Immunology
Introduction
The immune system is defined as the body’s defense system that protects against pathogenic microorganisms and noninfectious foreign substances.
The immune system is the third line of defense against infection. Innate and adaptive are the two types of immune response.
Duality Of Immune System
Humoral (Antibody-mediated) Immunity
- It involves the production of antibodies against foreign antigens, by a subset of lymphocytes called B-cells. These B cells are stimulated to form plasma cells and they secrete antibodies.
- Humoral immunity is involved in defense against bacteria, bacterial toxins, and viruses that circulate freely in body fluids before they enter cells. They are also responsible for certain reactions against transplanted tissue.
Read And Learn More: General Medicine Question And Answers
Cell-mediated Immunity
Question 1. Write a short note on cell-mediated immunity.
Answer:
It involves a specialized set of lymphocytes called T-cells that recognize foreign antigens on the surface of cells, organisms, or tissues: Helper T-cells and cytotoxic T-cells.
- Defense against:
- Bacteria and viruses lie inside the host cells and are not accessible to antibodies.
- Fungi, protozoa, helminths, cancer cells, transplanted tissue.
Cell-mediated Immunity Antigens
Most of the antigens are proteins or large polysaccharides from foreign microbes or nonmicrobes.
- Microbes: The antigens may be capsules, cell walls, toxins, viral capsids, flagella, etc.
- Nonmicrobes: These antigens include pollen, egg white, serum proteins, and surface molecules of red blood cells and transplanted tissue.
Lipids and nucleic acids become antigens only when combined with proteins or polysaccharides.

Cell-mediated Immunity Haptens
These are small foreign molecules that are not antigenic by themselves.
They become antigenic when coupled to a carrier molecule. Once antibodies are formed they will recognize haptens.
Cell-mediated Immunity Epitope
An epitope represents a small part of an antigen that interacts with an antibody. Each antigen may have one to several epitopes. Each epitope is recognized by a different antibody.
Antibodies
Question 2. Briefly discuss immunoglobulins and their functions.
Answer:
- Antibodies (immunoglobulins) are proteins that recognize and bind to a particular antigen with very high specificity.
- These are produced in response to exposure to the antigen.
- One virus or microbe may have several antigenic determinant sites. Each site may bind to different antibodies.
- Each antibody possesses at least two identical sites which bind to an antigen. These sites are called antigen-binding sites.
- The valence of an antibody: It is the number of antigen-binding sites in antibodies and most antibodies are bivalent.
Antibodies Antibody structure
- Monomer: It is an antibody molecule having a flexible Y-shape with four protein chains, i.e., two identical light chains and two identical heavy chains.
- Variable regions: These are two sections at the end of the Y’s arms. They contain antigen-binding sites (Fab).
- They are identical on the same antibody but vary from one antibody to another.
- Constant regions: They represent the stem of the monomer and lower parts of the Y arms.
- Fc regions: They are regions on the stem of monomers and are important because they can bind to complement cells.
- Fab region: For antigen binding.
Antibodies Immunoglobulin classes
Functions of immunoglobulins/antibodies
- Act as opsonins: Immunoglobulins coat the bacterial surfaces and act as opsonins. This facilitates phagocytosis by cells possessing Fc receptors (e.g., neutrophils).
- Antibody-dependent cell-mediated cytotoxicity (ADCC): In this, antibodies bind to microbes via their Fab region.
- Cytotoxic NK cells attach via Fc receptors and kill these organisms by the release of toxic substances called perforins.
- Activation of the complement system: The binding of antibodies to antigens can trigger activation of the classical complement pathway.
- Complement components can function as opsonins (C3b component), aid in phagocytosis, chemotaxis (recruitment of leukocytes by C3a and C5a), and cause the death of microbes (by MAC-membrane attack complex C5–9).
- Neutralization: Some antibodies may directly neutralize the biological activity of their antigen target or toxins released by bacteria.
- This is an important feature of IgA antibodies at mucosal surfaces.
- Processing of antigen: Antibodies present on B lymphocytes help in the internalization of antigens and further processing for presentation to other cells.
- Agglutination: IgM antibodies help in the agglutination of particulate matter including bacteria and viruses.
- Immobilization of microbes: Antibodies against bacterial cilia or flagella may immobilize their movement and ability to escape phagocytosis.
- Protection of mucosal surface: This is observed with IgA type antibodies.
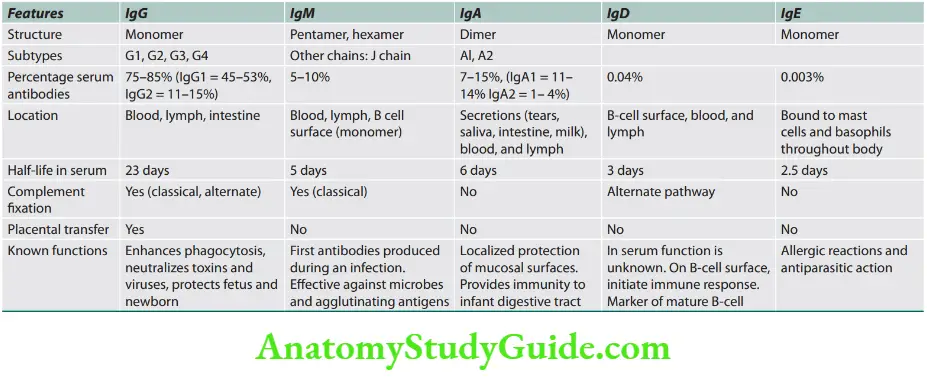
- Immune-complex formation: Antibodies combine with antigens to form immune complexes. The size of these immune complexes varies depending on the ratio between antigens and antibodies. Larger immune complexes can be removed by the phagocytic cells in the reticuloendothelial (RE) system.
- Transplacental passage: Maternal antibodies can pass through the placenta from mother to fetus conferring immunity to the fetus.
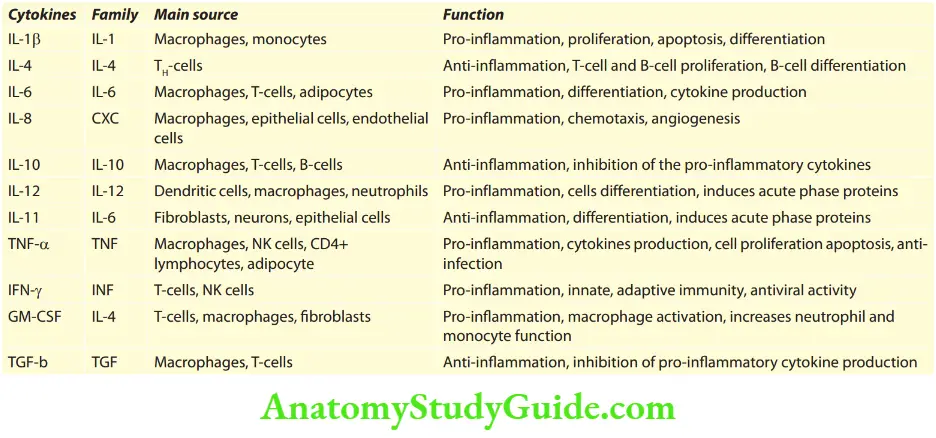
Cytokines
Question 3. Write a short note on cytokines.
Answer:
The immune response involves multiple interactions between many cells.
These cells are lymphocytes, dendritic cells, macrophages, other inflammatory cells (e.g., neutrophils), and endothelial cells. Few of these interactions are due to cell-to-cell contact.
However, many interactions and effector functions of leukocytes are mediated by short-acting soluble proteins:
multipurpose chemical messengers called cytokines. Cytokines which mediate communications between leukocytes are called interleukins.
Mode of action: Cytokines exert their effect by binding to specific receptors on target cells. They may act in three ways.
Autocrine is when the cytokines act on the same cells which secrete them.
Paracrine is when cytokines are produced by one cell type which acts on adjacent/nearby target cells.
Endocrine when the cytokines secreted into the circulation act on target cells at a site distant from their site of synthesis.
Cytokines Interleukins
They are produced by and signal between white cells which mediate endocrine communication at a site distant from their site of synthesis.
Source: Activated macrophages.
Cytokines Effects: Fever, bone marrow release of neutrophils into circulation (leukocytosis), B-cell proliferation, antibody production, and production of IL-2 by T-cells. Interleukin-2 (IL-2) stimulates the proliferation of activated B-cells, T-cells, and NK cells.
Tumor necrosis factor-α
- Source: TNF-α is produced by activated macrophages in response to infection by bacteria and other microbes.
- Effects: Fever, cachexia (pathologic state characterized by weight loss and anorexia seen in neoplastic diseases and some chronic infections), anorexia, shock, enhanced leukocyte cytotoxicity, enhanced NK-cell function, acute phase protein synthesis, proinflammatory cytokine induction, increases apoptosis, and expression of cytokines and adhesion molecules.
- Important cytokines and their functions are given below:
Interferons
Question 4. Write a short note on interferons.
Answer:
- They are critical cytokines that play a key role in defense against viral infections and other intracellular microbes (e.g., Toxoplasma gondii).
- This group includes interferon-α (IFN-α), interferon-b (IFN-b), and interferon-g (IFN-g).
Interferons Source:
- These are produced by a wide range of cells when attacked by viruses and other non-self-pathogenic antigens.
- IFN-γ is released by T cells and NK cells?
Erythrocyte Sedimentation Rate
Question 5. Write a short note on the erythrocyte sedimentation rate (ESR).
Answer:
It is the rate at which red blood cells (RBCs) settle down when anticoagulated whole blood is allowed to stand.
RBCs have a net negative charge on their surface and do not aggregate but tend to repel each other.
Plasma proteins have a positive charge and neutralize the surface negative charge called the Zeta potential of red cells.
Factors Affecting ESR
Plasma factors: An increase in certain plasma proteins (especially fibrinogen), reduces the repulsive forces between the red blood cells and causes them to stack together like tires or rouleaux.
Rouleaux has more mass/surface area ratio than single red cells, and therefore sediment faster and increases the ESR.
- An accelerated ESR is favored by elevated levels of fibrinogen, globulins, and cholesterol (which increase the positive charge of plasma) whereas albumin and lecithin retard ESR.
- Elevated fibrinogen and raised ESR are also observed in pregnancy, old age, and end-stage renal disease.
- In acute inflammation, ESR and acute phase reactants are raised.
- ESR is increased in conditions associated with monoclonal (multiple myeloma) or polyclonal (chronic infections like tuberculosis) increase in immunoglobulins.
In systemic lupus erythematosus (SLE), ESR is raised but CRP may be normal.
Red cell factors: The sedimentation rate is directly proportional to the weight of the cell aggregates and inversely proportional to the surface area.
ESR may be low if plasma proteins are low or red cell morphology is abnormal making rouleaux formation impossible.
- Anemia increases the ESR and polycythemia decreases it.
- Microcytes sedimentation is slower than macrocytes.
- Red cells with an abnormal or irregular shape, such as sickle cells or spherocytes, do not exhibit rouleaux formation and have low ESR.
Normal range: For men 1–10 mm 1st hour and for women 5–20 mm 1st hour.
Acute Phase Reactants/Proteins
Question 6. Write a short note on acute phase reactants.
Answer:
Acute phase proteins are produced by the liver in response to inflammatory stimuli.
- ESR (discussed above)
- C-reactive protein: C-reactive protein (CRP) plays an important role in host defense and stimulates repair and regeneration.
- CRP is an acute phase reactant produced in the liver.
- It opsonizes invading pathogens.
- CRP increases (maybe up to 1,000-fold) within 6 hours of acute inflammation. Levels fall within a few days after the inflammation subsides. Sequential measurement of CRP is useful in monitoring disease activity.
- In some inflammatory diseases, there may be normal or slight elevations of CRP concentration despite unequivocal evidence of active inflammation. These include SLE, systemic sclerosis, ulcerative colitis, and leukemia.
- However, existing infection in these conditions is associated with significantly raised levels of CRP.
- It is to be noted that intercurrent infection does not cause a significant CRP response in these conditions.
- Procalcitonin is a 14.5 kDa peptide, a sensitive marker of bacterial infections. It can guide the progression of infections, especially for pneumonia and sepsis. Levels of procalcitonin can be used to guide antibiotic therapy.
- Ferritin: During malignancy and infection, ferritin concentrations are elevated.
- Organisms like pseudomonas cause ferritin levels to drop because they have virulence factor siderophores (pyoverdine and pyocyanin) that chelate and import iron.
- Haptoglobin: It is a scavenger protein that has antioxidant,
antimicrobial, and anti-inflammatory properties. The levels of haptoglobin decrease during intravascular hemolysis and its levels increase during inflammation.
Serum amyloid A plays a role in host defense and stimulates repair and regeneration.
In chronic inflammation, it may contribute to the development of amyloidosis.

- Fibrinogen plays an essential role in wound healing.
- α 1-antitrypsin andα1-antichymotrypsin: They control inflammation by neutralizing the enzymes produced by activated neutrophils and preventing tissue destruction.
- Hepcidin increases during inflammation causing anemia of chronic disease.
- Albumin: Albumin is a negative APP, and its production is decreased to conserve amino acids for positive APPs.
- Transferrin: Transferrin is a negative APP. Macrophages internalize transferrin to sequester iron and inhibit microbial iron scavenging.
- Others: Ceruloplasmin, Alpha2-macroglobulin, Alpha1-antichymotrypsins, and manganese superoxide dismutase.
Complement System
Question 7. Write a short note on the complement system.
Answer:
- The complement system is a group of plasma proteins (b globulins), the major source being the liver. C1q is produced mainly by bone marrow-derived cells like macrophages and dendritic cells.
- They are proteases and are important in both inflammation and immunity.
Pathways of Complement System Activation
The decisive step in complement activation is the proteolysis of the third component, C3. Cleavage of C3 can occur by any one of three pathways:
1. Classical pathway: It is activated by antigen-antibody (Ag-Ab) complexes in which C1 binds to antibodies (IgM or IgG), acute phase proteins, charged molecules, and apoptotic or necrotic cell debris.
2. Alternative pathway: This pathway is antibody-independent.
- It is activated by binding of C3 directly to microbial surface molecules [e.g., endotoxin, or lipopolysaccharides (LPS)], complex polysaccharides, cobra venom, and “altered self” such as tumor cells, in the absence of antibodies.
- They cleave C3 to C3b.
- C3b in the presence of factors B and D generates C3bBb (alternative pathway C3 convertase).
- C3bBb is labile and degraded by factors I and H, but stabilized by properdin and can subsequently activate C3.
- This results in persistent and prolonged C3 activation and hypocomplementemia.
- In addition, decreased synthesis of C3 by the liver is also responsible for hypocomplementemia.
3. Lectin pathway: It directly activates C1 when plasma mannose-binding lectin (MBL) binds to mannose on microbes.

Complement System and Disease

Major Histocompatibility Complex
Question 8. Write a short note on the HLA system/major histocompatibility complex (MHC).
Answer:
All human cells express a series of molecules on their surfaces that are recognized by other individuals as foreign antigens.
It was found that major histocompatibility antigens are the important antigens involved in the rejection of transplanted organs.
These antigens are encoded by a segment of chromosome 6 (6p21.3) known as the major histocompatibility complex.
Significance of MHC/HLA System
Question 9. List diseases associated with HLA-B27.
Answer:
In transplantation: HLA typing is a prerequisite in selecting appropriate donor and recipient combinations for transplantation.
Regulations of cell-to-cell interaction: During immune response (immune response genes).
Defense against viral infections.
HLA and disease association: HLA alleles confer a state of susceptibility, or risk, for disease.
Many diseases are known to be associated with certain HLA alleles. The diseases associated with the HLA locus can be broadly categorized.



Immunodeficiency
Immunodeficiency may be primary or secondary.
Primary Immunodeficiency Diseases
Classification of Primary Immunodeficiency Diseases
Question 10. Classify primary immunodeficiency disorders.
Answer:
Primary immunodeficiency diseases (PIDs) are classified into eight major categories according to the component of the immune system primarily involved
Common components, their frequency, and their susceptibility in primary immunodeficiency are presented

Classification of primary immunodeficiency diseases.
- Combined T-cell and B-cell immunodeficiencies
- Predominantly antibody deficiencies
- Other well-defined immunodeficiency syndromes
- Diseases of immune dysregulation
- Congenital defects of phagocyte number and function
- Defects in innate immunity
- Autoinflammatory disorders
- Complement deficiencies

Warning Signs for Suspicion of Primary Immunodeficiency Disorders

Antibody Deficiencies
Question 11. Write a short note on B-cell (antibody) immunodeficiency disorders.
Answer:
- Constitute 70% of PIDs.
- Recurrent pyogenic infections start after 6–12 months. Main types of antibody deficiencies.
X-linked agammaglobulinemia (XLA) (Bruton agammaglobulinemia)
Question 12. Write short note on XLA.
Answer:
Described as the “prototypical antibody deficiency” and is the first immunodeficiency described.
- Bruton tyrosine kinase (Btk) is necessary for the growth and maturation of B-cell precursors. It is deficient in this disease.
- Xq21.3–22 (>300 mutations).
- B-lymphocytes are decreased, <1% CD19 or CD20-positive lymphocytes.
- Tonsils are small, with no palpable lymph nodes.
- Autosomal recessive transmission due to mutation in the genes of the surface-bound μ heavy chain, λ5, and Igα.
Clinical features: Little Boys With Big Infections!
- Symptoms appear at 6–9 months of age (after loss of maternal Ig).
- Sites of infection: Mucous membranes, ear (otitis media), lungs (bronchitis/pneumonia), blood (sepsis), gut (giardia, or
enterovirus), skin, eyes, and meningitis. - Typical bacterial infections include Haemophilus influenza and Streptococcus pneumoniae.
- Also seen: Neutropenia, malignancy in older patients, and renal and joint involvements.
Prevention of infections in patients with PID involves avoidance measures, vaccination, prophylactic antibiotics, and immune globulin therapy.
Common Variable Immunodeficiency (CVID)
Question 13. Write a short note on the common variable immunodeficiency.
Answer:
- Diagnosis depends on the exclusion of other causes of antibody deficiencies.
- Incidence: 1/10,000–1/50,000 (second or third decade)
- Decrease in IgG, A +/–M. B-Cells are normal or decreased.
Common Variable Immunodeficiency (CVID) Clinical features
- CVID has a tendency to autoantibody formation.
- Chronic pulmonary infections, chronic giardiasis, intestinal malabsorption, atrophic gastritis, and pernicious anemia.
- T-cell immunity may be deficient.
- Increased risk of lymphoreticular and gastrointestinal malignancies.
IgA deficiency
- Incidence: 1/700 Caucasians but symptoms in <33%.
- Presents with recurrent respiratory infections and chronic diarrhea.
- Associated conditions with IgA deficiency.
- Transient hypogammaglobulinemia of infancy
- Selective antibody deficiency
- Associated with other immunodeficiencies (Job syndrome, hyper-IgM)
Associated conditions with IgA deficiency.
- Common variable immunodeficiency (CVID)
- Deficiency of IgG2 (IgG4, E) and poor response to polysaccharide antigens
- Ataxia-telangiectasia
- Risk of asthma and autoimmune diseases (RA, vitiligo, thyroiditis)
- Auto and also anti-IgA antibodies (MG!).
IgG subclass deficiency
- Normal total serum IgG levels with subnormal levels of one or more IgG subclasses.
- Mutations in heavy chain genes on 14q32.3 (g1, 2, 3, 4).
- IgG1: Lead to a decrease in total serum IgG.
- IgG2: Most common IgG subclass deficient in children. Frequently associated with poor response to polysaccharide antigens and IgA deficiency.
- IgG3: Most common IgG subclass deficient in adults.
- IgG4: Levels vary widely in normal people.
Transient hypogammaglobulinemia of infancy
- Decrease of the maternal IgG transferred to the fetus during pregnancy but delayed Ig production (IgM, IgG, and IgA).
- Normal range after 36 months.
- The normal response to vaccination.
Selective antibody deficiencies
- Normal total serum levels of IgG and IgM but failure to respond to certain antigens (polysaccharide antigens).
- Asymptomatic or recurrent sinopulmonary infections.
- Found in sickle cell anemia, asplenia, Wiskott-Aldrich syndrome, and DiGeorge syndrome.
Hyper-IgM syndrome
- Deficit in IgM: IgG switch. Elevated IgM levels and a deficiency in IgG, IgA, and IgE.
- The most frequent deficit concerns CD154 on CD4+ T-cells (Xq26.3–27.1).
- The hyper-IgE syndrome (Job syndrome)
- Recurrent abscesses, eczema, dysmorphia, eosinophilia, and high serum levels of IgE.
- Autosomal dominant and sporadic cases.
- Cell-mediated (T-cell) Immunodeficiency (Box 20.4)
Question 14. Write a short note on T-cell (cell-mediated)
Answer:
- immunodeficiency disorders.
- Combined Immunodeficiencies
- Severe combined immunodeficiency (SCID)
Question 15. Write a short note on severe combined immunodeficiency.
Answer:
- Failure to thrive.
- The onset of infections in the neonatal period.
- Opportunistic infections, chronic or recurrent thrush, chronic rashes, and chronic or recurrent diarrhea.
- Lack of lymphoid tissue including thymus, tonsils, and lymph nodes.
- Characteristic abnormality in adenosine deaminase deficient SCID includes cupping at the end of the ribs demonstrated on a chest radiograph and an absent thymic shadow.
- Common: Pneumocystis jirovecii infection, or persistent oral or diaper candidiasis.
ADA (adenosine deaminase deficiency) SCID: Profound lymphopenia (<500/mm3); skeletal abnormalities, including chondro-osseous dysplasia (flared costochondral junctions and bone-in-bone anomalies in vertebrae); and deficiency of all types of lymphocytes.
Immunodeficiency Omenn syndrome
It is a rare autosomal recessive disease usually presenting in the neonatal period, characterized by symptoms of SCID associated with other findings like erythroderma, lymphadenopathy, hepatosplenomegaly, and eosinophilia.
Immunodeficiency Treatment
- Pneumocystis prophylaxis with trimethoprim-sulfamethoxazole.
- Replacement IgG therapy. Enzyme replacement therapy [e.g., polyethylene glycol-adenosine deaminase (PEG-ADA)].
- Patients should only be transfused with irradiated blood products and should not receive any live vaccines.
Question 16. List the clinical features of DiGeorge syndrome.
Answer:
Features of DiGeorge syndrome (congenital thymic aplasia) are
T-cell (cell-mediated) immunodeficiency disorders.
- DiGeorge syndrome
- Defect in CD3/TCR
- Defect in signaling
- Defects in cytokine production as IL-2 and IFN gamma
- Defect in cytokine response
Combined immunodeficiency disorders.
- Severe combined immunodeficiency (SCID)
- Omenn syndrome
- Adenosine deaminase deficiency (ADA)
- Ataxia-telangiectasia (AT) syndrome
- Wiskott-Aldrich syndrome (WAS)
- EBV-associated immunodeficiency (Duncan’s syndrome)
Wiskott-Aldrich syndrome (WAS)
Question 17. Write a short note on Wiskott-Aldrich syndrome.
Answer:
- X-linked recessive disease.
- Immunodeficiency, micro platelet thrombocytopenia, and eczema.
- Mutations of the gene encoding WASP at X11p.
- Low IgM, normal IgA and IgG, and high IgE.
Ataxia-Telangiectasia
Question 18. Write a short note on ataxia-telangiectasia.
Answer:
- Autosomal recessive.
- Progressive cerebellar ataxia, abnormal eye movements, oculocutaneous telangiectasias, and immune deficiency.
- Also characterized by recurrent sinopulmonary infections, bronchiectasis, and interstitial lung disease.
- Vascular malformations (telangiectasia).
- IgA and IgG2 deficiency. The T-cell function is variably depressed.
- Gene is responsible for chromosome 11q22.3.
- Increased serum alfa fetoprotein is diagnostic.
- Prone for various malignancies (lymphomas, breast cancer, and acute leukemias).
Phagocyte Deficiencies
- Chronic granulomatous disease (CGD)
- Leukocyte adhesion deficiency (LAD I)
- Chediak-Higashi syndrome, an autosomal recessive condition, is characterized by oculocutaneous albinism, easy bruising, abnormal functions of the natural killer cells, and recurrent pyogenic infections and is a result of a mutation in the lysosomal trafficking regulator (LYST) gene.
- IL-12/IFNg pathway deficiencies.
- Chronic or cyclic neutropenia.
Diseases of Immune Dysregulation
Four groups of diseases are included in this category.
- Familial hemophagocytic lymphohistiocytosis (FHL) includes perforin deficiency. HLH secondary to autoimmune
diseases is called macrophage activation syndrome. - UNC13D (Munc13–4) deficiency
- Syntaxin 11 deficiency and STXBP2 (Munc18–2) deficiency
- Autoimmune lymphoproliferative syndrome (ALPS).
Question 19. List the clinical features of IgG4-related diseases.
Answer:
Immunoglobulin G4-related Disease (IgG4-RD)
IgG4-RD is an immune-mediated fibroinflammatory condition affecting multiple organs.
The hallmarks of IgG4-RD are dense lymphoplasmacytic infiltrations with a predominance of IgG4-positive plasma cells in the affected tissue, as accompanied by some degree of fibrosis and obliterative phlebitis and an increased number of eosinophils.
IgG4-related diseases Clinical Manifestations
Four main clinical phenotypes have been identified.
- Group 1: Pancreatic-hepatobiliary disease
- Group 2: Retroperitoneal fibrosis/aortitis
- Group 3: Head and neck limited disease
- Group 4: Classic Mikulicsyndrome with systemic involvement.
Common manifestations of IgG4-RD include the following, type 1 autoimmune pancreatitis and IgG4-related sclerosing cholangitis, Mikulicdisease, Riedel’s thyroiditis, interstitial pneumonitis, and tubulointerstitial nephritis.
Tissue biopsy is the gold standard for the diagnosis which reveals tumor-like swelling of involved organs, a lymphoplasmacytic infiltrate enriched in IgG4-positive plasma cells, variable degree of fibrosis that has a characteristic “storiform” pattern.
In addition, elevated serum concentrations of IgG4 are found in 60–70% of patients.
IgG4-related diseases Treatment
- Glucocorticoids-prednisone
- Rituximab, azathioprine, and methotrexate.
DiGeorge syndrome (congenital thymic aplasia).
Defective development in the thymus and parathyroid develops from the third and fourth pharyngeal pouch.
- Thymic hypoplasia leads to variable immunodeficiency.
- Characteristic facies. Deletion in 22q11 in >80%.
- Acronym—CATCH 22
- Cardiac abnormality (commonly interrupted aortic arch, truncus arteriosus, and tetralogy of Fallot)
- Abnormal facies
- Thymic aplasia
- Cleft palate
- Hypocalcemia/hypoparathyroidism
Question 20. Discuss the clinical presentation and management of cytokine release syndrome.
Answer:
Cytokine Release Syndrome
Cytokine release syndrome (CRS) is an acute systemic inflammatory syndrome characterized by fever and multiple organ dysfunction that can be triggered by a variety of factors such as infections and certain drugs.
It is associated with chimeric antigen receptor (CAR)-T cell therapy, therapeutic antibodies, and haploidentical allogeneic transplantation.
CRS was reported in the setting of haploidentical donor stem cell transplantation, graft-versus-host disease.
Cytokine storm due to massive T cell stimulation is also a proposed pathomechanism of severe viral infections such as influenza, SARS 2-COVID-19.
Cytokine Release Syndrome Clinical Manifestations
- Include fever, which may be accompanied by fatigue, headache, diarrhea, and arthralgia.
- Mild CRS can progress to a more severe syndrome which may include hypotension, hypoxia, and uncontrolled systemic inflammatory response with circulatory collapse, vascular leakage, cardiac dysfunction, and multiorgan system failure.
Cytokine Release Syndrome Treatment
- Mild CRS of any cause, symptomatic treatment with antihistamines, antipyretics, and fluids
- For severe CRS caused by CART-T cell therapy, initial treatment with tocilizumab plus a corticosteroid rather than a single agent alone appears beneficial.
Tocilizumab should be given intravenously over 1 hour as follows, patients <30 kg 12 mg/kg, patients >30 kg 8 mg/kg; total tocilizumab dose should not exceed 800 mg.
Steroid therapy includes hydrocortisone 100 mg every 8 hours, dexamethasone 10 mg four times daily, or methylprednisolone 1 mg/kg/day until there is improvement in CRS.
Question 21. List the diagnostic criteria of hemophagocytic lymphohistiocytosis.
Answer:
Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and life-threatening syndrome of excessive immune activation.
It is most common in infants and young children but can affect patients of any age, with or without a predisposing familial condition.
The term macrophage activation syndrome (MAS) refers to a form of HLH that occurs primarily in patients with juvenile idiopathic arthritis or other rheumatologic diseases.
Genetic defect: Mutations at familial hemophagocytic lymphohistiocytosis (FHL) loci include PRF1/Perforin, UNC13D/ Munc13-4, STX11/Syntaxin11.
Hemophagocytic Lymphohistiocytosis Clinical Features
Diagnostic Criteria of HLH-2004
The diagnosis of HLH can be established if one of either 1 or 2 below is fulfilled:
- A molecular diagnosis consistent with HLH is made.
- Diagnostic criteria for HLH are fulfilled (5 of the 8 criteria below):
- Fever
- Splenomegaly
- Cytopenias (affecting >2–3 lineages in the peripheral blood):
- Hemoglobin <9 g/dL(in infants <4 weeks of age, hemoglobin <10 g/dL)
- Platelets <100 × 109/L
- Neutrophils <1.0 × 109/L
- Hypertriglyceridemia (fasting triglycerides >265 mg/dL) and /or hypofibrinogenemia (fibrinogen <150 mg/ dL)
- Hemophagocytosis in bone marrow, spleen, or lymph nodes
- Low or absent NK-cell activity
- Ferritin >500 ng/mL
- Elevated soluble CD25 (i.e., sIL2R) two standard deviations above age-adjusted laboratory-specific norms.
Hemophagocytic Lymphohistiocytosis Treatment
- Among patients who are acutely ill or deteriorating, HLH-94-based therapy includes etoposide and dexamethasone given as tapering doses over 8 weeks, with intrathecal methotrexate and hydrocortisone for those with central nervous system involvement.
- Patients should receive supportive care like transfusions of red blood cells, platelets, and fresh frozen plasma.
- Hematopoietic cell transplantation (HCT) following induction therapy is recommended.
- Refractory or recurrent HLH—emapalumab (interferon gamma blocking antibody) and alemtuzumab (anti-CD52 antibody) can be tried.
Hypersensitivity Reactions
Question 22. List the types of hypersensitivity reactions.
Answer:
Immunologically mediated reactions (hypersensitivity reactions) that may produce in tissue damage are summarized.
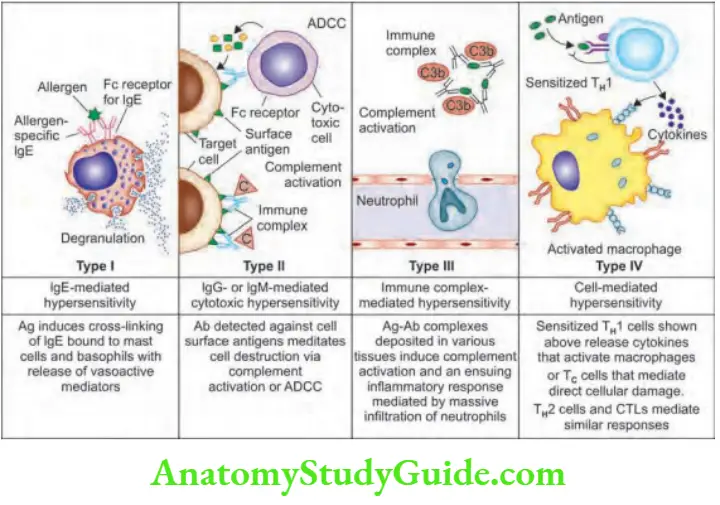
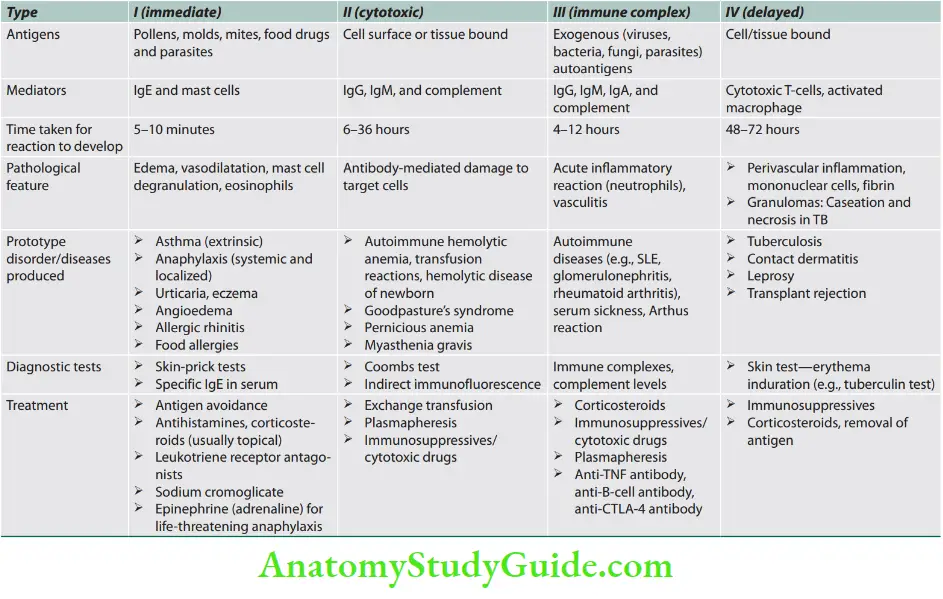
Urticaria (“Hives”)
Question 23. Write a short essay/note on urticaria or hives or nettle rash.
Answer:
Urticaria (also known as hives) is produced due to localized edema of the dermis secondary to a temporary increase in capillary permeability.
The term angioedema is used if edema involves subcutaneous or submucosal layers.
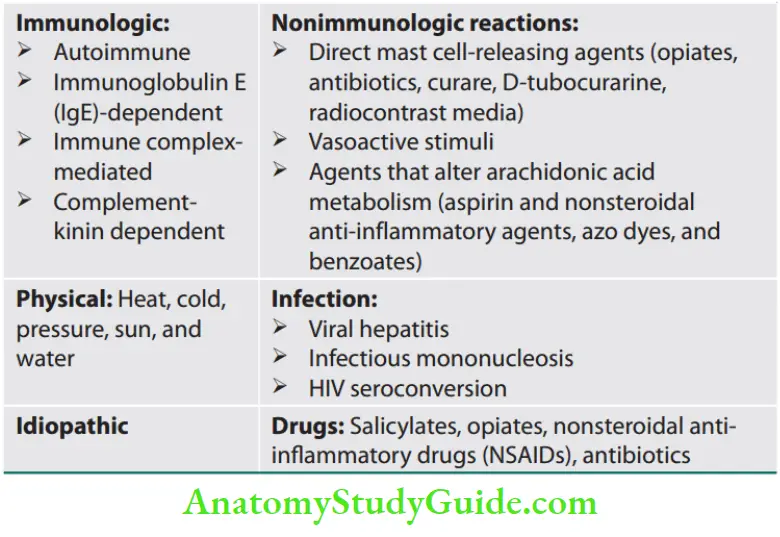
Classification (causes) of urticaria and/or angioedema is presented
Etiology and Pathogenesis
Types: Acute urticaria is the presence of urticaria for less than 6 weeks and chronic if it persists for more than 6 weeks.
- Urticaria may be brought out by either immunologic or nonimmunologic mechanisms. Urticaria is triggered by a wide variety of antigens or by physical stimuli, including cold, pressure, and sunlight. They produce local degranulation of mast cells by various mechanisms such as
- Type I hypersensitivity,
- Spontaneous mast cell degranulation (chronic urticarial),
- Chemical mast cell degranulation,
- Autoimmunity (chronic urticaria)
Etiology and Pathogenesis Clinical Manifestation
- Urticaria is common and can be seen in persons of all ages.
- The common triggers are cold, heat, sweating, exercise, pressure, vibration, and sunlight. Dermographism, literally “skin
writing” is the most common form of physical urticaria. - Urticarial eruptions may involve any area of the body from the scalp to the soles of the feet.
- Urticarial lesions represent a local area of edema involving only the superficial portions of the dermis.
- On certain body sites (e.g., the lips, and hands), the edema spreads deeper into subcutaneous tissue and is referred to as angioedema.
- They are well-circumscribed wheals, pink to light red in color, with erythematous raised serpiginous borders and a
blanched center. - The size of the lesions varies from one millimeter to several centimeters.
- Almost always pruritic and individual wheals come and go within 24 hours and those lesions lasting up to 6 weeks are called acute urticaria.
- If the urticaria recurs over a period of 6 weeks or more it is known as chronic urticaria.
- When individual lesions last more than 36–48 hours and leave postinflammatory hyperpigmentation or palpable purpura, it is called urticarial vasculitis.
- Urticaria may be associated with headache, dizziness, nausea, vomiting, abdominal pain, diarrhea, and arthralgias.
Etiology and Pathogenesis Management
- Management of urticaria depends on its severity and duration.
- Mild urticaria limited to the skin: Antihistamines (diphenhydramine) or the newer nonsedating agents (terfenadine, cetirizine, and
loratadine). - Severe urticaria: Short-term corticosteroids (up to 1 mg/kg).
- Chronic urticaria: Finding the cause and removing the causative antigen. Antihistamines, omalizumab, and cyclosporine for 8–16 weeks may be beneficial.

Allergy
Question 24. Define allergy. List atopic disorders.
Answer:
- Allergy is defined as a hypersensitivity reaction induced by exposure to an otherwise harmless exogenous substance (known as an allergen), generally environmental.
- In an allergic reaction, initial exposure to an allergen triggers the production of specific IgE antibodies by activated B-cells.
- These IgE antibodies bind to the surface of mast cells via high-affinity IgE receptors.
- The first dose of allergen (priming dose) sensitizes the immunologic system (B lymphocyte).
- On re-exposure, the allergen (shocking dose) binds to membrane-bound IgE which activates the mast cells, releasing vasoactive mediators (the early phase response), and causing a type I hypersensitivity reaction and the symptoms of allergy.
- This may be followed by late phase reaction and is mediated by basophils, eosinophils, and macrophages.
Examples: Asthma, anaphylaxis, rhinitis, urticaria, angioedema, eczema, and food hypersensitivity.
Angioedema
Question 25. Write a short essay/note on the clinical features and treatment of angioedema.
Answer:
Angioedema is defined as a well-demarcated localized edema involving the deeper layers of the skin, including the subcutaneous tissue and submucosal tissues.
Angioedema Etiology
It is an IgE-mediated reaction that causes the direct release of histamine from the mast cells.
It follows a variety of allergens. It may develop due to insect stings, drug reactions, food allergies, and exposure to other biological products.
Rarely, angiotensin-converting enzyme (ACE) inhibitors may produce angioedema due to increased levels of bradykinin. Most of the cases are idiopathic.
C1-esterase Inhibitor Deficiency
C1 esterase (C1INH) inhibitor is a complement protein that inhibits spontaneous activation of the classical complement pathway.
C1 esterase inhibitor also regulates kinin cascade, activation of which increases local bradykinin levels and produces local pain and swelling.
Both C1-esterase inhibitor and C1 levels are low. C1INH deficiency produces bradykinin.
A deficiency of C1INH may be a hereditary or acquired disorder.
- Hereditary angioedema: It is an autosomal dominant disorder, is due to C1 esterase inhibitor (C1INH) deficiency. Angioedema develops either spontaneously or following infection or injury (e.g., dental injury). Onset is usually in early childhood. The attacks become worse at puberty and usually, their frequency and severity decrease after the age of 50 years and may even disappear totally. Diagnosis is confirmed by low levels of C1-esterase inhibitor (in 85% of cases) or dysfunctional C1-esterase inhibitor (in 15% of cases).
- Mutations in chromosome 11; Autosomal dominant inheritance.
- Type I C1ID: 85% of patients have no detectable protein.
- Type II C1ID: 15% of patients have dysfunctional protein
- Acquired C1INH deficiency: It presents in a manner similar to the hereditary angioedema but the onset occurs in the fifth and sixth decades of life. It is due to the appearance of an autoantibody. It may occur with B-cell lymphoma, multiple myeloma, Waldenstrom’s macroglobulinemia, and chronic lymphocytic leukemia.
Angioedema Clinical Features
- It may occur at any age but is most common in young adults.
- It presents with well-defined, nonpitting swelling, usually nonpruritic.
- It may be associated with urticaria lesions.
- Angioedema up to 6 weeks is called acute and if it lasts beyond 6 weeks is called chronic.
- It may involve any area of the body but often affects the periorbital area, lips, and genital areas.
- Angioedema of the upper respiratory tract may cause laryngeal
obstruction which may be life-threatening. - Involvement of the gastrointestinal system may produce abdominal colic, with or without nausea and vomiting.
- Angioedema does not produce residual discoloration unless there is extravasation of RBCs.
Angioedema Treatment
- Identification of the etiologic factor(s) and removal of the offending agent if possible.
- Antihistamines to control the lesions, e.g., diphenhydramine cetirizine, desloratadine.

- Observe for any evidence of airway obstruction and if present manage in a fashion similar to those with anaphylaxis.
- Acute attacks should be controlled with epinephrine.
- Chronic angioedema not responding to maximal dosages of antihistamines, glucocorticosteroids and other immunomodulating agents (e.g., methotrexate, cyclosporine) may be considered.
- During severe attacks of hereditary angioedema due to C1INH deficiency, fresh frozen plasma is lifesaving as it provides a C1-esterase inhibitor.
- Danazol is useful to prevent episodes of hereditary angioedema.
- Newer therapy for hereditary angioedema: Purified C1 inhibitor concentrate, recombinant C1INH—Conestat alfa, bradykinin receptor antagonist—Icatibant, and kallikrein inhibitor—Ecallantide.
Systemic Anaphylaxis
Question 26. Write a short note on anaphylactic reactions (anaphylaxis).
(or)
Discuss the causes and treatment/management of anaphylactic shock.
Answer:
Systemic Anaphylaxis Definition: Systemic anaphylaxis is a life-threatening form of immediate (appears within minutes after systemic exposure to specific antigen) type l hypersensitivity reaction mediated
by IgE.
It is a systemic response that develops when mast cells (possibly basophils) are provoked to secrete mediators with potent vasoactive and smooth muscle contractile activities.
Systemic Anaphylaxis Causes
It occurs in sensitized individuals and requires prior sensitization to inciting antigen, either alone or in combination with a hapten.
It usually occurs when the allergen is administered parenterally and it is less likely after oral ingestion, inhalation, or cutaneous or ocular topical contact.
The causes of systemic anaphylactic reactions
Anaphylaxis may be triggered by extremely small doses of antigen, e.g., the minute dose used in skin testing for various forms of allergies.
Systemic Anaphylaxis Mechanism
Initial exposure to antigen: The sensitizing antigen (allergen) from its site of entry is presented to T-cells which differentiate into TH2 cells.
IL-13 secreted by TH2 cells enhances IgE production by B-cells. IgE gets attached to mast cells and basophils.

Causes of systemic anaphylactic reactions.
Anaphylaxis: IgE-mediated mast cell degranulation
1. Proteins:
- Foreign proteins (e.g., antisera)
- Foods (peanuts, fish and shellfish, egg, milk, and soya products)
- Food additives (aspartame, monosodium glutamate)
2. Drugs:
- Antibiotics (penicillins, cephalosporins tetracyclines, trimethoprim-sulfamethoxazole, vancomycin, and nitrofurantoin)
- Chemotherapeutic agents
- Hormones (e.g., insulin, vasopressin, and parathormone)
- Enzymes (chymotrypsin, penicillinase, and streptokinase)
- Intravenous anesthetic agents (suxamethonium and propofol)
- Latex
3. Biological agents:
- Blood
- Tetanus, rabies, and diphtheria antitoxins
- Antithymocyte globulin
- Vaccines
4. Insect bites and stings:
- Honey bee (bee venom)
- Wasps (wasp venom)
During subsequent exposure to antigen, the antigen (allergen) binds to the IgE antibodies previously bound to the mast cells and produces IgE-induced degranulation of mast cells and basophils.
It results in the liberation of a number of mediators.
Mediators released by mast cells include preformed mediators (stored in secretory granules) and secondary mediators (newly synthesized products).
They are responsible for the pathogenesis of an anaphylactic reaction.
Anaphylactoid Reactions (Pseudo-allergic Reactions/ Non-IgE-mediated)

Question 27. Write a short note on anaphylactoid reactions.
Answer:
- They are indistinguishable from anaphylactic reactions.
- Most non-IgE-dependent foreign agents do not require antigen processing (sensitization) and can elicit a mast cell activation response on first antigenic exposure itself.
However, they may be associated with IgG and IgM antibodies and not IgE.
These antibodies activate the complement system through the classical pathway and produce activated complement components and may cause direct release of preformed mediators from mast cells and basophils.
Short-lived because involves only the degranulation of the mast cells and not cytokine synthesis.
Causes:
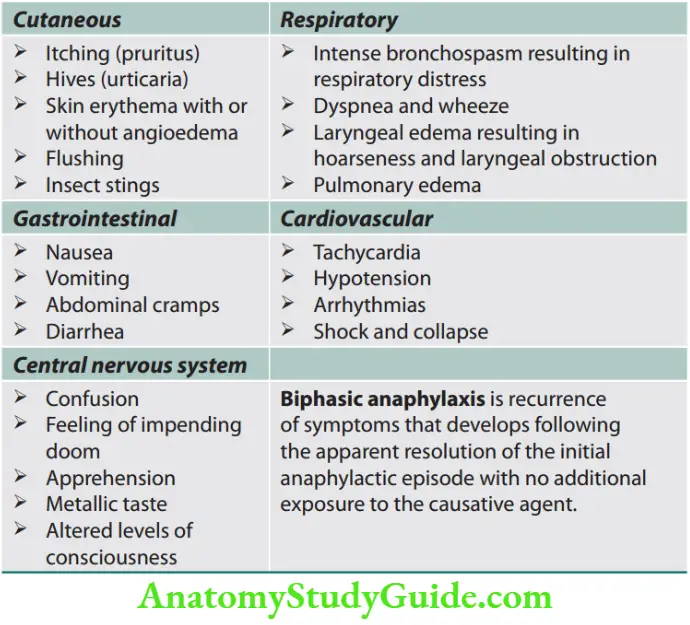
Anaphylactoid Reactions Clinical Features
The anaphylactic response appears within minutes after systemic exposure to a specific antigen.

Diagnostic Criteria for Anaphylaxis
Anaphylaxis is likely when one of the following three criteria occurs:
1. Acute skin and/or mucosal symptoms (e.g., hives, pruritus, flushing, lip/tongue/uvula swelling) and one of the following:
- Respiratory symptoms (e.g., wheezing, stridor, shortness of breath, and hypoxia)
- Hypotension or associated end-organ dysfunction (e.g., hypotonia, syncope, and incontinence)
2. Exposure to probable allergen and two or more of the following:
- Skin/mucosal tissue involvement
- Respiratory symptoms
- Hypotension or end-organ dysfunction
- Persistent gastrointestinal symptoms (e.g., emesis and abdominal pain)
3. Decreased blood pressure after exposure to known allergen for the patient:
- Adults: Systolic blood pressure <90 mm Hg or >30% decrease in systolic blood pressure
- Infants and children: Hypotension for age or >30% decrease in systolic blood pressure
Mast cell mediators in anaphylactic reactions.
Anaphylactoid reactions (Pseudo-allergic reactions/non-IgE-mediated).
Anaphylactoid: Non-IgE-mediated mast cell degranulation
1. Drugs:
- Aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs)
- Vancomycin
- Narcotics (e.g., codeine, morphine, opiates)
- Radiocontrast media/dye
2. Physical:
- Exercise
- Exposure to cold
3. Autoimmune:
4. Narcotics/vancomycin
5. Idiopathic
Question 28. Write a short note on the treatment of anaphylaxis. List lifesaving drugs used in anaphylactic shock.
Answer:
Anaphylactic Shock Treatment
Anaphylaxis is an acute medical emergency and its early recognition is imperative since death occurs within minutes to hours after the first symptoms.
- Prevent further contact with the allergen, e.g., removal of a bee sting.
- Ensure airway patency: Fatal outcomes in anaphylaxis are mainly due to either airway constriction or hypotension.
- Oxygen alone via a nasal catheter or with nebulized salbutamol/albuterol, oxygen 4–6 L/min.
- If progressive hypoxia develops, either endotracheal intubation or tracheostomy with intermittent positive ventilation is required for oxygen delivery.
- Administer adrenaline (epinephrine) intramuscularly into the thigh and is the most critical drug to administer.
Earlier administration during the course of an anaphylactic event is better.
- Adult: 0.3–0.5 mg (0.3–0.5 mL of a 1:1,000 solution) IM in the lateral thigh, repeated at 10- to 15-minute intervals if necessary.
- Child: 1:1,000 dilution at 0.01 mg/kg or 0.1–0.3 mL administered IM in the lateral thigh, repeated at 10- to 15-minute intervals if
necessary. - 0.5 mL of 1:1,000 solution sublingually in cases of major airway compromise or hypotension.
- 3–5 mL of 1:10,000 solution via central line.
- 3–5 mL of 1:10,000 solution diluted with 10 mL of normal saline via endotracheal tube.
- For protracted symptoms that require multiple doses of epinephrine, an IV epinephrine drip may be useful; the infusion is titrated to maintain adequate BP.
- Administer antihistamines: They may prevent the progression of urticaria and pruritus, but do not reverse hypotension or tissue edema by directly opposing effects of mast cell activation, e.g., chlorphenamine 10 mg IM or slow IV injection, diphenhydramine, 50–100
mg IM or IV. - Administer corticosteroids: Hydrocortisone 200 mg IV stat (not effective for the acute event as it takes 4 hours to act; but alleviates recurrence of bronchospasm, urticaria, and hypotension).
- Glucagon could reverse refractory bronchospasm and hypotension in patients who are taking b-adrenergic antagonists.
- The recommended dosage is 1–5 mg intravenously bolus slowly over 5 minutes followed by an infusion at 5–15 µg/min titrated to clinical response.
Provide supportive treatments:
Bronchodilators: They relieve bronchospasm, e.g., nebulized b2-agonists/salbutamol and aminophylline, 0.25–0.5 g IV.
Hypotension: Immediately put the patient in the Trendelenburg position (prevent progression to anaphylactic shock).
Shock is treated with intravenous fluids (if needed with dopamine) to restore or maintain blood pressure. ECMO has been tried.
Non-IgE-mediated (idiopathic) anaphylaxis: Usually presents slowly over a few hours. Begins with pruritus of the palms and soles, then progresses to general pruritus, erythema, and urticaria, with diarrhea, abdominal pain, and hypotension.
Respiratory symptoms are rare.
Allergen Immunotherapy or Desensitization
Subcutaneously incremental doses of allergen are given at 1–2 weekly intervals until the top dose is reached. Sublingual immunotherapy has been practiced too.
Useful for the prevention of atopy/anaphylaxis.
Acute Serum Sickness
Question 29. Write a short note on serum sickness.
Answer:
- It is a systemic immune complex-mediated disease (type III hypersensitivity reaction).
- It was a frequent sequela to the administration of large amounts of foreign serum (e.g., serum from immunized horses used for protection against diphtheria). But nowadays it is infrequent.
- IgG is produced when a foreign antigen is injected in large quantities. They form soluble immune (antigen-antibody) complexes.
Mechanism: Introduction of antigen → body’s immune system responds by synthesizing antibodies after 4–10 days → antibody reacts with antigen, forming soluble complexes that may diffuse into vascular walls and may initiate activation of complement→ complement-containing immune complexes generate an influx of leukocytes into vessel walls—proteolytic enzymes that can mediate tissue damage are released → immune complex deposition + inflammatory responses are responsible for vasculitic lesions seen.
Acute Serum Sickness Clinical Features
Fever, urticaria (at the site of injection), joint pains (arthralgias), lymph node enlargement, and proteinuria appear during this phase.
Wherever immune complexes deposit the tissue damage is similar.
TAcute Serum Sickness treatment
Treatment of acute serum sickness.
Autoimmunity
Question 30. Write a short essay/note on autoimmunity.
Answer:
Autoimmunity Definition: Autoimmunity is defined as an immune reaction in which the body produces autoantibodies and immunologically competent T-lymphocytes against self-antigens.
Mechanisms of Autoimmunity
Genetic Factors
Role of susceptibility genes: Most autoimmune diseases are complex multigenic disorders and genetic factors have an important role.
Runs in families: The incidence is greater in monozygotic than in dizygotic twins.
Association with HLA genes: Most significant.
Environmental Factors
Role of infections
A variety of microbes may trigger autoimmunity by several mechanisms.
Molecular mimicry: Few viruses and microbes may express antigens that have an antigenic structure similar to self-antigens.
Immune responses against them may cross-react with self-tissue and this phenomenon is known as molecular mimicry.
Example: Rheumatic heart disease, in which antibodies formed against streptococcal bacterial proteins, cross-react with myocardial proteins and cause myocarditis.
- Molecular mimicry between microbial proteins and host tissues is also found in Klebsiella and HLA-B27 in ankylosing spondylitis, Coxsackievirus, and glutamic acid decarboxylase
in insulin-dependent (type l) diabetes mellitus, rheumatoid arthritis, and multiple sclerosis.
Breakdown of energy: Tissue necrosis and inflammation produced by microbial infections can cause up-regulation of co-stimulatory molecules on APCs.
This may favor the breakdown of energy and activation of T-cells.
Immune dysregulation: Normally, T-cells regulate any autoreactive T- and B-cells that have survived clonal deletion.
Dysregulation of T-cell function can lead to loss of control and the development of autoimmune disease, e.g., immunodeficiency diseases such as hypogammaglobulinemia and HIV infection are commonly associated with autoimmune phenomena.
Other environmental factors
- Ultraviolet radiation
- Cigarette smoking
- Local tissue injury/damage: Some autoantigens are hidden within the cells or tissues in immunologically privileged sites (e.g., the brain or the anterior chamber of the eye).
- Therefore, the lymphoid cells remain in a state of immunologic ignorance.
- In this state, lymphoid cells are neither activated nor anergized to proteins expressed by these immunologically privileged sites.
- If the tissue in the privileged site is damaged by trauma or inflammation or tumor, the antigens may be released.
- These antigens can evoke an autoimmune response, e.g., Dressler’s syndrome is an acute pericarditis developing in a patient with myocardial infarction secondary to the production of anti-myocardial antibodies, development of insulin-dependent diabetes in association with Coxsackie virus infection, multiple sclerosis, and sympathetic ophthalmia.
- Drugs: T-cell bypass is a mechanism by which T-cells help auto reactivation of B-cells instead of suppressing them, e.g., include drug-induced autoimmune responses such as quinine-induced thrombocytopenia.
- Hormones.
Treatment of acute serum sickness.
- Corticosteroid creams or ointments: Relieve discomfort from itching and rash.
- Antihistamines: May reduce the length of illness and itching.
- NSAIDs: May relieve joint pain.
- Paracetamol: Helpful in relieving fever and muscle pain.
- Medications causing problems should be stopped and future use should be avoided.
Lesion produced in acute serum sickness.
- Vasculitis—inflammatory lesion in blood vessels.
- Glomerulonephritis—inflammatory lesion in renal glomeruli.
- Arthritis—inflammatory lesion in the joints.

Classification of autoimmune disorders is presented.
Therapy of Autoimmune Disease
- Control of symptoms (e.g., nonsteroidal anti-inflammatory drugs for joint pains, fever, etc., in SLE; b-blockers in patients with thyrotoxic features).
- Specific replacement therapy where the organ has been completely destroyed (e.g., thyroid hormone replacement in hypothyroidism).
- Immunosuppressive therapy includes corticosteroids, cytotoxic
drugs in different forms, different regimens, and different routes. - Newer and experimental forms of therapy include immunomodulation, e.g., plasmapheresis, intravenous immunoglobulin therapy, and cyclosporin.
Adult Immunization
Question 31. List common vaccines that are recommended for adults.
Answer:
Screening tools are to be checked before vaccination.
Contraindications for Immunization
- Previous anaphylaxis to a specific vaccine
- History of anaphylaxis to egg protein, neomycin (MMR)
- Live vaccines in pregnancy
- Live vaccines in patients with impaired immunity.
Adverse Effects of Active Immunization
- Mild fever
- Infection (live vaccines)
- Local reaction
- Allergy/anaphylaxis
- SSPE, demyelination, and brachial neuritis.
Documentation
- Provide a copy of the vaccine information statement (VIS) to the patient/documents to be maintained.
- Date of vaccination and next dose
- Vaccine manufacturer
- Lot number
- Dose and site of vaccine
- Vaccinator’s initials
The Advisory Committee on Immunization Practices (ACIP) adult immunization, age-based recommendations in India.

Utilize screening tools; H-A-L-O.
- Health condition
- Age
- Lifestyle
- Occupation
Adult immunization based on medical and other indications in India is presented.
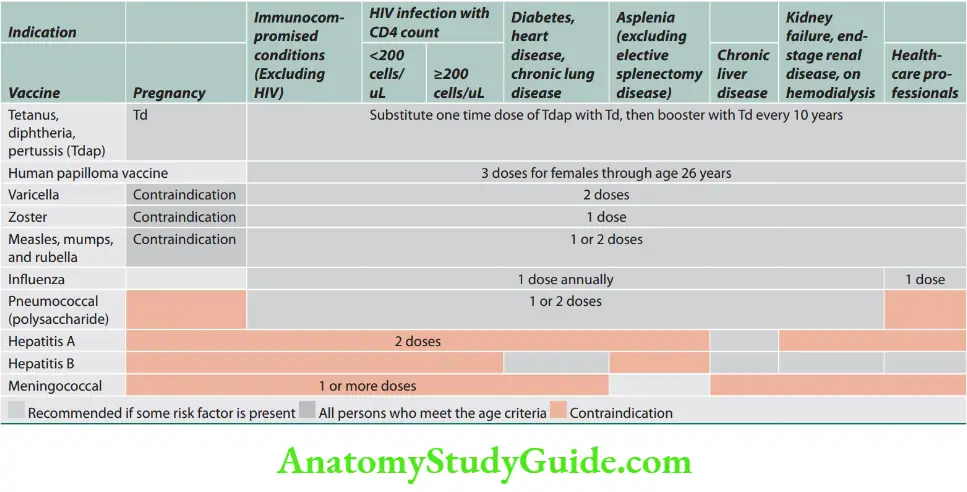
Vaccination details are presented

Amyloidosis
Question 32. Discuss the classification, clinical features, and management of amyloidosis.
Answer:
Amyloidosis Definition: Amyloidosis is a group of acquired and hereditary disorders characterized by the extracellular deposition of insoluble polymeric protein fibrils in tissues and organs.
Classification of Amyloidosis

Amyloidosis Clinical Features
- It is usually a rapidly progressive disease. The clinical features depend on the organs involved.
- Nonspecific symptoms: These include fatigue and weight loss.
- Renal amyloidosis: Kidneys are affected in about 70% of patients.
- It usually presents with proteinuria (often in the nephrotic range), hypoalbuminemia, secondary hypercholesterolemia, and edema or anasarca.
- Cardiac amyloidosis: The heart is the second most commonly (50%) affected organ and may show concentric thickening of ventricles and diastolic dysfunction, leading
to restrictive cardiomyopathy. It may present with heart failure. - Neuropathies: Autonomic dysfunction with gastrointestinal motility disturbances (diarrhea, constipation) and peripheral sensory neuropathies are relatively common.
-
- Carpal tunnel syndrome with weakness and paresthesia of the hands may be an early presenting feature.
- Macroglossia: Enlarged tongue is pathognomonic of AL amyloidosis that may be found in ~10% of patients.
- Amyloidosis of the liver: It causes cholestasis and hepatomegaly.
- Amyloidosis of the spleen: It may produce functional hyposplenism in the absence of significant splenomegaly.
- Cutaneous amyloidosis: It can produce:
- Easy bruising due to amyloid deposits in capillaries or due to a deficiency of clotting factor X, which can bind to amyloid fibrils.
- Cutaneous ecchymosis especially around the eyes and can produce a “raccoon-eye” sign.
- Other findings: These include nail dystrophy, alopecia, and amyloid arthropathy with thickening of synovial membranes in wrists and shoulders.

Diagnosis
- A key feature for the diagnosis of AL amyloidosis is the identification of the underlying B-lymphoproliferative process and clonal light chain.
- Serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) are NOT useful screening tests. However, more than 90% of cases show serum or urine monoclonal LC or whole immunoglobulin by immunofixation electrophoresis of serum (SIFE) or urine (UIFE).
Heredofamilial Amyloidosis
1. Familial Mediterranean Fever
- Autosomal recessive disorder.
- Characterized by recurrent attacks of fever accompanied by inflammation of serosal surfaces (peritoneum, pleura, and synovial membrane).
2. Familial Amyloidotic Neuropathies
- Familial amyloidotic neuropathies are characterized by deposition of amyloid in peripheral and autonomic nerves and the fibrils are made up of mutant TTRs.
- ATTR (transthyretin-associated) amyloidosis is transmitted as an autosomal dominant disease.
- Clinically, they present with peripheral sensorimotor and autonomic neuropathy and symptoms of autonomic dysfunction, diarrhea, and weight loss. Renal involvement is less common than with AL amyloidosis. Macroglossia does not develop.
Hemodialysis-associated (Dialysis Related) Amyloidosis
Patients with chronic renal failure (ESRD) on long-term hemodialysis have high levels of b2-microglobulin in the serum because it cannot be filtered through the dialysis membrane and gets deposited as amyloid.
Its incidence appears to be decreasing with newer high-flow dialysis techniques.
It produces rheumatologic manifestations and usually presents with carpal tunnel syndrome, persistent joint effusions, spondyloarthropathy, or cystic bone lesions.
Other Amyloids
Cerebral amyloidosis: The brain is a common site of amyloid deposition, though it is not directly affected in any form of acquired systemic amyloidosis.
Alzheimer’s disease: Intracerebral and cerebrovascular amyloid deposits are found in Alzheimer’s disease. Most cases are sporadic.
Transmissible spongiform encephalopathy: In hereditary spongiform encephalopathies, several amyloid plaques are observed.
Treatment
- Aim of treatment: To support the function of affected organs and, in acquired amyloidosis, treatment of the associated primary disorder to prevent further amyloid deposition.
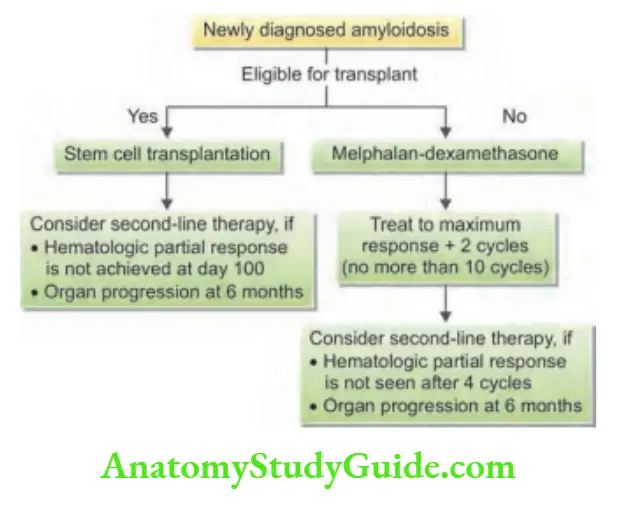
- Treatment of underlying disorder: Treatment of any underlying inflammatory source or infection may produce regression of existing amyloid deposits. Chemotherapy with melphalan plus dexamethasone or stem cell therapy is useful in AL amyloidosis. Guidelines for the treatment of newly diagnosed primary amyloidosis are presented in
- Second-line chemotherapy includes thalidomide/lenalidomide, bortezomib, carfilzomib plus dexamethasone.
- Colchicine may be useful in familial Mediterranean fever.
- Nephrotic syndrome and congestive cardiac failure are treated with the relevant therapies.
- Hereditary transthyretin amyloidosis: In ATTR amyloidosis, where transthyretin is mainly produced in the liver, liver transplantation is the definitive therapy.
Leave a Reply