow orbits with ocular proptosis, strabismus, hypertelorism, and parrot-nose.
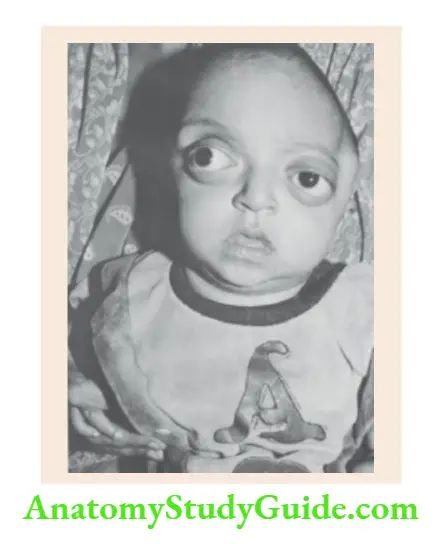
Syndromes with Multiple Hamartomas
- Sturge-Weber syndrome
Portwine hemangioma (nevus flammeus) involving facial area innervated by ophthalmic division of trigeminal nerve, ipsilateral meningeal hemangiomata with the rail-track type of calcification, and ipsilateral choroid plexus hemangioma. - Incontinentia pigmenti
Irregular linear streaks and plaques of vesicles and verrucous patches followed by pigmented skin lesions over the trunk and extremities, alopecia, abnormal dentition, seizures, spasticity, and mental retardation. The pigment is distributed in macular whorls, reticulated patches, flecks, splashes, and linear streaks. - Tuberous sclerosis
Adenoma sebaceous on the face, ash-leaf macules, shagreen patches, seizures, retinal pakoras, pit-shaped defects in tooth enamel, phalangeal cysts, and renal angiomyolipomata. - LEOPARD syndrome
The mnemonic stands for lentigines, electrocardiographic conduction defects, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness. It has features of Noonan syndrome with multiple lentigines. - Neurofibromatosis type 1
Multiple cafe-au-lait spots (>5 with a size of at least 5 mm), multiple neurofibromata, and Lisch nodules (pigmented hamartomata of the iris). - Klippel-Trenaunay-Weber syndrome
Asymmetric hypertrophy of limb with phlebectomy and hemangiomata, port wine stains, lymphangial-mates anomalies, syndactyly, polydactyly or macrodactyly, dislocated hips, kyphoscoliosis, and visceral hemangiomata.
Leave a Reply