Endocrinology Disorders Of Pituitary And Hypothalamus
Pituitary Hormones and Their Principal Actions:
Table of Contents
Question 1. Write short essay/note on:
- Pituitary hormones and their principal actions.
- List the hormones of anterior pituitary.
Answer:

Read And Learn More: General Medicine Question And Answers
Hypopituitarism:
Hypopituitarism Defination:
- Hypopituitarism is defined as combined deficiency (partial or complete) of any of the anterior pituitary hormones.
- Panhypopituitarism is defined as deficiency of all anterior pituitary hormones. It may be due to selective or multiple deficiencies of pituitary hormones or diseases of the hypothalamus.
Hypopituitarism Etiology:
Hypopituitarism Various causes of hypopituitarism:
“Nine I’s”: Invasive, Infarction, Infiltrative, Injury, Immunologic, Iatrogenic, Infectious, Idiopathic, and Isolated.
Hypopituitarism Isolated hormone deficiencies
- Invasive tumors: Pituitary adenomas, cysts, metastasis, and hypothalamic tumors
- Injury: Surgery, irradiation, and stalk section
- Infarction: Sheehan’s syndrome (postpartum pituitary necrosis), diabetic antepartum necrosis, and carotid aneurysm
- Cerebrovascular accident (CVA): Ischemic stroke, subarachnoid hemorrhage, and pituitary apoplexy
- Inflammatory diseases: Granulomatous disease and autoimmune (lymphocytic) hypophysitis
- Infiltrative diseases: Hemochromatosis, amyloidosis, sarcoidosis, Langerhans’ cell histiocytosis
- Injury: Head trauma
- Immunologic: Lymphocytic hypophysitis
- Infections: Meningitis, tuberculosis, syphilis, fungal infection such as Candida and Hantavirus. More likely immunocompromised patients such as HIV and high-dose steroids
- Idiopathic
- Developmental defects (Kallmann syndrome) and genetic diseases such with mutations detected in:
- ROBOS guidance receptors (ROBOS) determine axonal guidance in the central nervous system.
- BMP4 induces Rathke’s pouch formation, which is necessary for anterior pituitary development.
- HESX1, LHX3, and LHX4 transcription factors that are important for pituitary organogenesis and early differentiation of pituicytes.
- PROP-1, which is necessary for the differentiation of a cell type that is a precursor to somatotroph, lactotroph, thyrotroph, and
gonadotroph cells. - PIT-1 (called POU1F1 in the human), which acts temporally just after PROP-1 and is necessary for the differentiation of a cell type that is a precursor of somatotroph, lactotroph, and, to a lesser degree, thyrotroph cells.
- TPIT, which is required for specific differentiation of the corticotroph cells.
Hypopituitarism Clinical Features:
- The presentation is highly variable and depends on the underlying cause/lesion.
- Symptoms and signs depend on the degree of hypothalamic and/or pituitary deficiencies. Mild deficiencies may be asymptomatic.
Hypopituitarism secondary to pituitary tumors:
- Symptoms are due to mass effects (e.g., headache, visual impairment, electrolyte alterations, and disorders of the autonomic nervous system produced by hypothalamic involvement). As the lesions progress, there is a sequential loss of hormone secretion.
- Prolactin (PRL) deficiency is rare, except for complete destruction of pituitary or genetic syndromes.
- The order of diminished trophic hormone reserve function by pituitary compression usually follows the order
GH > FSH > LH > TSH > ACTH. - Longstanding panhypopituitarism produces a classic picture of pallor with hairlessness (“alabaster skin”).
Hypopituitarism Laboratory Investigations:
- Demonstration of low levels of trophic pituitary hormones: Biochemical diagnosis of pituitary insufficiency is made by demonstration of low levels of trophic pituitary hormones along with low levels of target hormones.
- Low free thyroxine (T4) with low or inappropriately normal TSH level suggests secondary hypothyroidism.
- Low early morning (8‒9 AM) cortisol, about ≤3 µg/dL, with low or inappropriately normal ACTH.
- Low testosterone level without elevation of gonadotrophins (LH and FSH) suggests hypogonadotropic hypogonadism.
- Low IGF-1 (insulin-like growth factor-1) level indicates GH deficiency

Hypopituitarism Provocation Tests:
These may be necessary to assess pituitary reserve:
- GH reserve: GH response to insulin-induced hypoglycemia, L-dopa, arginine, growth hormone-releasing hormone (GHRH), or growth hormone-releasing peptides (GHRPs). Insulin-induced hypoglycemia (also known as insulin tolerance test) is the gold standard test.
- Thyrotropin-releasing hormone (TRH) reserve: TRH stimulates prolactin secretion. In the presence of optimal levels of TRH, exogenous administration of TRH will not raise levels of prolactin. In the absence of a TRH reserve, exogenous administration of TRH will elevate serum prolactin levels.
- ACTH reserve: It is assessed by measuring ACTH and cortisol levels during insulin-induced hypoglycemia.
- Corticotropin-releasing hormone (CRH) administration induces ACTH release, and administration of synthetic ACTH (cosyntropin) causes adrenal cortisol release. It is an indirect indicator of pituitary ACTH reserve.
- Metyrapone test: Metyrapone blocks 11-beta-hydroxylase, the enzyme that catalyzes the conversion of 11-deoxycortisol to cortisol, resulting in a reduction in cortisol secretion. If the hypothalamo-pituitary-adrenal axis is normal, the ensuing fall in serum cortisol should cause an increase in ACTH secretion and therefore an increase in adrenal steroidogenesis and 11-deoxycortisol.
- In normal subjects, administration of 750 mg of metyrapone orally every 4 hours for 24 hours results in a decline in 8 AM serum cortisol to <7 µg/dL and an elevation in 8 AM serum 11-deoxycortisol to ≥10 µg/dL at the end of the 24 hours. In patients who have decreased ACTH reserve due to hypothalamic or pituitary disease, the serum 11-deoxycortisol concentration will be <10 µg/dL and the serum cortisol < 7 µg/dL at the end of 24 hours.
- TSH reserve: TSH response to TRH.
Surgical biopsy of tumor: Usually done as part of a therapeutic operation of tumors. Histological examination of pituitary tumors will help inidentifying the type of tumor. Immunohistochemistry useful in confirming their secretory capacity.
- Chromophobe (usually nonfunctioning)
- Acidophil (typically prolactinor growthhormone secreting)
- Basophil (typically ACTH-secreting).
- Management: It is by replacement of deficient hormones.
Sheehan’s Syndrome
Question 2. Write a short note on Sheehan’s syndrome.
Answer:
Sheehan’s syndrome is a potentially life-threatening complication due to infarction of pituitary gland following postpartum hemorrhage.
Sheehan’s Syndrome Mechanism:
During pregnancy, the pituitary gland is enlarged and is more vulnerable to ischemia. Postpartum hemorrhage and consequent systemic hypotension can cause pituitary infarction.
Sheehan’s Syndrome Clinical Features:
- Earliest symptom is failure to lactate.
- Failure to regain menstruation after delivery
- Other symptoms of hypopituitarism: They appear over months or years. Few patients may present acutely (hypotension, hyponatremia, and hypothyroidism).
- Coma and death can occur in severe cases.
Sheehan’s Syndrome Diagnosis:
- Refer laboratory findings of hypopituitarism.
- MRI: In early stages, it may show hypertrophied pituitary. Later stages, atrophic pituitary and empty sella.
Sheehan’s Syndrome Replacement therapy for adult hypopituitarism:
Sheehan’s Syndrome Adrenocorticotropic hormone:
Hydrocortisone 15–25 mg daily in divided doses. Mineralocorticoid replacement not needed.
Sheehan’s Syndrome Follicle-stimulating hormone/Luteinizing hormone:
Sheehan’s Syndrome Female Patients (any one):
- Conjugated estrogen 0.65 mg/day
- Micronized estradiol 1 mg/day
- Ethinyl estradiol 0.02–0.05 mg/day
- Estradiol skin patch 4–8 mg twice weekly
- Estradiol plus testosterone
Sheehan’s Syndrome Male Patients (any one):
- Testosterone enanthate 200 mg IM every 2–3 weeks
- Testosterone skin patch 2.5–5.0 mg/day; can increase dose up to 7.5 mg/day
- Testosterone gel 3–6 g daily
Sheehan’s Syndrome Growth hormone:
Adults: Recombinant human GH (rhGH) starting dose 2–5 µg/kg subcutaneously daily. This weight-based recommendation is for the starting dose only. The goal should be to start with low doses and increase gradually until the serum IGF-1 concentration is normal.
Children: Recombinant human GH (rhGH) is 0.16–0.24 mg/kg/week, divided into once daily injections. Further adjustments of GH dose based on growth response, serum insulin-like growth factor-1 (IGF-1) levels, and body weight
Sheehan’s Syndrome Thyroid-stimulating hormone:
L (levo)-thyroxine dose of 1.6 µg/kg daily and adjusting dose according to serum free T4 levels in upper half of reference range.
Sheehan’s Syndrome Vasopressin:
- Intranasal desmopressin-rhinal tube 5–20 μg twice daily
- Oral DDAVP (desmopressin) 300–600 μg daily, usually in divided doses
Sheehan’s Syndrome Prolactin:
Recombinant human prolactin (r-hPRL), although not commercially available, has been used experimentally.
Empty Sella Syndrome:
Question 3. Write a short note on empty sella syndrome.
Answer:
- Often an incidental MRI finding.
- In this condition, herniation of arachnoid diverticulum through an incomplete diaphragm sellae results in symmetrically ballooned sella, which gets filled up with cerebrospinal fluid (CSF).
- Usually have normal pituitary function, implying that the surrounding rim of pituitary tissue is fully functional.
- An empty sella can be a primary congenital weakness of the diaphragm or secondary subsequent to infarction of a pituitary adenoma or to surgical or radiation-induced damage to the sellar diaphragm.
- Hypopituitarism may develop insidiously.
- Common in obese, multiparous women with chronic headache.
- Rarely, functional pituitary adenomas may arise within the rim of pituitary tissue, and these are not always visible on MRI.
Kallmann Syndrome:
Question 4. Write a short note on Kallmann syndrome.
Answer:
- It is due to defective hypothalamic gonadotropin-releasing hormone synthesis.
- Olfactory bulb agenesis or hypoplasia is associated with anosmia or hyposmia.
- Conditions associated with this are color blindness, optic atrophy, cleft palate, renal abnormalities, cryptorchidism, and neurologic abnormalities like synkinesis or mirror movements.
- The deficiency prevents progression through puberty due to low LH and FSH levels and sex steroids.
- Males: Delayed puberty and hypogonadism, including micropenis. Replacement by human chorionic gonadotropin (hCG) or testosterone for a long-term is needed.
- Female: It manifests as primary amenorrhea and failure of secondary sexual development. Long-term treatment with estrogen and progestin is warranted.
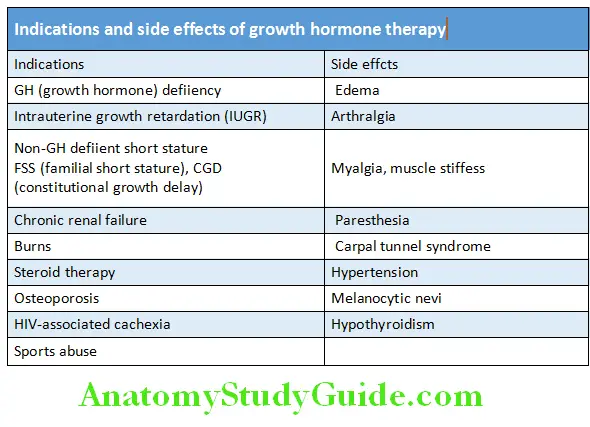
Kallmann Syndrome Growth Hormone Therapy:
Kallmann Syndrome Lists the indications and side effects of growth hormone therapy.
Route of administration of GH: Subcutaneously after 8.00 PM, 3‒7 times a week (0.15‒0.3 mg/kg/week).
Effect is dose-dependent and the response is better if started earlier. Average increment in height = 10 cm/year.
Better response in classic growth hormone deficiency (GHD).
Pituitary Apoplexy:
Question 5. Write a short note on pituitary apoplexy.
Answer:
Pituitary apoplexy is a rare life-threatening endocrine emergency characterized by a sudden onset of headache, visual symptoms, altered mental status, and hormonal dysfunction due to acute hemorrhage or infarction of a pituitary gland.
Pituitary Apoplexy Clinical Features:
- Sudden severe headache, double vision, and sudden severe visual loss.
- Cardiovascular collapse, change in consciousness, neck stiffness, and sometimes hypoglycemia. Sometimes acute lifethreatening hypopituitarism.
- GnRH deficiency is most common. Acute adrenal insufficiency is common due to loss of ACTH. TSH deficiency occurs in half of the patients.
- The condition can evolve over 1‒2 days.
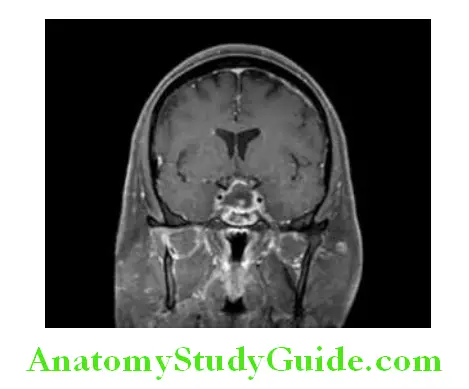
Pituitary Apoplexy CT/MRI findings: Pituitary imaging without contrast (CT or MRI) usually reveals signs of intrapituitary or intra-adenoma hemorrhage, “pituitary ring sign” on T1-weighted MRI, stalk deviation and compression of normal pituitary tissue and, in severe cases, signs of parasellar subarachnoid hemorrhage.
Pituitary Apoplexy Management of Pituitary Apoplexy:
Initial management: Conservative with careful monitoring of fluid and electrolyte balance along with immediate replacement of deficient hormones, in particular corticosteroids. Close monitoring of vision. High-dose corticosteroids and supportive treatment needed.
Surgical decompression: If there is a rapid deterioration in visual acuity and fields, surgical decompression of the optic chiasm may be required.
Pituitary Tumors:
Question 6. Write short essay/note on clinical manifestation, investigations, and management of pituitary tumors.
Answer:
Pituitary tumors are the most common cause of pituitary disease, and most of them are benign pituitary adenomas.
Pituitary Tumors Classifiation:
- Pituitary tumors are classified depending on the tumor size into microadenomas (<1 cm in diameter) and macroadenomas (exceed 1 cm in diameter).
- Pituitary adenomas may be functional (i.e., associated with hormone excess and clinical manifestations thereof ) or nonfunctional (i.e., without clinical symptoms of hormone excess). Nonfunctional tumors detected incidentally during MRI/CT examination or at autopsy are called as pituitary incidentalomas.
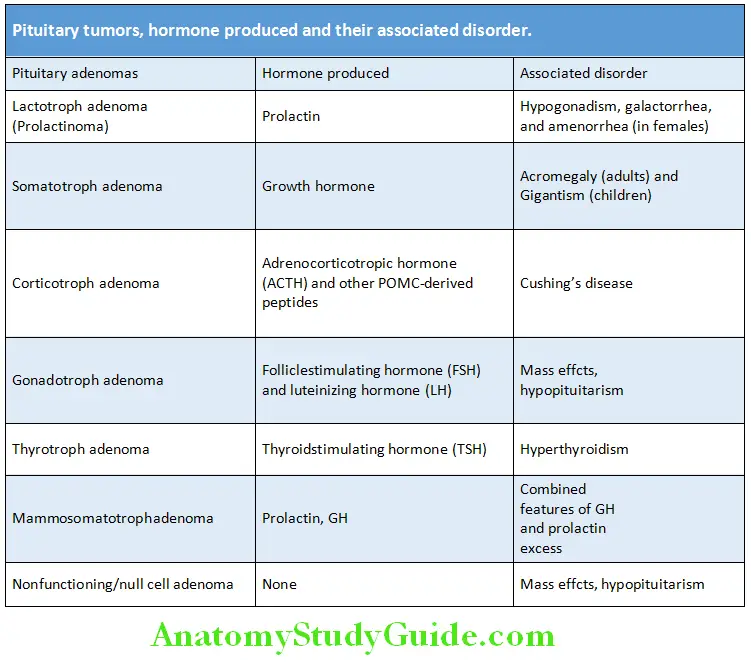
Various tumors of pituitary are listed in Table.
Other tumors of pituitary are listed in Box.
Pituitary Tumors Clinical Features:
The signs and symptoms may be due to mass effects and endocrine abnormalities.
Pituitary Tumors Mass effects: Mass effects of the enlarging tumor can produce specific signs and symptoms of hypofunction by pressure effect on surrounding normal pituitary tissue (see hypopituitarism). The mass effect on the neighboring structures causes:
- Due to stretching of the diaphragm sellae or by invasion of bone It causes headache and is common (especially in patients with macroadenomas) but non-specific.
Pituitary Tumors Due to pressure effects on optic chiasma, nerve or tract: Visual field abnormalities which include loss of acuity and optic atrophy (superior temporal quadrantanopia or temporal hemianopia).
- Due to lateral extension into cavernous sinus with subsequent compression of cranial nerve which produces palsies of cranial nerves III, IV, and VThis results in diplopia and strabismus and facial numbness.
- Due to mass effects on hypothalamus Obesity, disturbances of sleep, thirst, appetite, temperature regulation, and diabetes insipidus (DI).
Pituitary Tumors Others: Anosmia (frontal lobe involvement), vomiting, papilledema (raised intracranial tension).
- Occasionally, pituitary tumors infarct or bleeding into produces “pituitary apoplexy”.
Pituitary Tumors Clinical features due to secretion of hormones.
Pituitary Tumors Investigations:
Plain radiograph of the pituitary fossa (skull) It may show one or more of the following:
- Enlargement of sella turcica
- Calcification of suprasellar region
- Erosion of clinoid process
- Double floor of the sella.
Pituitary Tumors CT scan with contrast enhancement: More sensitive to detect bony erosions and presence of calcification.
Pituitary Tumors Magnetic resonance imaging (MRI) of the pituitary: MRI with gadolinium (imaging method of choice) is superior to CT scanning and shows pituitary mass.
In situations in which the use of gadolinium is contraindicated, such as renal impairment or pregnancy, MRI without gadolinium may still be helpful. Small lesions within the pituitary fossa (small pituitary microadenomas) are very common.
Such small lesions are sometimes detected during MRI scanning of the head for other reasons and are called “pituitary incidentalomas”.
Visual field plotting by automated computer perimetry or Goldmann perimetry.
Pituitary Tumors Functional assessment of the pituitary gland: By hormonal assays and include prolactin (lactotroph adenomas), IGF-1 (somatotroph adenomas), 24-hour urinary-free cortisol (corticotroph adenomas), plasma corticotropin (ACTH), FSH, LH, free T4, and thyroid function tests.
Pituitary Tumors Other tumors of pituitary:
- Craniopharyngioma
- Metastatic tumors
- As a component of multiple endocrine neoplasia type I (MEN-I), which includes parathyroid, pancreatic, and pituitary (usually prolactinoma) tumors
Pituitary Tumors Treatment of Pituitary Tumors:
Pituitary Tumors Surgery:
- Surgery (except for prolactinomas) is the primary mode of treatment for pituitary tumors that warrant intervention.
- Surgery via the trans-sphenoidal adenomectomy or hypophysectomy is the treatment of choice.
- Very large tumors are removed via the open transcranial (usually transfrontal) route. It is usually performed by an endoscopic or
endonasal approach.
Pituitary Tumors Medical therapy:
Somatostatin analogs and/or dopamine agonists: Octreotide is administered postresection. Drug therapy with dopamine agonists such as bromocriptine and cabergoline are effectively used in the management of prolactinomas. They induce a rapid fall in PRL levels and can decrease the size of tumor and possibly avoid surgery.
Pituitary Tumors Radiation therapy:
It is usually used as adjunctive therapy after surgery or when surgery is impracticable or incomplete or in combination with medical therapy. Includes external radiotherapy, or implantation of Yttrium in the pituitary fossa, or gamma knife or a modified linear accelerator.
- It suppresses the tumor growth and reduces its secretory capacity.
- Gamma knife (stereotactic radiosurgery) involves precise delivery of large single high-energy dose directly to the tumor under stereotactic surgery and is particularly useful for residual tumor in the cavernous sinus.
Acromegaly:
Question 7. Write short essay/note on acromegaly and its major clinical signs.
Answer:
Growth hormone (GH) is required for proper growth and development. It directly affects metabolism of fat and indirectly effects bone growth.
Acromegaly results from persistent hypersecretion of growth hormone (GH). Excess GH stimulates hepatic secretion of insulin-like growth factor-1 (IGF-1), which causes most of the clinical manifestations of acromegaly.
Excess secretion of GH prior to closure of epiphyseal growth plates in long bone before onset of puberty causes pituitary gigantism. Excess secretion after puberty causes acromegaly. Males and females are equally affected.
Acromegaly Etiology of Acromegaly:
Acromegaly Excess of growth hormone (GH excess) after puberty may be due to: Pituitary tumor (somatotrope pituitary adenoma) is the most common cause. Acromegaly is caused by growth hormone (GH) secretion usually from a macroadenoma of pituitary gland. Few adenomas secrete both GH as well as prolactin.
Acromegaly Other tumors: In a few patients, acromegaly is caused by tumors of the pancreas, lungs, and adrenal glands (either because they produce GH themselves or, more frequently they produce GHRH, the hormone that stimulates the pituitary to make GH).
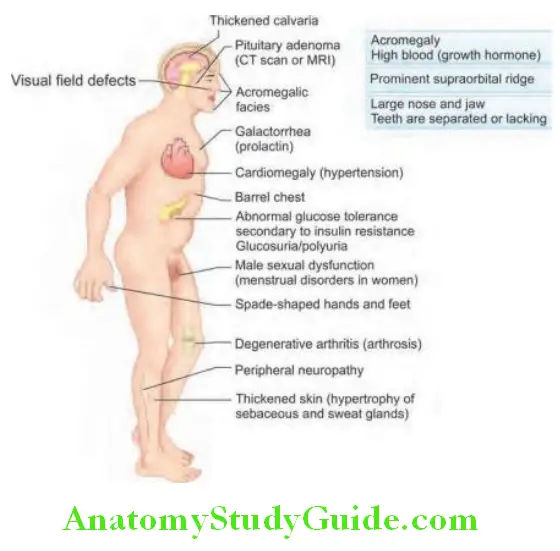
Acromegaly Clinical Features:
Clinical features are due to increased GH (growth hormone) and IGF-1 (insulin-like growth factor-1).
Acromegaly Investigations:
Acromegaly Biochemical investigations:
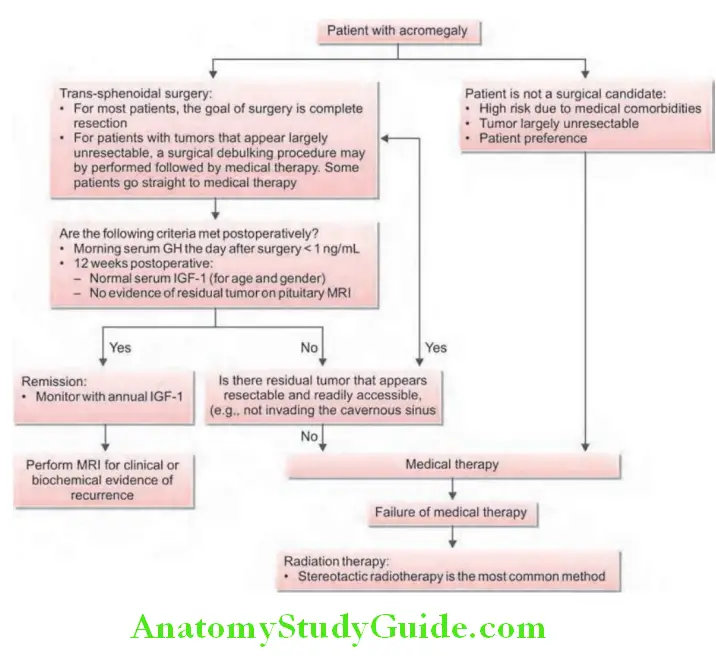
Insulin-like growth factor-1 (IGF-1) levels: It is the single best test useful in diagnosis. IGF-1 is almost always elevated in acromegaly (Flowchart 2.1).
GH levels: Confirmed by assessment of GH secretion. Basal fasting GH levels are >10 ng/mL (465 pmol/L) (normal, 1–5 ng/mL, 46–232 pmol/L) in >90% of patients and range from 5 ng/mL (232 pmol/L) to >500 ng/mL (23,000 pmol/L), with a mean of approximately 50 ng/mL (2,300 pmol/L).
Oral glucose tolerance test: The most specific dynamic test for establishing the diagnosis of acromegaly. GH levels are measured before and 2 hours after an OGTT. In normal subjects, serum GH concentrations fall to 1 ng/mL or less within 2 hours of ingestion of 75 g glucose. The criterion for the diagnosis of acromegaly is a GH concentration of >1 ng/mL. Some patients may show a paradoxical rise and about 25% of patients with acromegaly have a positive diabetic glucose tolerance test.
Serum insulin-like growth factor binding protein-3 (IGFBP-3) concentrations are elevated in patients with acromegaly


- Thyrotropin-releasing hormone (TRH), in a dose of 500 µg intravenously, raises serum GH concentrations by 50% or more in approximately one-half of patients with acromegaly, with peak values occurring at 20‒30 minutes of administration.
- L-DOPA (500 mg orally) reduces serum GH concentrations by 50% or more in approximately one-half of patients with acromegaly, while it raises the GH concentration in normal subjects.
- Postprandial plasma glucose may be elevated and serum insulin is increased in 70% of patients.
Acromegaly Prolactin: Shows mild-to-moderate elevation in about 30% of patients due to cosecretion of prolactin from the tumor.
- Others: Elevated serum phosphorus.
Acromegaly Radiological investigations:
Acromegaly MRI of pituitary: If the biochemical tests are abnormal and GH hypersecretion is confirmed, MRI will almost always reveal and localize the pituitary adenoma usually a somatotroph adenoma (most common cause of acromegaly). If the MRI is normal, abdominal, chest imaging or DOTATATE positron emission tomography (PET) scan should be performed to look for an ectopic source of hormone secretion.
Acromegaly X-ray:
Plain films of skull: Shows sellar enlargement in 90% of cases. Other findings may be thickening of the calvarium, enlargement of the frontal and maxillary sinuses, and enlargement of the jaw.
Radiographs of the hand: Shows increased soft tissue bulk, “arrowhead” tufting of the distal phalanges, increased width of intra-articular cartilages, and cystic changes of the carpal bones.
Radiographs of the feet: Shows similar changes to that of hand, and there is increased thickness of the heel pad (normal heel pad thickness < 22 mm).
X-ray of spine: Scoliosis, calcification of spinal ligaments.
Pituitary function: Partial or complete anterior hypopituitarism is common.
Visual field examination: Defects are common (e.g., bitemporal hemianopia)


Acromegaly Treatment of Acromegaly:
- Untreated acromegaly is associated with markedly reduced survival.
- Most deaths are due to heart failure, coronary artery disease, and hypertension related causes. Patients with acromegaly have an increased risk for cancer (i.e., carcinoma colon).
- Treatment is indicated in all except the elderly or those with minimal abnormalities.
Acromegaly Aim of therapy:
- To lower the serum IGF-1 concentration to within the normal range for the patient’s age and gender.
- Control adenoma size and reduce mass effects.
- Alleviate symptoms such as headaches, vision loss without causing hypopituitarism.
- Reverse metabolic abnormalities such as diabetes mellitus.
- To lower the serum growth hormone (GH) concentration to <1 µg/L
1. Surgery: Trans-sphenoidal surgical removal of pituitary adenoma is the first-line therapy.
2. Medical therapy: Indication for primary medical management: Patients
- With no risk of visual impairment from the tumor
- Unfit candidates for surgery and those who decline surgery
- Tumors that are unlikely to be controlled by surgery
- Persistence of acromegaly after surgery
- Require preservation of intact pituitary function (especially fertility).
Acromegaly Drugs used: There are three receptor targets for the treatment of acromegaly.
Acromegaly Somatostatin receptor ligands (SRL):
About 90% growth-hormone secreting adenomas express somatostatin receptor subtypes (SSTR), SSTR2, and SSTR5.
Mode of action: Somatostatin analogs (Octreotide, Pasireotide or Lanreotide) are more effective than dopamine agonists and act on pituitary somatostatin receptors to produce inhibition of GH and IGF-1.
Limitations: Costly, transient diarrhea, nausea, and abdominal discomfort due inhibition of motor activity, gallstones, and hair loss < 10%.
Acromegaly GH receptor antagonist:
Pegvisomant is a GH receptor antagonist, blocks peripheral IGF-1 action in almost all patients, and is indicated in patients who are inadequately controlled with other modalities or in patients experiencing clinically significant drug side effects.
Limitations: Daily injection, costly, acts on peripheral tissues, and neither affects pituitary tumor nor secretion of GH, GH raises 76% due to loss of negative feedback by lower IGF-1 levels, and LFTs to be monitored due to elevated levels of AST.
Acromegaly Dopamine receptor agonists:
Act on D 2 receptors. Bromocriptine or cabergoline are dopamine receptor agonists and are useful in those with mildly elevated IGF-1.
3. Radiotherapy:
Indications: It is usually employed as second-line treatment
- If acromegaly persists after surgery
- In patients who are not fit candidates for surgical therapy
- In whom medical therapy fails.
External radiotherapy or implantation of Yttrium into the gland.
Stereotactic radiosurgery (gamma knife and cyber knife).
4. Others: Treatment of associated conditions such as diabetes, hypertension, and hyperlipidemia.
Prolactinoma:
Question 8. Write short essay/note on
Answer:
Prolactinoma and hyperprolactinemia.
Prolactinoma Causes, clinical features, investigations, and treatment of hyperprolactinemia.
- Prolactinoma is a pituitary tumor that produces prolactin.
- Most common functional pituitary tumor. Most of these tumors are microadenomas.
- Elevated level of plasma prolactin is known as hyperprolactinemia.
Prolactinoma Causes of hyperprolactinemia:
- Physiological
- Pregnancy
- Stress
- Nursing
- Nipple stimulation
Prolactinoma Pathological:
Prolactinoma Drug-induced:
Estrogens, opiates, dopamine-receptor antagonists (phenothiazines, butyrophenones, metoclopramide), and dopaminedepleting agents (reserpine and methyldopa)
Prolactinoma Disease states:
- Pituitary adenomas (lactotroph, somatotroph-lactotroph, and stalk compression by chromophobe tumors)
- Hypothalamic and stalk disease (craniopharyngiomas, irradiation, granulomas, and stalk section/compression)
- Primary hypothyroidism
- Miscellaneous (cirrhosis, chronic renal failure, and seizures)
Prolactinoma Causes of Hyperprolactinemia:
Prolactinoma Clinical features:
Hyperprolactinemia stimulates milk production in the breast and inhibits GnRH and gonadotropin secretion.
Prolactinoma It usually presents with:
Hypogonadism, decreased libido, infertility, and galactorrhea (spontaneous or expressible) in both sexes.
- In females: Amenorrhea, oligomenorrhea, and osteoporosis.
- In males: Hypogonadotropic hypogonadism leading to loss of libido, impotence, infertility, gynecomastia, and rarely galactorrhea.
- A sufficiently large macroadenoma usually produces visual field defects and headache.
Prolactinoma Investigations:
The investigation of prolactinomas is the same as for other pituitary tumors discussed above.
- Normal range for serum prolactin is approximately 5‒20 µg/L.
- The diagnosis of hyperprolactinemia is made by a serum prolactin concentration that is well above the normal range > 20 µg/L in men and postmenopausal women and >30 µg/L in premenopausal women.
- Serum PRL over 150 µg/L in a nonpregnant woman is generally due to pituitary adenoma; a level of over 300 µg/L is almost diagnostic of tumor (even in a nursing mother).
Prolactinoma Hook effect: Caution should be exercised in interpreting serum prolactin concentrations between 20 and 200 µg/L in the presence of a macroadenoma because of possible artifactually low values due to the «hook effect».
This effect occurs when a very high serum prolactin, e.g., 5,000 µg/L, saturates both the capture and signal antibodies used in immunoradiometric and chemiluminescent assays, preventing the binding of the two in a «sandwich».
The result is an apparent prolactin concentration that is only modestly elevated, suggesting that the macroadenoma is clinically nonfunctioning. The artifact can be avoided by repeating the assay using a 1:100 dilution of serum.
Prolactinoma Macroprolactin: Macroprolactin is native prolactin that is bound to IgG. This causes hyperprolactinemia through decreased prolactin clearance.
- Visual fields should be checked.
- Primary hypothyroidism must be excluded.
- Anterior pituitary function should be assessed if there is evidence of hypopituitarism or radiological evidence of a pituitary tumor.
- MRI or contrast-enhanced CT scan of the pituitary: It is needed if there are any clinical features suggestive of a pituitary tumor. It is desirable when prolactin is significantly elevated (above 1,000 mU/L). It can easily delineate macroprolactinoma (tumors above 10 mm diameter), but microprolactinoma (smaller ones) may be more difficult to delineate.
Prolactinoma Treatment of Prolactinoma:
Hyperprolactinemia is usually treated to prevent the long-term effects of estrogen or testosterone deficiency.
Prolactinoma Medical treatment by dopamine agonists:
- Cabergoline is the first choice with a dose of 0.25–0.5 mg twice a week.
- Bromocriptine (1.25–2.5 mg twice daily) reduces the secretion of prolactin as well as size of pituitary tumor.
Quinagolide: Initial: 0.025 mg once daily for 3 days followed by 0.05 mg once daily for 3 days. Maintenance (beginning on day 7): 0.075 mg once daily
Treatment of men: Testosterone.
Treatment of women: Estradiol.
Trans-sphenoidal removal: It is indicated if dopamine agonists fails or in the presence of a large, invasive tumor.
External radiotherapy: Rarely necessary.
Asymptomatic patients who do not require restoration of pregnancy give estrogens to prevent bone loss and should be regularly monitored.
Diabetes Insipidus (DI):
Question 9. Write a short note on diabetes insipidus (DI) and its diagnosis.
Answer:
- Diabetes insipidus (DI) is a disorder in which polyuria due to decreased collecting tubule water reabsorption is induced by either decreased secretion of antidiuretic hormone (ADH; central DI) or resistance to its renal effects (nephrogenic DI).
- It is characterized by the persistent passage of excessive amounts of dilute urine and thirst.
Diabetes Insipidus Types of Diabetes Insipidus (DI):
- Primary deficiency (neurogenic, pituitary, hypothalamic, cranial or central DI): It is due to agenesis or destruction of neurohypophysis.
- Secondary deficiency: It is due to inhibition of ADH secretion (primary polydipsia).
- Deficient action of ADH (nephrogenic DI)
- Transient diabetes insipidus of pregnancy produced by accelerated metabolism of vasopressin by vasopressinases released from the placenta (gestational DI).
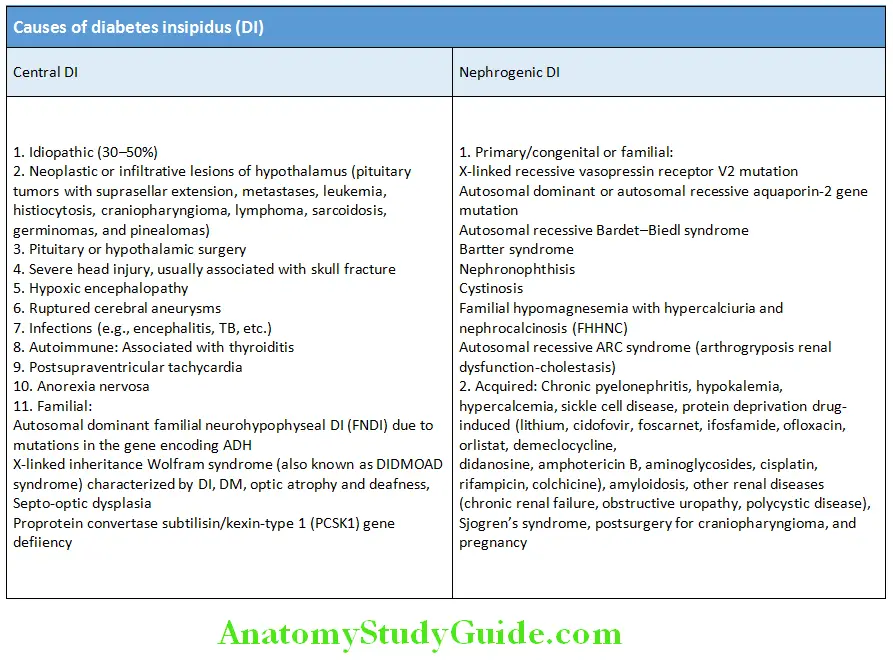
Diabetes Insipidus Causes of Diabetes Insipidus:
Diabetes Insipidus Clinical features:
Polyuria and polydipsia: The urine output may range from 2 L/day with mild partial DI to over 10‒15 L/day in patients with severe disease, nocturia, and compensatory excessive thirst (polydipsia) are the most marked symptoms.
Other features: Change in mentation, insomnia, and weight loss. Skin, mucous membranes cool.
If left untreated changes in LOC (loss of consciousness), tachycardia, tachypnea, and hypotension (shock-like symptoms), but unlike hypovolemic shock, urine output is increased.
Patients with nephrogenic DI may also have manifestations related to underlying cause such as lithium toxicity, hypercalcemia, and hypokalemia.
It can lead to hypernatremia, restlessness, agitation, diminished deep tendon reflexes, and seizures.
Diabetes Insipidus Complications:
- Hypernatremia, dehydration, and their neurological sequelae
- Growth retardation
- Hydronephrosis (due to excessive urine output)
Diabetes Insipidus Investigation and diagnosis:
- Careful history, examination
- Document presence of polyuria (usually 2‒15 L/24 hours).
- Measurement of osmolality of plasma and urine: It establishes the diagnosis.
- Normally, plasma osmolality is <295 mOsml/kg and urine osmolality (random specimen) is 50‒1,200 mOsml/kg.
- In patients with DI and excess urine free water, there is high or high normal plasma osmolality (>295 mOsml/kg)
and low urine osmolality (50–150 mOsml/kg). - In primary polydipsia, plasma osmolality tends to be low and may be lower than urinary osmolality
Diabetes Insipidus Urine:
- High 24 hours urine volumes. If the volume is <2 L, there is no need for further investigation.
- Clear and of low specific gravity of urine.
- Low urine osmolality and usually less than that of plasma osmolality.
- Serum sodium is high or high normal (hypernatremia) and indicates loss of water.
- MRI of pituitary and hypothalamus to identify hyperintensities in the posterior pituitary or thickening of the pituitary stalk can help determine the cause of CDI.
Question 10. Write a short note on water deprivation test.
Answer:
Water deprivation test (Miller‒Moses Test)
Diabetes Insipidus Indication:
- Diagnosis or exclusion of diabetes insipidus
- To differentiate CDI and NDI
- To differentiate diabetes insipidus from primary polydipsia
Diabetes Insipidus Procedure:
- Should be done in the morning under observation with 8 hours fasting. No fluids (water deprivation) from 07:30 hours.
- Measure plasma and urine osmolality, urine volume and weight hourly for up to 8 hours.
- Abandon fluid deprivation if weight loss is >5%.
- If plasma osmolality > 300 mOsm/kg and/or urine osmolality < 600 mOsm/kg, ADH (vasopressin) is given in the dose of 2 μg IM at end of test. Allow free intake of fluids but measure urine osmolality for 2–4 hours.
Diabetes Insipidus Normal response:
- Withholding fluid in normal individuals, the plasma osmolality remains within normal range (275–295 mOsm/kg) while urine osmolality rises above 600 mOsm/kg (800–1,200 mOsm/kg).
- Urine osmolality is greater than plasma osmolality after restriction of water.
- Urine osmolality increases minimally (<10%) after exogenous ADH.
Diabetes Insipidus Primary polydipsia:
- Patients with primary polydipsia start with low normal plasma osmolality (280 mOsm/kg).
- Urine/plasma osmolality ratio rises to >2 after dehydration (water deprivation).
Diabetes Insipidus Central diabetes insipidus:
- After water restriction in patients with DI, the plasma osmolality rises above normal (>300 mOsm/kg) without rise in urine osmolality (<600 mOsm/kg) or specific gravity of urine. (Normally urine osmolality rises to 1,000–1,200 mOsml/kg after water restriction). Urine osmolality remains less than plasma osmolality and urine/plasma osmolality ratio remains <1.5.
- After ADH is given, urine osmolality increases 100% in complete CDI and over 50% in partial CDI.
Diabetes Insipidus Nephrogenic diabetes insipidus:
- Urine osmolality remains less than plasma osmolality.
- After ADH, urine osmolality increases by <50%.
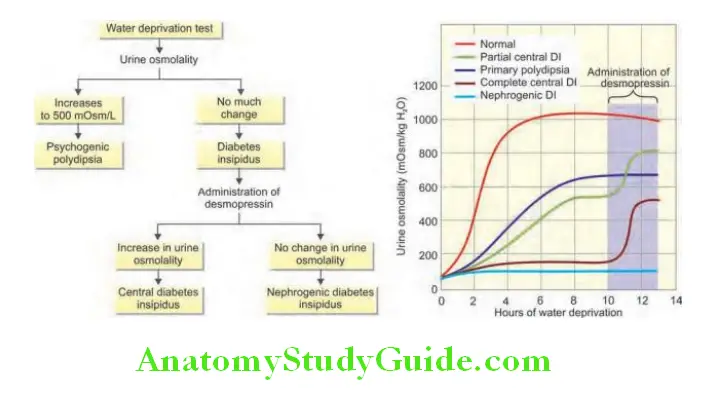
- Alternative to water deprivation test is by infusing hypertonic (5%) saline and measures ADH secretion in response to increasing plasma osmolality.
Diabetes Insipidus Treatment:
Diabetes Insipidus Goals of treatment:
- Balance fluid intake with output: In acute cases, rapidly replace fluid and in chronic cases with slow replacement to prevent cerebral edema.
- Daily weights, accurate intake/output monitoring, urine specific gravity, and osmolality.
Diabetes Insipidus Drugs:
Diabetes Insipidus Desmopressin (DDAVP): Drug of choice, initial dose of 5 µg (rhinal tube) or 10 µg (metered spray) by intranasal route is given at bedtime. This dose is titrated up, in 5 µg increments as needed, depending on the response of the nocturia and then additional daytime doses are added. The typical daily maintenance dose is 5–20 µg once or twice daily.
Diabetes Insipidus Chlorpropamide: Increases the renal responsiveness to vasopressin. Hypoglycemia may develop.
Diabetes Insipidus Carbamazepine: It is an alternate drug which enhances ADH release and raises the sensitivity of the collecting duct to it.
Diabetes Insipidus Clofibrate: It is a lipid lowering agent which stimulates residual ADH production in the hypothalamus, therefore, increasing ADH release from the posterior pituitary.
Diabetes Insipidus Nonsteroidal anti-inflammatory drugs (NSAIDs): Work by inhibiting renal prostaglandin synthesis which are ADH antagonists and also, they decrease the glomerular filtration rate by prostaglandin-mediated effect of afferent arteriole dilation.
Treatment of nephrogenic DI:
- Provision of adequate fluids especially in cases of impaired thirst, calorie and decreased dietary solute such as low sodium diet.
- Correct the underlying cause.
- Thiazide diuretics: These (e.g., hydrochlorothiazide 25 mg once or twice daily with a low sodium diet) are the first-line therapy in nephrogenic DI.
- Amiloride (potassium-sparing diuretic): Additive effect with thiazide diuretic and may be particularly beneficial in patients with reversible lithium nephrotoxicity by possibly allowing lithium to be continued.
- Exogenous ADH (DDAVP)
NSAIDs: Indomethacin
Thyroid Disorders
Thyroid Function Tests (TFTs):
Question 11. Write short essay/note on various thyroid function tests.
Answer:
Thyroid Function Tests Indications for thyroid function tests:
Screening for thyroid dysfunction
Thyroid Function Tests Surveillance: Women with postpartum thyroiditis, postneck irradiation
Thyroid Function Tests Monitoring: Treatment of hyperthyroidism and hypothyroidism
Various Thyroid Function Tests:
Question 12. Write short essay/note on thyroid stimulating hormone.
Answer:
Serum thyroid-stimulating hormone/thyrotropin (TSH):
- It is measured currently by TSH chemiluminometric assays.
- Normal range for serum TSH is approximately 0.4–5.0 mU/L.
Various Thyroid Function Tests Interpretation
Various Thyroid Function Tests Thyroid diseases: As a single test of thyroid function TSH is the most sensitive index of thyroid function.
- TSH levels can help in differentiating hyperthyroidism, hypothyroidism, and euthyroidism (normal thyroid gland function) in most cases.
- Raised/elevated levels indicate primary hypothyroidism.
- Low/suppressed levels indicate primary thyrotoxicosis.
Various Thyroid Function Tests Nonthyroid diseases: Other conditions affecting TSH levels include:
- TSH-secreting tumors of pituitary
- Severe nonthyroidal illness (e.g., sick euthyroid syndrome)
- Low TSH may be observed in first trimester of pregnancy with high doses of corticosteroids and patients ingesting biotin supplements due to assay interference.
- Secondary hypothyroidism due to hypothalamic-pituitary disease may produce low, normal, or normal-high levels of TSH that are inappropriate for the very low free T4 level.
Various Thyroid Function Tests Thyrotropin releasing hormone (TRH) stimulation test:
- It may be used in the investigation of hypothalamic pituitary dysfunction.
- Serum TSH is measured before and after the intravenous administration of TRH.

Various Thyroid Function Tests Use: To differentiate secondary or tertiary hypothyroidism.
- Secondary hypothyroidism (pituitary disease): TRH administration does not produce increase in TSH.
- Tertiary hypothyroidism (hypothalamic disease): TRH administration produces delayed increase in TSH.
- In primary hypothyroidism, TRH administration produces a prompt increase in TSH.
Various Thyroid Function Tests Serum free T3 (tri-iodothyronine T3) and free T4 (free thyroxine/fT4):
- Advantage of this test compared to the measurement of total T3 and T4 is that these are not influenced by changes in the thyroid hormone-binding globulins (TBG), prealbumin and albumin. Its level reflects secretory activity.
- Primary thyrotoxicosis: fT3 and fT4 levels are elevated.
- T3-thyrotoxicosis: fT3 levels are normal and fT3 levels are elevated.
- On T4 therapy: fT4 levels are elevated and fT3 levels are normal.
Various Thyroid Function Tests Total serum thyroxine (tT4)
- Measured by automated competitive binding chemiluminometric assays.
- Its levels are altered by factors that affect the concentration of TBG.
Various Thyroid Function Tests Normal ranges: 4.6–11.2 µg/dL.
Various Thyroid Function Tests Increased: In hyperthyroidism, during pregnancy, estrogen therapy, tamoxifen use and as a congenital anomaly.
Various Thyroid Function Tests Decreased: In hypothyroidism, nephritic syndrome, androgen therapy, liver failure, or drugs (e.g., salicylates, sulfonylureas, and phenytoin).
Various Thyroid Function Tests Total serum tri-iodothyronine (tT3):
- Measured by automated competitive binding chemiluminometric assays.
- Its levels are subject to the same limitations as for tT4 in relation to TBG.
Various Thyroid Function Tests Normal range: 75–195 ng/dL.
- T3 resin uptake, free T4 index (FTI), effective T4 ratio Nowadays, above three tests (4, 5, and 6) are not used.
Various Thyroid Function Tests Reverse T3 (rT3):
- Reverse T3 (rT3) is mainly an inactive metabolite of T4 in peripheral tissues.
- It has extremely limited utility for assessing rare conditions such as consumptive hypothyroidism, MCT8 or SBP2 mutations, or possibly distinguishing central hypothyroidism from nonthyroidal illness in critically ill hospitalized patients.
Various Thyroid Function Tests Thyroglobulin (Tg):
It is synthesized by follicular cells, secreted into the lumen of the thyroid follicle, stored as a colloid and is involved in iodination/synthesis of thyroid hormones.
Various Thyroid Function Tests Use: To predict the outcome of therapy for hyperthyroidism.
- Increased: Well-differentiated thyroid carcinoma, hyperthyroidism
- Decreased: Total thyroidectomy or destruction of thyroid by radiation.
Various Thyroid Function Tests Uptake of radioactive iodine (RAIU) or technetium:
The iodine uptake activity of thyroid can be measured by administering orally a low/trace dose of radioactive iodine 131I or 121I and the radioactivity over the thyroid is measured after 4 hours, using a counter over the neck.
Pregnancy and breastfeeding are absolute contraindications to radionuclide imaging. However, in the unusual instance where radioiodine uptake measurement is felt to be essential for a definitive diagnosis in a lactating woman, breast milk can be pumped and discarded for 5 days after ingestion of I123, then breastfeeding may be resumed; breastfeeding should not be resumed if the I131 isotope is used for determining the uptake.
The amount of radioactivity that is taken up by the thyroid gland is known as radioactive iodine uptake (RAIU).
Alternatively, thyroid uptake is measured by giving technetium-99m (99mTc) intravenously.
Various Thyroid Function Tests Uses of radioactive iodine uptake (RAIU):
- Evaluation of hyperthyroidism
- Differentiate Graves’s disease from toxic goiter
- Function of a thyroid nodule as hot or cold
Various Thyroid Function Tests Interpretation: Normal uptake ranges from 10% to 35% in 24 hours.
Various Thyroid Function Tests Uptake increased: Overactive thyroid gland synthesizing excess T3 shows increased uptake of iodine. A very high RAIU is seen in hyperthyroidism (e.g., Graves’ disease, toxic multinodular goiter, toxic adenoma and early thyroiditis). Iodine/enzyme deficiency may show increased uptake even in the absence of thyrotoxicosis.
Various Thyroid Function Tests Uptake decreased: Low RAIU is seen in hypothyroidism and late thyroiditis. Excess iodine may show diminished uptake even in the presence or thyrotoxicosis. Acute autoimmune thyroiditis may manifest as low iodine uptake thyrotoxicosis.
Various Thyroid Function Tests Use: To determine the functional activity and morphology of the thyroid gland. Very useful in determining the activity of a solitary thyroid nodule. Functional nodule appears as a “hot” nodule, and a nonfunctional appears as a “cold” nodule.
Various Thyroid Function Tests Useful in follow-up of patients with treated thyroid cancer.
Detection of ectopic thyroid tissue: Confirmation of a mass on the tongue as lingual thyroid in the midline of the neck as thyroglossal duct, or in the mediastinum as substernal goiter.
Ultrasound of thyroid gland: Look for nodularity, vascularity, shape, microcalcifications, lymph nodes status, and for guided FNAC.
Thyroid auto-antibody tests: The different types of thyroid autoantibodies responsible for the autoimmune thyroid disorders are:
Anti-microsomal antibody:
- Antithyroid peroxidase (TPO) antibody (TPOAb): They are involved in the tissue destructive process associated with hypothyroidism in Hashimoto and atrophic thyroiditis.
Antithyroglobulin (Tg) antibody: May be present in Hashimoto’s thyroiditis and Graves’ disease.
TSH receptor (TR) antibody (TRAb): Classified as stimulating, blocking, or neutral.
- Stimulating antibodies (thyroid-stimulating immunoglobulins, TSI) cause Graves’ disease.
- Thyroid receptor-blocking antibodies can cause hypothyroidism. Neutral antibodies bind the receptor but do not stimulate or block function.
Various Thyroid Function Tests Tests to determine etiology of thyroid disease:
Various Thyroid Function Tests Calcitonin: It is secreted by parafollicular C-cells and is increased in medullary carcinoma of thyroid.
Fine-needle aspiration cytology/excision biopsy: It is helpful in diagnosis thyroid diseases.
Thyrotoxicosis:
Discuss the etiology, clinical features, investigations (laboratory diagnosis), complications and management of hyperthyroidism/thyrotoxicosis.
Discuss the etiopathogenesis, clinical features, investigations, diagnosis, and management/treatment modalities of Graves’ disease.
Thyrotoxicosis Defiition:
- Thyrotoxicosis is a state of circulating thyroid hormone excess (with hypermetabolic state) caused by exposure to excessive levels of thyroid hormone (free T3 and T4).
- This increase in circulating hormone may be either from destruction of thyroid gland or from ectopic source.
- Hyperthyroidism (thyroid overactivity): It is the clinical consequence due to the excessive circulating thyroid hormone due to excessive thyroid function/hyperfunction and is the most common cause of thyrotoxicosis. Its causes are listed in Table
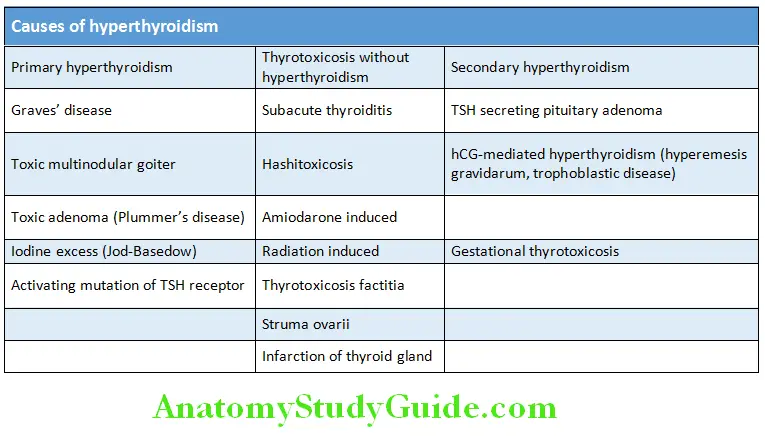
Etiology and Pathogenesis of Hyperthyroidism:
Question 13. Write short essay/notes on causes of hyperthyroidism.
Answer:
Few of the causes of hyperthyroidism/thyrotoxicosis are discussed below.
1. Graves’ disease:
Graves’ disease is the most common form of thyrotoxicosis. It is characterized by one or more of the following features:
- Thyrotoxicosis
- Goiter
- Orbitopathy (exophthalmos)
- Dermopathy (pretibial myxedema).
Etiology and Pathogenesis of Hyperthyroidism Age, gender, and genetic susceptibility:
- It may occur at any age with a peak incidence between 20 and 40 years of age.
- The diseases cluster in families and are more common in women.
- The concordance rate in monozygotic twins is 20‒40%.
- Associated with certain alleles of CTLA-4
- Associated with certain alleles of HLA on Chromosome 6 namely, HLA-DRB1*08 and DRB3*0202.
- HLA-DRB1*07 is found to be protective.
Etiology and Pathogenesis of Hyperthyroidism Autoantibodies:
It is an autoimmune disorder with autoantibodies.
TSI or TSH-receptor antibodies of the stimulating type (TRAb): IgG type of antibodies directed against the TSH receptors on the follicular cell of thyroid. They stimulate thyroid hormone production and enlargement of thyroid.
T cells: Activated T cells release cytokines and increase the secretion of thyroid-specific autoantibodies from B cells. The current concept is that thyroid-specific T cells in Graves’ disease primarily act as helper (CD4+ Th1) cells.
Histology: Characterized by follicular hyperplasia, intracellular colloid droplets, cell scalloping, a reduction in follicular colloid, and a patchy (multifocal) lymphocytic infiltration.
Ophthalmopathy and dermatopathy: Observed in Graves’ disease is due to immunologically mediated activation of fibroblasts in the extraocular muscles and skin. This along with accumulation of glycosaminoglycans and trapping of water causes edema initially. Later fibroblasts cause fibrosis.
Hyperthyroidism may be triggered by viral or bacterial infections:
E. coli and Yersinia enterocolitica possess cell membrane TSH receptors. The antibodies produced against these micro-organisms can cross-react with the\ TSH receptors (molecular mimicry).
Other mediating mechanisms: Bystander activation and thyroid cell HLA-antigen expression.
Etiology and Pathogenesis of Hyperthyroidism Other precipitating causes:
- Thyroid injury by radiation and drugs
- Stress
- Smoking
- Sex steroids
- Pregnancy
- Fetal microchimerism
- Iodine and iodine-containing drugs such as amiodarone.
- Alemtuzumab, a monoclonal antibody against CD52, has been associated with a 10‒15% incidence of new-onset Graves’ disease.

Etiology and Pathogenesis of Hyperthyroidism Thyroid orbitopathy (eye signs):
- An autoimmune disease of the retro-ocular tissues occurring in patients with Graves’ disease. Although it has often been referred to as Graves’ ophthalmopathy, or thyroid eye disease (TED), it is primarily a disease of the orbit and is better termed Graves’ orbitopathy.
- The ophthalmopathy causes abnormal protrusion of the eyeball (exophthalmos).
- Sympathetic overactivity may produce the characteristic wide, staring gaze, and can also be the reason for lid lag.
- It is observed in about 50% of the patients when first seen. It may precede Graves’ disease by many years or may develop even after successful treatment of Graves’ disease.
- The volume of both the extraocular muscles and retro-ocular connective tissue is increased due to fibroblast proliferation, inflammation and the accumulation of hydrophilic glycosaminoglycans (GAG), mostly hyaluronic acid.
Etiology and Pathogenesis of Hyperthyroidism Pathogenesis of orbitopathy:
- The main autoantigen is the thyroid-stimulating hormone receptor (TSHR), which is expressed primarily in the thyroid but is also expressed in adipocytes, fibroblasts, and a variety of additional sites.
- TSHR antibodies and T-cells activate retro-ocular fibroblast and adipocyte TSHR and IGF-1 receptors, initiating a retro-orbital inflammatory environment.
- GAG secretion by fibroblasts is increased by thyroid-stimulating antibodies and activated T cells (via cytokine secretion), implying that both B and T cell activation are integral to this process.
- The accumulation of hydrophilic GAG in turn leads to fluid accumulation, muscle swelling, and an increase in pressure within the orbit.
- These changes, together with retro-ocular adipogenesis, displace the eyeball forward leading to extraocular muscle dysfunction and impaired venous drainage.
Etiology and Pathogenesis of Hyperthyroidism Symptoms: There may not be any ocular symptoms, but the patient may be distressed by the appearance. However, the following symptoms can prevail:
- A gritty or foreign object sensation in the eyes
- Excessive tearing, often made worse by exposure to cold air, wind, or bright lights
- Eye or retro-ocular discomfort or pain
- Blurring of vision
- Diplopia
- Color vision desaturation
- Loss of vision in severe cases
Etiology and Pathogenesis of Hyperthyroidism Signs:
- Exophthalmos (proptosis) often asymmetric but can be symmetric
- Lid lag
- Lid retraction
- Periorbital edema
- Corneal ulcers
- Chemosis
- Ophthalmoplegia
- Visual field defects
- Papilledema.
Etiology and Pathogenesis of Hyperthyroidism Treatment:
Etiology and Pathogenesis of Hyperthyroidism Reversal of hyperthyroidism:
- Rapid amelioration of symptoms, with a beta-blocker and decreasing thyroid hormone synthesis with a thionamide or surgery.
- Reduction of thyroid hormone synthesis decreases eyelid retraction and stare. (If hypothyroidism develops during the course of therapy, more fluid retention can occur and may have an adverse effect on the orbitopathy).
- For patients with active and moderate-to-severe or sight-threatening orbitopathy, thionamides or surgery are the preferred treatment modalities, as this is usually followed by a fall in serum thyrotropin receptor antibody (TRAb) concentrations, suggesting a waning of autoimmunity. Radioiodine therapy leads to the development or worsening of orbitopathy as it is associated with a sustained increase in TRAb, hence contraindicated.
- Patients with active and mild orbitopathy may still be candidates for thionamides, radioiodine, or surgery. If radioiodine is chosen, glucocorticoids should be administered concurrently for those with risk factors for progression.
Etiology and Pathogenesis of Hyperthyroidism Smoking cessation: Increases the incidence of symptomatic Graves’ orbitopathy, increases the risk of worsening orbitopathy after radioiodine therapy and also renders patients more refractory to anti-inflammatory therapy.
Etiology and Pathogenesis of Hyperthyroidism Local measures: Eye shades, artificial tears, and lubricants such as methylcellulose or petroleum jelly, raising head end of bed at night, eye patching and prisms for diplopia.
Etiology and Pathogenesis of Hyperthyroidism Medical:
- Selenium 100 µg twice daily may improve symptoms in patients with mild Graves’ orbitopathy. It has an antioxidant role and also decreases inflammatory activity.
- Glucocorticoids are the mainstay of immunomodulatory therapy for moderate-to-severe Graves’ orbitopathy. For patients with moderate symptoms, a trial of oral (prednisone, 30 mg/day for 4 weeks) or IV (methylprednisolone, 500 mg once weekly for weeks 1–6, then 250 mg once weekly for weeks 7–12 with cumulative dose 4.5–5 g over 12 weeks) can be tried. For more severe or progressive cases, initial IV therapy is appropriate.
- Teprotumumab, an IGF-1 receptor inhibitor, was approved for the treatment of Graves’ orbitopathy by the Food and Drug Administration (FDA) in February 2020. It is administered every 3 weeks as an IV infusion (10 mg/kg initial dose, then 20 mg/kg) for a total of eight infusions. Teprotumumab is contraindicated during pregnancy.
- Mycophenolate mofetil is under investigation for the treatment of Graves’ orbitopathy, either alone or in combination with glucocorticoids.
- Tocilizumab targets IL-6 is being investigated for the treatment of patients with Graves’ orbitopathy who are not improving with glucocorticoids.
- Rituximab reported to be as effective as glucocorticoids without the glucocorticoid-related side effects induces a fall in thyrotropin receptor antibody (TRAb) levels and depletion of B cells in the retro-orbital tissues.
Etiology and Pathogenesis of Hyperthyroidism Orbital decompression surgery: Indicated in optic neuropathy caused by enlarged extraocular muscles not responsive to highdose corticosteroids, severe orbital inflammation, excessive proptosis leading to exposure keratitis, corneal ulceration, or debilitating cosmetic defect, pain relief, and progressive orbitopathy not responding to other measures.
Etiology and Pathogenesis of Hyperthyroidism Other surgeries:
- Fat decompression surgery, bilateral lateral tarsorrhaphy, surgical recession of Müller’s muscle and the levator to correct upper lid retraction.
- For patients requiring both strabismus surgery and orbital decompression, the decompression should be performed first, followed by strabismus surgery.
2. Treatment-induced hyperthyroidism.
Drugs include iodine, amiodarone, iodine-containing contrast media, interferon-alpha, and rarely, lithium.
Etiology and Pathogenesis of Hyperthyroidism Iodine-induced hyperthyroidism:
- It develops following excess intake of iodine in diet or exposure to radiographic contrast media or iodine medication.
- Jod-Basedow phenomenon: In states of iodine deficiency, thyroid nodules can develop, some of which can be autonomous functioning nodules. On supplementation of iodine, there is an increase in production of thyroxine, leading to thyrotoxicosis. Usually develops in patients with autonomously functioning thyroid gland (e.g., nodular goiter/Graves’ disease). It can also occur in endemic goiter and with iodine. Takes place due to a loss in the negative feedback mechanism of the circulating hormones on the thyroid gland. It is characterized by suppressed serum TSH level with normal levels of circulating thyroid hormone.
Etiology and Pathogenesis of Hyperthyroidism Wolff‒Chaikoff effect: Autoregulation by the follicular cells protects the gland from the wide variations in the amount of iodide intake. Sometimes, excess exposure and uptake of iodide by the gland can inhibit organification of iodide, thereby diminishing hormone biosynthesis, resulting in hypothyroidism or myxedema.
Etiology and Pathogenesis of Hyperthyroidism Amiodarone-induced hyperthyroidism (AIT):
Amiodarone is a class III antiarrhythmic drug. A 200-mg formulation of the drug contains 75 mg iodine. It can also induce hyperthyroidism which can be table.

3. Thyrotoxicosis factitia:
- Exogenous hyperthyroidism is due to the surreptitious ingestion of thyroid hormone, it is termed as “thyrotoxicosis factitia”.
- Patients are clinically thyrotoxic without eye signs of Graves. High doses of thyroxine lead to TSH suppression and causes shrinkage of the thyroid.
- Find cause or contamination and treat symptomatically.
4. Toxic multinodular goiter (Plummer’s disease):
- Result of diffuse hyperplasia of thyroid follicular cells whose functional capacity is independent of regulation by TSH.
- Goiter will be nodular.
- Constitutes 14% of thyrotoxicosis cases.
- Commonly occurs in elderly women (>50 years).
- T3 (greater), T4 raised, and TSH undetectable.
5. Toxic solitary adenoma/nodule:
- Constitutes <5% of all thyrotoxicosis (hyperthyroidism) cases and the solitary nodule is follicular adenoma.
- Usually occurs in female >40 years of age.
- Suspected thyroid nodules merit close attention in the pediatric population because such nodules are much more likely to be malignant in children than they are in adults.


Clinical Features of Thyrotoxicosis:
Question 14. Write short essay/notes on clinical features/signs/manifestations of thyrotoxicosis/hyperthyroidism.
(or)
Write a short note on cardiac complications of hyperthyroidism.
Answer:
Classic symptoms include heat intolerance, tremor, palpitations, anxiety, weight loss, increased appetite increased frequency of bowel movements, and shortness of breath.

- Elderly patients present with anorexia, apathy, fatigue, weight loss, and dominant cardiovascular and myopathic features (apathetic hyperthyroidism).
- Younger patients present predominantly with neurological manifestations.
- Children present with excessive height or excessive growth rate, or with behavioral problems (e.g., hyperactivity), and weight gain rather than loss.
Pretibial myxedema (infiltrative dermopathy):
Question 15. Write short essay/notes on pretibial myxedema.
Answer:
- Formerly used to occur in 5% of patients with only Graves’ disease and 15% of patients with both Graves’ disease and ophthalmopathy.
- However, the incidence has declined due to early diagnosis and treatment of Graves’ disease.
- Characterized by bilateral, asymmetric, nonpitting, scaly thickening, and induration of the skin.
- Lesion may be violaceous or slightly pigmented (yellow-brown) and often have a peau d’orange appearance.
- The most frequent location is over the lower limbs, especially the pretibial areas or the dorsum of the foot. Rarely, the fingers and hands, elbows, arms, or face are affected (Fig. 2.7).
- Rarely, lesions progress to involve the legs, feet, or hands completely, resulting in a form reminiscent of elephantiasis.
- Occurs due to deposition of glycosaminoglycans especially hyaluronic acid, secreted by fibroblasts under the stimulation of local cytokines arising from a lymphocytic infiltration.
- Treatment of pretibial myxedema
- Some patients do not seek treatment as they are asymptomatic.
- Indications Pruritus, local discomfort, the unsightly appearance of the lesions, or progression of lesions.
- Nonpharmacologic includes minimizing risk factors, such as avoiding tobacco, reducing weight, normalizing thyroid function and in severe cases, physiotherapy. Normalization of thyroid function does not necessarily improve pretibial myxedema.
Pretibial myxedema Pharmacologic:
- Topical 0.025% fluocinolone acetonide under plastic wrap.
- Intralesional corticosteroids if there is no improvement with topical treatment after 4–12 weeks, using triamcinolone acetate dose calculated according to 8 mg per 2 cm—diameter circle area at each session, but the total dose was not >100 mg at each session in a patient.
- Pentoxifylline may be helpful in resistant cases

Pretibial myxedema Other disorders associated with Graves’ disease.
Pretibial myxedema Autoimmune disorders
Pretibial myxedema Endocrine: Addison’s disease, type 1 diabetes mellitus (DM), Hashimoto’s thyroiditis, primary gonadal failure, hypophysitis
Pretibial myxedema Nonendocrine: Celiac disease, pernicious anemia, myasthenia gravis, immune thrombocytopenic purpura, rheumatoid arthritis, vitiligo, and alopecia areata
Pretibial myxedema Others: Hypokalemic periodic paralysis and mitral valve prolapse
Pretibial myxedema Subclinical hyperthyroidism:
Pretibial myxedema Clinical manifestations:
Pretibial myxedema Clinical manifestations of subclinical hyperthyroidism.
Pretibial myxedema Bone disease
- Decreased bone density, especially in postmenopausal women
- Increased fracture risk
- Biochemical markers of increased bone resorption
- Increased urinary pyridinoline and deoxypyridinoline excretion
- Increased urinary hydroxyproline excretion
Pretibial myxedema Heart disease
- Increased incidence of atrial fibrillation
- Increased heart rate and incidence of atrial premature beats
- Increased cardiac contractility
- Increased left ventricular mass index and septal and posterior wall thickness
Pretibial myxedema Laboratory abnormalities:
- Decrease in serum total and low-density lipoprotein lowdensity lipoprotein (LDL) cholesterol concentrations
- Increased serum concentrations of hepatic enzymes and creatine kinase
- Increased serum concentration of sex hormone-binding globulin (SHBG)
Pretibial myxedema Other:
- Decreased time asleep at night
- Improved mood
Pretibial myxedema Laboratory findings: Serum levels of free T4 and T3 are within normal limits. Serum TSH levels is subnormal (<0.5 mU/L).
Pretibial myxedema Investigations of Graves’ Disease:
- TSH levels: Very low or undetectable. This is performed as the primary test and normal level excludes thyrotoxicosis.
- Serum T3 and T4 levels: Raised in most cases. T3 thyrotoxicosis is associated with raised T3 levels and normal T4 level.
- Absence of TSH response following intravenous TRH.
- I uptake by the thyroid gland: It may be increased but not necessary to perform in most of the patients.
- TSH receptor antibody (TRAb): Present in most cases.
- Few patients may show minor LFT abnormalities, mild hypercalcemia, and glycosuria.
Question 17. Write short essay/notes on drugs used in thyrotoxicosis.
(or)
Write short essay/notes on adverse effects of antithyroid drugs.
Answer:
Management of Hyperthyroidism of Graves’ Disease
The therapeutic approach to Graves’ hyperthyroidism consists of both rapid amelioration of symptoms with a beta-blocker and measures aimed at decreasing thyroid hormone synthesis: the administration of a thionamide, radioiodine ablation, or surgery.
Graves’ Disease Symptom control: Beta-blocker, Propranolol (drug of choice) in the dose of 20–40 mg 6 hourly or Atenolol 25–50 mg/day to reduce symptoms for immediate relief due to sympathetic overactivity such as anxiety, palpitation, increased bowel activity, lid retraction, finger tremors.
Graves’ Disease Decrease thyroid hormone synthesis: There are 3 treatment options for Graves’ disease. All three options are effective, but all three options have significant side effects.
Graves’ Disease There is no consensus as to the “best” treatment:
- Antithyroid drugs (thionamides),
- Radioiodine
- Surgery
1. Antithyroid drugs (ATD): May be used initially to control hyperthyroidism (in addition to beta-blockers) prior to definitive therapy with radioiodine or surgery; they may be prescribed for 1–2 years to attain a remission, or may be used long-term.
Indications: Primary therapy in pregnancy, in children and adolescents and severe Graves’ disease with eye changes.
The drugs include: Thionamides: Methimazole, Propylthiouracil, and Carbimazole
Mechanism of action: Inhibit the function of thyroid peroxidase (TPO) enzyme and prevent binding of iodine to tyrosine (prevents iodination and organification).
Graves’ Disease Methimazoleis the primary drug used to treat hyperthyroidism Dose:
- Free T4 1–1.5 times upper limit of normal: begin treatment with 5–10 mg once daily.
Free T4 1.5–2 times upper limit of normal: begin treatment with 10–20 mg once daily. - Free T4 2–3 times upper limit of normal: begin treatment on 20–40 mg daily in divided doses.
- The dose is tapered to maintenance levels (5–10 mg/day) as the patient improves.
Graves’ Disease Propylthiouracil: Preferred during the first trimester of pregnancy.
Graves’ Disease Dose: 300 mg daily in 3 equally divided doses; 400 mg daily in patients with severe hyperthyroidism and/or very large goiters; usual maintenance: 100–150 mg daily in 3 divided doses.
Graves’ Disease Carbimazole: It has additional immunosuppressive action.
Graves’ Disease Dose: Initially 20–60 mg daily given in 2–3 divided doses and maintenance 5–15 mg daily or alternatively 20–60 mg daily.
Graves’ Disease Total duration of treatment: 18–24 months.
Graves’ Disease Adverse effects: Rashes, urticaria, fever, arthralgia, blood dyscrasias (agranulocytosis), hepatotoxicity, aplasia cutis in neonates.
Graves’ Disease Antithyroid drugs (ATD) Advantages:
- No surgical risk, scar, or chances of injury to parathyroid or recurrent laryngeal nerve.
- Hypothyroidism, if induced, is reversible.
- Can be used even in children and young adults.
Graves’ Disease Antithyroid drugs (ATD) Disadvantages:
- Prolonged (often lifelong) treatment is needed and relapse rate is high.
- Not practicable in uncooperative patient.
2. Radioiodine ablation:
Preferred as definitive therapy of hyperthyroidism in nonpregnant patients except in patients with moderate or severe orbitopathy and chronic smokers. Administered as a capsule I 131 , induces extensive tissue damage by emitting gamma-radiation from within the follicles, resulting in ablation of the thyroid within 6–18 weeks.
Radioiodine ablation Dose: 185–555 MBq
Radioiodine ablation Indications:
- Patients Above 40 Years Of Age.
- Young Patients With A Short Life-Span Due To Some Other Reason.
- Young Individuals Who Are Sterilized.
Radioiodine ablation Complication: Hypothyroidism, infertility, and secondary cancers.
3. Surgery:
Subtotal thyroidectomy is the treatment of choice. Surgical treatment is reserved for multinodular goiter (MNG) with following features:
- Severe hyperthyroidism in children.
- Pregnant women who cannot tolerate antithyroid drugs.
- Large goiters with severe ophthalmopathy or with pressure symptoms.
- Patients who require quick normalization of thyroid function
Surgery Preparation for thyroidectomy: It includes pretreatment with propranolol or atenolol, methimazole 5–40 mg depending on severity and potassium iodide (to decrease vascularity and make the gland firm) 8 mg iodide per drop, 5–7 drops three times daily for 2 weeks before surgery.
Surgery Postoperative complications: Hypothyroidism, hypoparathyroidism, hypocalcemia, and damage to recurrent laryngeal nerve.
Surgery Adjunctive therapies:
Oral radiocontrast agents: Sodium ipodate and iopanoic acid are potent inhibitors of the peripheral conversion of T4 to T3.
Surgery Dose: A dose of 500–1,000 mg/day in combination with methimazole can rapidly ameliorate severe hyperthyroidism and can also be used to prepare a hyperthyroid patient for early surgery.
Surgery Iodine elixirs: Up to 10 drops of saturated solution of potassium iodide [SSKI, 50 mg iodide per drop (0.05 mL)] daily for 7–10 days is used perioperatively to reduce gland vascularity.
Glucocorticoids inhibit peripheral T4 to T3 conversion and in patients with Graves’ hyperthyroidism reduce thyroid secretion.
Cholestyramine, given in a dose of 4 g four times daily with methimazole, lowers serum T4 and T3 concentrations more rapidly than methimazole alone.
Surgery Potassium perchlorate: It reduces uptake of iodine. It is more toxic and produces red cell aplasia. It is used only as temporary measure in iodine-induced thyrotoxicosis or when other therapy is not acceptable.
Surgery Lithium: Blocks thyroid hormone release, but its use has been limited by its toxicity.
Surgery Rituximab: May induce a sustained remission in patients with Graves’ disease and induces low TSH-receptor antibodies (TRAb) levels, but its cost and side effects limit its utility
Surgery Skeletal Health: Advised to ingest 1,200–1,500 mg elemental calcium daily through diet or supplements.

Approach for the hyperthyroidism is presented in Flowchart:

Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm:
Question 18. Write short essay/note on hyperthyroid crisis/thyrotoxic crisis/thyroid storm/thyroid crisis.
Answer:
It is a rare life-threatening medical emergency that develops as complication of thyrotoxicosis which is associated with a mortality of 10‒30%.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Causes of Hyperthyroid Crisis:
Thyroid storm can develop in patients with longstanding untreated hyperthyroidism (Graves’ disease, toxic multinodular goiter, and solitary toxic adenoma), it is often precipitated by an acute event.
- Severe infections in a patient with previously undetected or inadequately treated hyperthyroidism/thyrotoxicosis.
- Following surgery May develop following subtotal thyroidectomy in an ill-prepared patient or some other surgery in an undetected hyperthyroidism/thyrotoxicosis.
- Following radiotherapy May occur within a few days of 131I therapy in an inadequately prepared patient. This is because of a transient rise in serum thyroid hormone levels caused due to acute damage by irradiation.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Other precipitating factors:
- Cerebrovascular accidents
- Diabetic ketoacidosis
- Acute coronary syndrome
- Use of iodine contrast agent
- Acute iodine load
- Parturition
- Sudden withdrawal of antithyroid medications
- Stress
- Major trauma.
It is unclear why certain factors result in the development of thyroid storm. Hypotheses include a rapid rate of increase in serum thyroid hormone levels, increased responsiveness to catecholamines, or enhanced cellular responses to thyroid hormone. One study found that while the total T4 and T3 levels were similar to those seen in uncomplicated patients, the free T4 and free T3 concentrations were higher in patients with thyroid storm.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Clinical Features:
- The clinical manifestations are due to marked hypermetabolism and excessive adrenergic response.
- Hyperpyrexia from 104°F to 106°F is common and is associated with flushing and sweating.
- Marked tachycardia rates that can exceed 140 beats/min, often with atrial fibrillation and high pulse pressure, hypotension, occasionally congestive heart failure occurs. A fatal outcome is associated with heart failure and shock.
- Central nervous system symptoms include marked agitation, restlessness, delirium, psychosis, stupor, and coma.
- Gastrointestinal symptoms include nausea, vomiting, diarrhea, abdominal pain, and hepatic failure with jaundice.
- Physical examination may reveal goiter, ophthalmopathy (in the presence of Graves’ disease), lid lag, hand tremor, and warm and moist skin.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Laboratory Findings:
Low TSH and high free T4 and/or T3 concentrations Nonspecific laboratory findings may include—mild hyperglycemia secondary to a catecholamine-induced inhibition of insulin release and increased glycogenolysis, mild hypercalcemia due to hemoconcentration and enhanced bone resorption, abnormal liver function tests, leukocytosis, or leukopenia.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Treatment of Hyperthyroid Crisis:
Patients should be admitted in ICU, rehydrated, and given antibiotics if there is infection.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Control hyperthermia: Aggressively by external cooling. Acetaminophen should be used instead of aspirin since the latter can increase serum free T4 and T3 concentrations by interfering with their protein binding
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Propranolol: Either orally (60–80 mg every 4–6 hours) or intravenously (1–5 mg 4 times daily). It also blocks peripheral conversion of T4 to T3. The Japanese guidelines recommend esmolol over propranolol because of increased mortality in patients with congestive heart failure treated with propranolol. In patients with reactive airways disease, cardio-selective beta-blockers such as metoprolol or atenolol could be considered.
Propylthiouracil 200 mg orally every 4 hours (preferred as it decreases T4 to T3 conversion) or methimazole (20 mg orally every 4–6 hours) or carbimazole (inhibit the synthesis of new thyroid hormone) 15–30 mg stat followed by 15 mg TID. In an unconscious or uncooperative patient, carbimazole can be administered rectally with good effect.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Lugol’s iodine: 10 drops TID about 1 hour after the first dose of thionamide.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Sodium iopodate: 500 mg/day orally will restore serum T3 levels to normal within 48–72 hours. This is a radiographic contrast medium which inhibits the release of thyroid hormones and also reduces the conversion of T4 to T3. Hence, more effective than potassium iodide or Lugol’s solution.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm IV hydrocortisone: 100 mg every 8 hours.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Bile acid sequestrant, cholestyramine: 4 g orally QID to reduce enterohepatic recycling of thyroid hormones.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Benzodiazepines: For agitation.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Digoxin: To control cardiac failure and atrial fibrillation (AF).
Plasmapheresis has been tried when traditional therapy has not been successful, it removes cytokines, antibodies, and thyroid hormones from plasma.
Lithium has also been given to acutely block the release of thyroid hormone. However, its renal and neurologic toxicity limit its utility.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid StormHypothyroidism:
Describe the etiology, clinical features, diagnosis, and management of primary hypothyroidism/spontaneous atrophic hypothyroidism?
Hypothyroidism is a clinical syndrome resulting from a deficiency of thyroid hormones. It results in a generalized slowing down of metabolic processes.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Infants and children: Hypothyroidism results in marked slowing of growth and development with serious permanent consequences including mental retardation, when it occurs in infancy (cretinism).
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Adults: Hypothyroidism causes a generalized decrease in metabolism with slowed heart rate, diminished oxygen consumption, and deposition of glycosaminoglycans in intracellular spaces, particularly in skin and muscle, producing in extreme cases the clinical picture of myxedema.
Hyperthyroid Crisis/Thyrotoxic Crisis/Thyroid Storm Classifiation and Etiology:
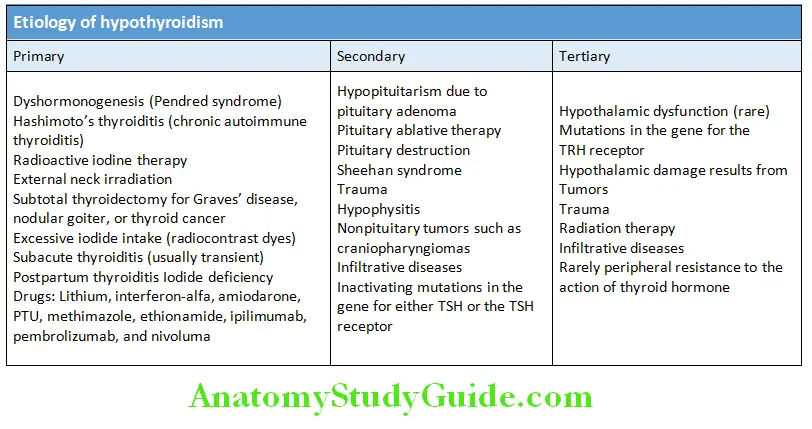
Question 19. Write short essay/note on causes of hypothyroidism and treatment.
Answer:
Primary hypothyroidism: Due intrinsic disorder of the thyroid gland. Accounts for over 95% of cases of hypothyroidism.
There are two degrees of primary hypothyroidism:
1. Subclinical hypothyroidism, defined as a high serum TSH concentration in the presence of normal serum free T4 and T3 concentrations.
2. Overt hypothyroidism, defined as a high serum TSH concentration in the presence of a low serum free T4 concentration.
Primary hypothyroidism Central hypothyroidism: It is rare and is caused due to failure of TSH and TRH production due to disease of anterior pituitary (secondary hypothyroidism) or hypothalamus (tertiary hypothyroidism).
Primary hypothyroidism Hashimoto’s thyroiditis (chronic autoimmune thyroiditis);
- Most common cause of primary hypothyroidism in iodine sufficient areas of the world.
- Organ-specific autoimmune disorder of thyroid characterized gradual thyroid failure with or without goiter formation by lymphoid infiltration of thyroid leading to apoptosis, fibrosis, and atrophy.
- The two major forms of Hashimoto’s thyroiditis are goitrous autoimmune thyroiditis and atrophic autoimmune thyroiditis (often called primary myxedema), with the common pathologic feature being lymphocytic infiltration, and follicular destruction.
- Patients have TRAb that block the effects of endogenous TSH, antibodies to TPO and thyroglobulin.
- The cause of Hashimoto’s thyroiditis is thought to be a combination of genetic susceptibility and environmental factors.
- Several mechanisms have been proposed for the pathogenesis of Hashimoto’s thyroiditis. These include molecular mimicry and bystander activation including the involvement of thyroid cell expression of HLAs and activation of thyroid cell apoptosis by a Fas ligand-Fas interaction.
- It may be observed in some patients of Graves’ disease treated with antithyroid drugs 10–20 years earlier.
- Patients have high-risk of developing other autoimmune disorders such as type 1 diabetes mellitus, pernicious anemia and Addison’s disease.
- Postpartum thyroiditis
- It is a destructive thyroiditis induced by an autoimmune mechanism within 1 year of parturition.
- It may produce transient hyperthyroidism alone, transient hypothyroidism alone or the two sequentially and then recovery.
- Thyroid biopsies in women with postpartum thyroiditis show lymphocytic infiltration, with occasional germinal centers, and disruption and collapse of thyroid follicles (lymphocytic thyroiditis).
- Laboratory assessment of thyroid function should be performed at 3 and 6 months postpartum in women at high-risk for developing postpartum thyroiditis, including women with type 1 diabetes mellitus, a history ofpostpartum thyroiditis after a previous pregnancy and history of high serum antithyroid peroxidase antibody concentrations prior to pregnancy

Hypothyroidism Clinical Features:
Question 20. Write short essay/note on clinical features of hypothyroidism.
Answer:
Depends on the duration and severity of the hypothyroidism.
Clinical Features Consequence of prolonged hypothyroidism:
- Infiltration of many body tissues by the mucopolysaccharides, hyaluronic acid and chondroitin sulfate.
- Infiltration of the dermis produces nonpitting edema (myxedema).
- The term myxedema refers to the accumulation of mucopolysaccharides in the subcutaneous tissues. It is most marked in the skin of the hands, feet, and eyelids.
Clinical Features Myxedema facies It is a peculiar facial appearance characterized by:
- Striking periorbital puffiness (due to myxedema)
- Scanty eyebrows
- Facial pallor (due to vasoconstriction and anemia), or a lemon-yellow tint to the skin (due to carotenemia produced by reduced conversion of carotene to vitamin A)
- Purplish lips and malar flush.
- Most cases of hypothyroidism are not clinically obvious and should be kept in mind when individuals complain of nonspecific symptoms such as tiredness, weight gain, depression or carpal tunnel syndrome.
- Many cases are diagnosed on biochemical screening.
Clinical Features Investigations of Primary Hypothyroidism:
Majority of hypothyroidism results from an intrinsic disorder of the thyroid gland (primary hypothyroidism).
Serum TSH: It is the investigation of choice. High TSH level confirms primary hypothyroidism.
Serum T4 levels: Low level confirms the hypothyroid state.

Thyroid and other organ-specific antibodies may be found.
Clinical Features Other abnormalities:
- Increased serum aspartate transferase from muscle and/or liver
- Increased serum lactate dehydrogenase (LDH) and creatine kinase (CK): With associated myopathy
- Hypercholesterolemia and hypertriglyceridemia
Clinical Features Hyponatremia: Due to an increase in ADH and impaired free water clearance.
Clinical Features Anemia: Usually normochromic and normocytic
Clinical Features Electrocardiogram (ECG): Demonstrates sinus bradycardia, low voltage QRS complexes, and ST segment and T wave abnormalities.
Chest radiograph: May reveal enlarged cardiac shadow.
Question 21. Write short essay/note on treatment of hypothyroidism (drug, dose, and duration of therapy).
Answer:
Treatment of Hypothyroidism:
Hypothyroidism is treated with T4.
Replacement therapy with levothyroxine sodium is given for life as a once daily dosage (1.6–2.3 µg/kg/day).
Treatment of Hypothyroidism Initial dose: It depends upon the severity of the deficiency as well as on the age and fitness of the patient.
- For young healthy patients—1.6 µg/kg/day.
- For older patients or those with coronary heart disease—25–50 µg/day
Treatment of Hypothyroidism Timing: Should be taken on an empty stomach with water, ideally an hour before breakfast.
The patient with symptomatic improvement should be re-evaluated with serum TSH measured in 4–6 weeks. If the TSH remains above the reference range, the dose of T4 can be increased by 12–25 µg/day in older patients and in younger patients, it can be increased by a higher dose. The patient will require a repeat TSH measurement in 6 weeks.
Treatment of Hypothyroidism For patients with heart disease: 12.5–25 µg/day and increase by 12.5–25 µg/day, if needed, at 6–8 weeks intervals. Few patients with ischemic heart disease may develop angina or worsen with therapy. They require β-blockers, vasodilators or coronary artery bypass graft (CABG) or angioplasty.
Dosage adjustments:
Age: In elderly start with half dose.
Severity and duration of hypothyroidism: Increase the dose in severe cases
Weight: 0.5 µg/kg/day increase up to 3.0 µg/kg/day
Malabsorption: Requires increased dose
Concomitant drug therapy: Thyroxine only to be taken on empty stomach
Pregnancy: 25–50% increase in dose, safe in lactating mother
Presence of cardiac disease: Start low dose or alternate day treatment.
Monitoring:
- Goal: It is to normalize TSH level regardless of cause of hypothyroidism and to restore T4 within the normal range.
- Adequacy of replacement: Assessed clinically and by thyroid function tests after 6 weeks on a steady dose.
- Complete suppression of TSH should be avoided because it may cause atrial fibrillation and osteoporosis.
- Lifelong therapy is needed.
Myxedema Coma:
Question 22. Write short essay on clinical features and management of myxedema coma and myxedema madness.
Answer:
- Myxedema coma is a very rare and life-threatening medical emergency with a high mortality rate that develops as a complication of severe hypothyroidism.
- The hallmarks of myxedema coma are decreased mentation and hypothermia.
- Other features are bradycardia, hyponatremia, hypoglycemia, hypotension, puffiness of the hands and face, puffy nose, swollen lips, enlarged tongue, hypoventilation, and features of the precipitating cause/illness.
- Epidemiology and precipitating factors:
- It can occur as the culmination of severe longstanding hypothyroidism with older women being most affected.
- It can be precipitated by an acute event in a poorly controlled hypothyroid patient, such as infection, myocardial infarction, hypothermia, surgery, drugs (amiodarone, β-blockers, diuretics, anesthetic agents, barbiturates, lithium, narcotics, and phenothiazines), cardiac failure, hypoxia, hyponatremia, and hypercapnia.
Myxedema Coma Warning signs: Presence of cool pale skin (due to hypothermia—body temperature may be as low as 25°C), absence of mild diastolic hypertension, and altered sensorium.
Laboratory findings:
- Serum free T4 is low, serum TSH is usually high but sometimes only slightly elevated.
- Serum cortisol, maybe low in patients with central hypothyroidism with associated hypopituitarism and secondary adrenal insufficiency.
- Serum creatine phosphokinase mostly markedly raised.
- Hyponatremia

Laboratory findings Myxedema Madness:
Patient presents with hypothermia and neuropsychiatric manifestations.
Laboratory findings Neuropsychiatric manifestations:
Laboratory findings Include:
- Depression is common in hypothyroidism.
- Rarely, elderly patient with severe hypothyroidism may become frankly demented, psychotic, confused, disoriented, sometimes with striking delusions and hallucinations. It may develop shortly after starting T4 replacement.
Laboratory findings Laboratory findings:
- Hypoglycemia and hyponatremia, increased CO2, decreased WBC count and Hct, increased CPK.
- Reduced ECG voltage, blood gases often reveal respiratory acidosis, hypoxia and hypercapnia.
EEG: Slow waves with decreased amplitude, triphasic waves may be present.
CSF analysis: Elevated protein levels (<100 mg/dL).
Sick Euthyroid Syndrome/Nonthyroid Illness State (NTIS):
Question 23. Write a short note on sick euthyroid syndrome.
Answer:
- Nonthyroidal illness influences thyroid hormone production and action at multiple levels, including H-P-T axis, thyroid hormone transport, and metabolism.
- Changes in thyroid function in the setting of systemic illness, surgery, or fasting not caused by primary thyroid or pituitary dysfunction are referred to as the nonthyroidal illness syndrome (also called “sick euthyroid syndrome”). Also called “low-T3 syndrome”, due to the most common abnormality, a decreased level of serum total T3.
- Measurement of serum T4 concentration can be normal, low or elevated.
- Serum TSH, is usually normal but can be influenced by nonthyroidal illness.
- Conditions associated with euthyroid sick syndrome include malnutrition, HIV, anorexia nervosa, trauma, myocardial infarction, chronic renal failure, diabetic ketoacidosis, cirrhosis, and sepsis.
- Treatment with thyroxine is not recommended.
Subclinical Thyroid Diseases:
Write a short note/essay on subclinical thyroid diseases.
Write a short note on subclinical hypothyroidism.
Subclinical Thyroid Diseases Subclinical Hypothyroidism
- Subclinical hypothyroidism is defined as an increased serum TSH in the presence of a normal serum free T4.
- Prevalent in women and elderly persons.
- Common causes of subclinical hypothyroidism.
Subclinical Thyroid Diseases Clinical consequences:
- Progression to overt hypothyroidism (2–4% yearly)
- Risk ofCV disease especially when TSH > 10 mU/L: Diastolic dysfunction, diastolic hypertension, increase in LDL-C, increased hsCRP, alteration in coagulation parameters, and endothelial dysfunction
Subclinical Thyroid Diseases Decreased fertility:
Subclinical Thyroid Diseases Nonalcoholic fatty liver disease (NAFLD):
- Neuropsychiatric manifestations and increased risk for Alzheimer disease.
Subclinical Thyroid Diseases Indication for treatment with levothyroxine:
- Patients with serum TSH > 10.0 mU/L
- Patients <65–70 years, asymptomatic with TSH ≥ 7 mU/L
- Patients <65–70 years, symptomatic with TSH from upper limit of normal to 6.9 mU/L.
- Patients who are infertile, ovulatory dysfunction or planning for pregnancy.
- Symptoms or signs of hypothyroidism
- Goiter
- High serum anti-TPO antibodies.
- High vascular risk including ischemic heart disease, diabetes, dyslipidemia.
Subclinical Hyperthyroidism:
Question 24. Write a short note on subclinical hyperthyroidism.
Answer:
- Defined as normal serum free T4 and T3 in the presence of subnormal TSH (<0.5 mU/L).
- Prevalence between 0.7 and 12.4%
- Natural course: 40–60% normalize. About 0.5‒8% progresses to overt hyperthyroidism.
- Effects of subclinical hyperthyroidism
- Cardiac effects of subclinical hyperthyroidism.
Subclinical Hyperthyroidism Noncardiac effects:
- Osteoporosis
- Decreased Bone mineral density
- Increased risk of fractures
- Muscle weakness
- Dementia, etc.
Thyroiditis:
Question 25. Write a short note on causes of thyroiditis.
Answer:
- It is a heterogeneous group of inflammatory disorders involving the thyroid gland.
- Etiologies range from autoimmune to infectious origins.
- The clinical course may be acute, subacute, or chronic.
Thyroiditis Causes of Thyroiditis
Thyroiditis Common causes of subclinical hypothyroidism.
- Iatrogenic
- Hashimoto’s thyroiditis
- Postpartum thyroiditis, painless thyroiditis
- Thyroid infiltration (amyloidosis, sarcoidosis, hemo chromatosis, etc.)
- Medications: Amiodarone, lithium, interferon, and sorafenib
- Partial thyroidectomy
- Head and neck radiation
Thyroiditis Causes of subclinical hyperthyroidism:
- Iatrogenic (exogenous subclinical hyperthyroidism)
- Autonomously functioning thyroid adenomas and multinodular goiters (endogenous subclinical hyperthyroidism)
- Graves’ disease (endogenous subclinical hyperthyroidism)
- Thyroiditis
- Euthyroid patients with Graves’ orbitopathy
- Early Graves’
- Graves’ disease in remission
- High hCG
Thyroiditis Cardiac effects of subclinical hyperthyroidism:
- Resting tachycardia
- Coronary heart disease
- Heart failure
- Atrial fibrillation
- Left ventricular hypertrophy
- Increase LV mass index
- Increase cardiac workload
- Diastolic dysfunction (impaired relaxation)
- Increased systolic function at rest
- Impaired systolic response to exercise
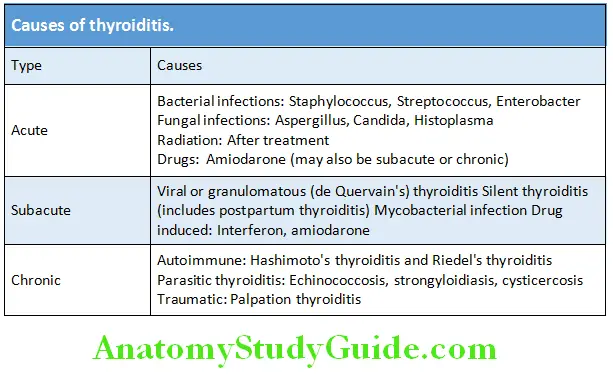
Subacute Thyroiditis (De Quervain’s Thyroiditis)
Question 26. Write a short note on subacute thyroiditis (de Quervain’s thyroiditis/granulomatous thyroiditis).
Answer:
- A spontaneously remitting, painful, subacute inflammatory disease of the thyroid characterized by transient inflammation of the thyroid gland.
- Most prevalent in the temperate zone.
- Strongly associated with HLA-B35
- Gender and age: Affect women more often than men (3 to 5:1). It is most common in young adults and middle age.
Subacute Thyroiditis Clinical Features
Subacute Thyroiditis Prodromal viral symptoms: These are often preceded by a viral infection (e.g., Coxsackie, mumps, adenovirus, measles) of upper respiratory tract. Prodromal symptoms include fever, malaise and pain in the neck with tachycardia, and local thyroid tenderness.
Anterior neck pain in the region of thyroid is the presenting symptom is majority of cases and occurs abruptly and may be sometimes unilateral.
- It may radiate to the ear, mandible, or occiput.
- Pain may shift to the contralateral lobe (creeping thyroiditis).
- Pain may be aggravated by moving the head, swallowing, or coughing.
Subacute Thyroiditis Functional impairment: Initially there is hyperthyroidism followed by euthyroidism and later followed by a period of hypothyroidism. Finally, full recovery occurs in 4–6 months. In about 5% of cases hypothyroidism may persist.
Subacute Thyroiditis Signs: Enlarged diffusely or asymmetrically and tender thyroid gland.
Subacute Thyroiditis Laboratory Findings:
- Mildly elevated serum free T4 and T3 and low serum TSH
- Erythrocyte sedimentation rate (ESR): Elevated (>50 mm/h may exceed 100 mm/h)
- CRP may be elevated
- High serum thyroglobulin
- Mild anemia
- Leukocyte counts: Normal or slightly elevated
- Serum IL-6 and Tg concentrations: Increased during the thyrotoxic phase.
- Thyroid radionuclide uptake is reduced or absent.
- Thyroid antibodies are transiently detectable at low titers in a minority of patients.
Subacute Thyroiditis Treatment:
Subacute Thyroiditis Mild cases: Salicylates (aspirin 2,600 mg daily) or NSAIDS (naproxen 500–1000mg daily in 2 divided doses or ibuprofen 1,200–3,200mg daily in 3–4 divided doses) relieve pain and tenderness
Subacute Thyroiditis Severe cases: Corticosteroids (prednisone 40 mg/day) have a more dramatic and rapid effect.
Symptoms of thyrotoxicosis should be managed with β-adrenergic blocking agents (propranolol 40–120 mg, propranolol LA 80 mg daily or 25–50 mg atenolol daily)
If TSH > 10 mU/L or associated with symptoms, 50–100 mg of levothyroxine for 6–8 weeks.
Hashimoto’s Thyroiditis:
Question 27. Write short essay/note on Hashimoto’s (autoimmune) thyroiditis.
Answer:
Organ-specific autoimmune disorder of thyroid characterized gradual thyroid failure with or without goiter formation by lymphoid infiltration of thyroid leading to apoptosis, fibrosis and atrophy with high titers of circulating:
Antithyroid peroxidase (TPO) antibody (TPOAb) in 95% patients.
Antithyroglobulin (Tg) antibody in 60–80% patients.
Most common cause of primary hypothyroidism in iodine sufficient areas of the world.
The two major forms of Hashimoto’s thyroiditis are goitrous autoimmune thyroiditis and atrophic autoimmune thyroiditis (often called primary myxedema).
Hashimoto’s Thyroiditis Age and gender: Most often diagnosed between 50 and 60 years of age, more frequent in women than in men with a sex ratio of approximately 7:1.
Association with other autoimmune diseases: Often associated with ulcerative colitis or type 1 diabetes mellitus, pernicious anemia and Addison’s disease.
Hashimoto’s Thyroiditis Pathology:
- Lymphocytic infiltration (lymphoid follicles with germinal center)
- Fibrosis
- Follicular cell hyperplasia
- Presence of oxyphil cells (Ashkenazy cells/Hürthle cells).
Hashimoto’s Thyroiditis Clinical Features:
- Most patients are asymptomatic.
- Some may have a feeling of tightness or fullness in the neck.
- Neck pain and tenderness are rare.
- Hypothyroidism, the most common cause of goitrous hypothyroidism. About 25% present with hypothyroidism and remaining are at a higher risk of developing in future years.
- Hashitoxicosis is seen in the acute phase.
- Chronic autoimmune thyroiditis is a component of type 2 autoimmune polyglandular syndrome
- Physical examination: Diffuse enlargement of thyroid with firm or rubbery consistency.
Hashimoto’s Thyroiditis Investigations:
Investigations and treatment of Hashimoto’s thyroiditis.
- Thyroid function tests: Show features of hypothyroidism
- Antithyroid peroxidase (TPO) antibody (TPOAb) in 95% patients
Antithyroglobulin (Tg) antibody in 60–80% patients - Antinuclear factor (ANF) may be found in patients below the age of 20 years
- Ultrasound of thyroid: Reduced echogenicity
- Fine-needle aspiration cytology (FNAC) of thyroid.
Hashimoto’s Thyroiditis Complication:
- An increased risk for thyroid lymphoma (rare).
Hashimoto’s Thyroiditis Treatment:
Levothyroxine (150–200 µg/day) is indicated for the treatment of hypothyroidism and it may produce shrinkage of goiter. The dose of thyroxine should be sufficient enough to suppress serum TSH to low but detectable levels.
Riedel’s Thyroiditis (Sclerosing Thyroiditis/Ligneous Thyroiditis/Invasive Fibrous Thyroiditis):
- Rare, chronic inflammatory disorder of unknown etiology.
- Characterized by dense fibrosis of the thyroid gland and adjacent perithyroidal tissues (parathyroid glands, the recurrent laryngeal nerve and trachea), and extra cervical areas (fibrous mediastinitis, retroperitoneal fibrosis, retroorbital fibrosis, sclerosing cholangitis, and pancreatitis).
- Women are four times more likely than men to be affected, and it most commonly occurs between the ages of 30 and 50 years.
Hashimoto’s Thyroiditis Clinical Features:
- Usually present with a long history of a painless, progressively increasing anterior neck mass.
- Most patients are euthyroid.
Hashimoto’s Thyroiditis Pressure symptoms:
- Dysphagia
- Cough
- Hoarseness
- Stridor
- Attacks of suffocation may be present.
- Local obstructive pneumonia
- Superior vena cava syndrome
Hashimoto’s Thyroiditis Physical examination:
- A stony-hard or woody thyroid mass which varies in size from small to very large. It may involve one or both lobes and is fixed to surrounding structures.
Hashimoto’s Thyroiditis Investigations:
Hashimoto’s Thyroiditis Thyroid function test: 25‒67% have subclinical or overt hypothyroidism.
Hashimoto’s Thyroiditis Thyroid antibodies: May be found in about 45% of patients.
Hashimoto’s Thyroiditis Serum calcium: May be low due to parathyroid invasion.
Hashimoto’s Thyroiditis Imaging:
- USG shows heterogeneous hypoechoic lesions that may infiltrate the perithyroid muscles
- Doppler shows absence of vascular flow in the Riedel’s regions.
- CT/MRI shows extent of fibrosis.
- Riedel’s thyroiditis is hypermetabolic on FDG-PET.
The differential diagnosis based on FNAC includes subacute thyroiditis, a fibrosing variant of Hashimoto’s thyroiditis, radiation-induced thyroiditis, and malignancy, specifically a paucicellular variant of anaplastic cancer.
Definitive diagnosis can be established pathologically only by open biopsy, since FNAC may be difficult to interpret due to insufficient thyroid epithelial cells but may reveal mononuclear cells and fibrous tissue.
Hashimoto’s Thyroiditis Treatment:
- If untreated, this condition may stabilize or regress.
- Glucocorticoids has reduced enlargement and induced softening of mass in a few patients; however, therapy is longterm and it recurs
when steroids are tapered. - Tamoxifen 10–20 mg twice daily
- Rituximab intravenous 375 mg/m 2 monthly for 3 months
- Mycophenolate mofetil 1 g twice daily
Hashimoto’s Thyroiditis Surgery: Indicated to relieve tracheal and esophageal compression or to rule out malignancy. Surgery should be limited to relieve the obstruction and extensive resection is not indicated to avoid injury to adjacent adhering structures.
Hashimoto’s Thyroiditis Radiation: Low dos in cases refractory to other treatment.
Hashimoto’s Thyroiditis Goiter:
Goiter is the enlargement of thyroid gland.
Hashimoto’s Thyroiditis Classifiation:

Hashimoto’s Thyroiditis Simple (Nontoxic) Goiter:
Diffuse nontoxic (simple) goiter is characterized by the diffuse enlargement of the thyroid gland without any nodularity.
Hashimoto’s Thyroiditis This includes: Simple hyperplastic goiter (colloid goiter)
Hashimoto’s Thyroiditis Causes:
- Physiological in pregnancy, puberty
- Iodine deficiency.
Hashimoto’s Thyroiditis Appearance: Large, smooth firm, and nontender goiter
Hashimoto’s Thyroiditis Effect: Euthyroid and pressure effect.
Hashimoto’s Thyroiditis Risk Factors for Malignancy in Goiter:
- Solitary thyroid nodules in patients >60 or <30 years of age
- Irradiation of the neck or face during infancy or teenage years
- Past history of surgery
- Presence of cervical lymphadenopathy
- Symptoms of pain or pressure (especially a change in voice)
- Male sex
- Large nodules (>3 or 4 cm)
- Growth of nodule
Diffuse Nontoxic (Simple) Goiter
Question 28. Write short essay/note on clinical features, investigations and treatment of goiter/multinodular goiter.
Answer:
Diffuse nontoxic (simple) goiter is characterized by the diffuse enlargement of the thyroid gland without any nodularity.

Diffuse Nontoxic Goiter Etiology:
Diffuse Nontoxic Goiter Types:
- Endemic
- Sporadic
Diffuse Nontoxic Goiter Endemic goiter:
This term is used when goiters are present in >10% of the population in a given region.
Diffuse Nontoxic Goiter The causes are:
Diffuse Nontoxic Goiter Deficiency of iodine: This may be due to low iodine in the soil, water and food.
Diffuse Nontoxic Goiter Consequences of iodine deficiency: Decreased synthesis of thyroid hormone and causes compensatory increase in TSH. This leads to follicular cell hypertrophy and hyperplasia and goitrous enlargement.
Diffuse Nontoxic Goiter Goitrogens: These are substances ingestion of which interferes with thyroid hormone synthesis. Goitrogenic substances include vegetables which belong to Brassicaceae (Cruciferae) family such as cabbage, cauliflower, Brussels sprouts, and turnips and cassava root which contain a thiocyanate that inhibits iodide transport within the thyroid. Consumption of this may worsen the concurrent iodine deficiency.
Diffuse Nontoxic Goiter Sporadic goiter:
Less frequent than endemic goiter
Age: Puberty or in young adult life
Sex: Female preponderance.
Causes: In most cases of sporadic goiter the cause is not known.
Diffuse Nontoxic Goiter Clinical Features:
Diffuse Nontoxic Goiter Endemic goiter:
- Diffuse enlargement of the thyroid with euthyroid state in the initial stage. This is called as simple goiter.
- But later they may develop hypothyroidism.
- In children and adults, endemic goiter may be present with features of hypothyroidism and mental retardation.
Diffuse Nontoxic Goiter Cretinism:
- Iodine deficiency may produce severe hormone-induced physiological damage to fetus and newborn and cause cretinism.
- Stunting
- Deaf-mutism
- Malformed limbs
- Spastic motor disorders
- Goiter
- Mental impairment.
Diffuse Nontoxic Goiter Investigations:
Serum inorganic iodide: Reduced
Urinary iodide excretion: Low < 50 µg/day.
Serum T4: Normal
Serum T3: May be normal or raised due to increased conversion of T4 to T3
TSH: May be normal or mildly raised.
Treatment:
Early stages: Supplementation with iodine.
Later may need suppressive therapy with T4.
Parathyroid Disorders
Calcium Homeostasis:
Question 29. Write a short note on calcium homeostasis and its importance.
Answer:
Calcium Homeostasis Distribution of Calcium:
- Calcium weight is 400 mg/kg in infants and 950 mg/kg in adults.
- Blood Ca++ level: 8.5–10.2 mg/dL. Usually 10mg/100 mL (so 500 mg total in plasma = 0.5 g).
- About 99% of total body calcium in the bone.
- Remaining 1% in intracellular fluid (ICF), extracellular fluid (ECF), and cell membranes.
Calcium Homeostasis It can be divided in three components:
- 45% ionized
- 40% bound to albumin
- 15% complex with anions (citrate and phosphate).

Calcium Homeostasis Importance of Ionized Calcium:
Ionized calcium (Ca ++) is physiologically important because:
- One of the major intracellular messengers
- Precise levels are necessary for muscle contraction (cardiac and skeletal)
- Responsible for exocytosis of secretory granules in neuronal synapses.
- Serves as second messenger in many cells.
- Necessary for blood clotting.
Calcium Homeostasis Regulation of Calcium Levels:
Calcium Homeostasis It occurs in three different organs namely:
Small intestine: Dietary Ca++ is absorbed in an active transcellular pathway in the duodenum and proximal jejunum. Paracellular calcium transport occurs throughout the length of the intestine.
Kidney: Ionized Ca++ is filtered through the nephron, and can be excreted in the urine.
Bone: Major storage site for Ca++.
Calcium Homeostasis Calcium cycling in bone tissue: Two processes that go on continuously and include bone formation and bone resorption.
- Calcium phosphate crystals are called “hydroxyapatite”. The surfaces of crystals can exchange Ca++ and phosphate ions with extracellular fluid.

Calcium Homeostasis Osteoblasts: Synthesize a collagen matrix that holds calcium phosphate in crystallized form. Once surrounded by bone, osteoblast becomes osteocyte.
Calcium Homeostasis Osteoclasts: They break down bone (removes Ca++ from bone). Change local pH, causing Ca++ and phosphate to dissolve from crystals into extracellular fluids.
Hyperparathyroidism:
Question 30. Write a short essay on clinical features, investigations, and management of primary hyperparathyroidism.
(or)
Write a short essay on classification of hyperparathyroidism.
Answer:
Classifiation and Causes of Hyperparathyroidism:

Clinical Features of Hyperparathyroidism:
The most common clinical presentation of primary hyperparathyroidism (>70%) is asymptomatic hypercalcemia detected by routine biochemical screening Classical symptoms of primary hyperparathyroidism are described by the adage “moans, bones, stones, abdominal groans”. However, nowadays only few patients present in this way.
Mobans: Psychiatric manifestations—lethargy, fatigue, depression, memory loss, psychoses, neuroses, paranoia, confusion, stupor, and coma
Bones: Arthritis, osteomalacia, and osteitis
Stones: Renal stones, uremia, polydipsia, and polyuria
Abdominal groans: Constipation, nausea, vomiting, peptic ulcers, indigestion, and pancreatitis
Nonspecific symptoms: About 50% of patients are asymptomatic while others have nonspecific symptoms.
These include:
Anorexia, nausea, vomiting, constipation, weakness, fatigue, lassitude, tiredness, generalized aches, weight loss, pain, drowsiness, poor concentration, memory loss, and depression.
Manifestations of hyperparathyroidism: Involve primarily the kidneys and the skeletal system.
Renal manifestations: Due either to deposition of calcium in the renal parenchyma or to recurrent nephrolithiasis.
- Recurrent renal calculi (usually composed of either calcium oxalate or calcium phosphate)
- Nephrocalcinosis: Deposition of calcium salts in the renal parenchyma.
- Polyuria and polydipsia
- Loss of renal function with uremia, hypokalemia, hyperuricemia, hyperchloremic acidosis, and dilute urine.
Hyperparathyroidism Skeletal manifestations:
- Bone pain, osteopenia, osteoporosis, fractures, and deformity due to osteitis fibrosa cystica (10–25% of patients)
- Localized bone swelling/brown tumor (e.g., mandible).
Hyperparathyroidism Other manifestations:
- Hypertension is a common feature.
- Calcification of cornea (observed by slit-lamp examination), arterial walls, and soft tissues of hand.
- Peptic ulcers
- Myopathy
A family history of hypercalcemia or primary hyperparathyroidism secondary to a parathyroid adenoma raises the possibility of multiple endocrine neoplasia (MEN) syndrome. Features of multiple endocrine neoplasia syndromes are presented in Box.
Hyperparathyroidism Investigations:
Hyperparathyroidism Biochemical investigations: Estimation of several fasting serum calcium and phosphate samples should be done.
Hyperparathyroidism Hallmark of primary hyperparathyroidism:
- Hypercalcemia and hypophosphatemia with detectable or elevated intact PTH levels during hypercalcemia. When this combination is present in an asymptomatic patient then further investigation is usually unnecessary. Established hypercalcemia in more than one serum measurement accompanied by elevated immunoreactive PTH (iPTH) is characteristic.
- Serum concentrations of 1,25 dihydroxy vitamin D may therefore be at upper limits of normal or elevated.
Hyperparathyroidism Correction of serum calcium concentrations: It should be
corrected to the prevailing serum albumin concentration.
- Calcium level is corrected for low albumin levels by adding 0.8 mg/dL to the total serum calcium level for every 1.0 g/dL by which the serum albumin concentration is lower than 4 g/dL.
- May be associated with mild hyperchloremic acidosis.
- Calcium/creatinine (Ca/Cr) clearance ratio [24-hour urine Ca x serum Cr] ÷ [serum Ca x 24-hour urine Cr]
Hyperparathyroidism Urine investigations:
Hypercalciuria (24 hours urinary calcium) (>200‒300 mg/24 hours): Observed in ~30% of patients.
Increased markers of bone resorption: These include urinary pyridinoline, deoxypyridinoline, and N-telopeptide of collagen.
Hyperparathyroidism ECG findings:
- Shortened QT interval
- Rarely cardiac arrhythmias
- Rarely ST-segment elevation
Hyperparathyroidism Radiological abnormalities:
- Most sensitive and specific radiologic finding of osteitis fibrosa cystica is subperiosteal resorption of cortical bone, best seen in high-resolution films of the phalanges.
- A similar process in the skull leads to a salt-and-pepper appearance.
- Bone cysts or brown tumors may be evident as osteolytic lesions.
- The other important skeletal consequence of hyperparathyroidism is osteoporosis. Unlike other osteoporotic disorders, hyperparathyroidism often results in the preferential loss of cortical bone.
- Dental films may disclose loss of the lamina dura of the teeth, but this is a nonspecific finding also seen in periodontal disease.
Hyperparathyroidism Nephrocalcinosis: Appears as scattered opacities within the renal outline.
Hyperparathyroidism Soft tissue calcification: For example, calcification of arterial wall.
Dual-energy X-ray absorptiometry (DEXA) and CT scan: Reveal reduced bone density.
Hyperparathyroidism Investigations for localization of the tumor: Parathyroid imaging is generally indicated only for patients who have undergone previous parathyroid surgery.
Hyperparathyroidism Investigations to localize the tumor include:
High-resolution ultrasonography, CT scanning and subtraction imaging and scintigraphy with technetium 99m sestamibi.
Selective neck vein catheterization with PTH estimation.
Hyperparathyroidism Features of multiple endocrine neoplasia (MEN) syndromes.
Hyperparathyroidism MEN 1 (Wermer’s syndrome):
- Parathyroid hyperplasia (very common)
- Pancreatic tumors (benign or malignant)
- Gastrinoma
- Insulinoma
- Glucagonoma
- VIPoma
- Pituitary tumor
- Growth hormone-secreting
- Prolactin-secreting
- Adrenocorticotropic hormone (ACTH)-secreting
- Other tumors: Lipomas, carcinoids, adrenal, and thyroid adenomas
Hyperparathyroidism MEN 2A (Sipple syndrome):
- Medullary carcinoma of the thyroid
- Pheochromocytoma (benign or malignant)
- Parathyroid hyperplasia
Hyperparathyroidism MEN 2B:
- Medullary carcinoma of the thyroid
- Pheochromocytoma
- Mucosal neuromas and ganglioneuromas
- Marfanoid habitus
- Hyperparathyroidism (very rare)


Hyperparathyroidism Treatment:
Hyperparathyroidism Primary hyperparathyroidism:
Patients with symptomatic primary hyperparathyroidism should undergo parathyroid surgery which is the only definitive therapy.
Guidelines for surgery in asymptomatic primary hyperparathyroidism: Any ONE of the following criteria:
Hyperparathyroidism Serum calcium: >1 mg/dL above the upper limit of normal.
Hyperparathyroidism BMD by DXA: T-score <–2.5 at lumbar spine, total hip, femoral neck, or distal one-third radius.
- Vertebral fracture by radiograph, CT, MRI, or VFA.
- Creatinine clearance <60 mL/min
- 24-hour urine for calcium >400 mg/day and increased stone risk by biochemical stone risk analysis. 74 Endocrinology
- Presence of nephrolithiasis or nephron-calcinosis by radiograph, ultrasound, or CT.
- Age < 50 years
- For patients who are unable to have surgery and whose primary indication for surgery is symptomatic and/or severe hypercalcemia,
- Cinacalcet is preferred over bisphosphonates.
- Cinacalcet: In primary hyperparathyroidism, initially: Oral, 30 mg twice daily; increase dose incrementally every 2–4 weeks (to 60 mg twice daily, 90 mg twice daily, and 90 mg 3 or 4 times daily) as necessary to normalize serum calcium levels.
Hyperparathyroidism Secondary hyperparathyroidism:
- Treatment of chronic kidney disease (CKD)
- According to Kidney Disease Outcomes Quality Initiative (KDOQI) guidelines, in all dialysis patients, serum phosphate should be
maintained between 3.5–5.5 mg/dL, serum calcium should be maintained <9.5 mg/dL and PTH values should be maintained less
than two to nine times the upper limit for the PTH assay. - Treatment options for increased PTH include calcimimetics, calcitriol, or synthetic vitamin D analogs. In a recent update, oral
calcitriol is comparable to IV vitamin D analog for hemodialysis patients with secondary hyperparathyroidism.
Hyperparathyroidism Cinacalcet: In secondary hyperparathyroidism, initially orally: 30 mg once daily; increase dose incrementally every 2 to 4 weeks as necessary to maintain intact parathyroid hormone (iPTH) level between 150 and 300 pg/mL. May be used alone or in combination with vitamin D and/or phosphate binders.
Hyperparathyroidism Tertiary hyperparathyroidism: Parathyroidectomy
Hyperparathyroidism Refractory Hyperparathyroidism:
- Defined as persistent and progressive elevations of serum PTH, that cannot be lowered to levels < 600 pg/mL despite treatment with vitamin D derivatives and cinacalcet and without causing significant hyperphosphatemia or hypercalcemia.
- Patients with severe disease may require parathyroidectomy.
Hypercalcemia
Question 31. Write short essay/note on hypercalcemia.
Answer:
- Normal serum calcium level is 8–10 mg/dL (2.0–2.5 mmol/L)
- Normal ionized calcium levels are 4–5.6 mg/dL (1–1.4 mmol/L).
- Raised calcium level is known as hypercalcemia.
- Hypercalcemia is considered as mild if the total serum calcium level is between 10.5 and 12 mg/dL (2.6–3 mmol/L), moderate 12–14 mg/dL and severe when the level is above 14 mg/dL.
- Hypercalcemia is one of the most common biochemical abnormalities. It is often detected incidentally during routine biochemical investigation in asymptomatic patients. However, it can present with chronic symptoms and occasionally as an acute emergency.
- In patients with hypoalbuminemia or hyperalbuminemia, the measured calcium concentration should be corrected in view of the albumin abnormality.
Causes of Hypercalcemia:
Write short essay/note on causes of hypercalcemia

Clinical Features:
Question 32. Write a short essay/note on the management of hypercalcemia.
Answer:
Clinical Features Management: Treatment of acute hypercalcemia:
Adequate rehydration and volume expansion are essential, usually at least 4–6 L of 0.9% saline on day 1, and 3–4 L for several days thereafter.
Diuretics after correction of volume (not recommended in the present day due to potential complications and the availability of drugs that inhibit bone resorption, which is primarily responsible for the hypercalcemia). For example, furosemide 40–160 mg/day or ethacrynic acid 50–200 mg/day. Forced diuresis with 4–6 L of intravenous fluid/day and furosemide 2 hourly.
Sodium, potassium, and magnesium should be supplemented. Saline decreases concomitant reabsorption of sodium and calcium in
both the proximal and distal renal tubules, and enhances urinary excretion of calcium.
Intravenous bisphosphonate is the treatment of choice for hypercalcemia of malignancy or of undiagnosed cause. Concurrent administration of zoledronic acid 4 mg IV over 15 minutes or pamidronate 60–90 mg over 2 hours.
Calcitonin (4 IU/kg IV 6-hourly), tachyphylaxis develops after 24–48 hours. Prednisolone (20–40 mg daily) is effective in few instances (e.g., in myeloma, lymphoma, sarcoidosis, and vitamin D excess). Oral phosphate (sodium cellulose phosphate) 5 g three times daily.
Denosumab: For hypercalcemia refractory to ZA or in patients in whom bisphosphonates are contraindicated due to severe renal impairment. Starting dose 120 mg every 4 weeks.
Cinacalcet: Reduces the serum calcium in patients with severe hypercalcemia due to parathyroid carcinoma and in hemodialysis patients with an elevated calcium-phosphorous product and secondary hyperparathyroidism.
Dialysis: Treatment of last resort, HD with little or no calcium in the dialysis fluid.
Hypoparathyroidism
Question 33. Write a short note on causes and general management of hypoparathyroidism.
Answer:
Deficient secretion of PTH which manifests itself biochemically by:
- Hypocalcemia
- Hyperphosphatemia
- Diminished or absent circulating iPTH and
- Clinically the symptoms of neuromuscular hyperactivity.
Hypoparathyroidism Causes of Hypoparathyroidism:
Surgical hypoparathyroidism: It is the most common. It may be due to the removal of the parathyroid glands or due to interruption of blood supply to the parathyroid glands. Postparathyroidectomy (hungry bone syndrome)
Hypoparathyroidism Idiopathic hypoparathyroidism:
- Occurs at an early age (genetic origin) with autosomal recessive mode of transmission “multiple endocrine deficiency– autoimmune-candidiasis (MEDAC) syndrome”.
- “Juvenile familial endocrinopathy”—“Hypoparathyroidism—Addison’s disease—mucocutaneous candidiasis (HAM) syndrome”
- Circulating antibodies for the parathyroid glands and the adrenals are frequently present.
Hypoparathyroidism Other associated disease: Pernicious anemia, ovarian failure, autoimmune thyroiditis, and diabetes mellitus.
Infantile hypoparathyroidism:
Hypoparathyroidism Infiltrative diseases: Granulomatous, iron overload, and metastases
Hypoparathyroidism Radiation-induced destruction:
Hypoparathyroidism Human immunodeficiency virus (HIV):
Pseudohypoparathyroidism (resistance to PTH)
Functional hypoparathyroidism: In patients who has chronic hypomagnesemia of various causes. Magnesium is necessary for the PTH release from the glands and also for the peripheral action of the PTH.
DiGeorge syndrome is a familial condition where the hypoparathyroidism is associated with intellectual impairment, cataracts and calcified basal ganglia, and occasionally with specific autoimmune disease.
Hypoparathyroidism Malabsorption syndrome: Presumably secondary to decreased calcium level and may lead to steatorrhea with longstanding untreated disease.
Hypoparathyroidism Clinical Features:
Hypoparathyroidism Acute:
- Neuromuscular irritability (Tetany)
- Paresthesia (perioral and extremities)
- Muscle twitching
- Carpopedal spasm
- Trousseau’s sign
- Chvostek’s sign
- Seizures
- Laryngospasm
- Bronchospasm
- Prolonged QT interval
- Hypotension, heart failure, and arrhythmia
- Papilledema
Hypoparathyroidism Chronic:
- Ectopic calcification (basal ganglia)
- Extrapyramidal signs
- Parkinsonism
- Dementia
- Subcapsular cataracts
- Abnormal dentition
- Dry skin
Hypoparathyroidism Treatment:
To effectively treat hypocalcemia in patients with magnesium deficiency, hypomagnesemia should be corrected first.
Hypoparathyroidism Acute cases:
Hypoparathyroidism Symptomatic: IV calcium (initially, 10 mL ampule of 10% calcium gluconate in 50 mL of 5% dextrose infused over 10–20 minutes, followed by an intravenous infusion of calcium gluconate) with oral calcitriol (0.5 µg two times daily).
Hypoparathyroidism Asymptomatic: IV calcium if serum corrected calcium is ≤7.5 mg/dL. Oral calcium (1–4 g of calcium carbonate daily in divided doses) should be initiated as soon as the patient is able to take supplements orally.
Hypoparathyroidism Chronic cases:
Calcium: Calcium carbonate or calcium citrate 1,000–2,000 mg daily in divided doses.
Hypoparathyroidism Vitamin D:
Hypoparathyroidism Calcitriol (drug of choice): Initial: 0.25–0.5 µg daily and maintenance: 0.5–2 µg daily.
Hypoparathyroidism Alfacacidol: Initial 0.25 µg daily and maintenance 0.5–1 µg daily Dihydrotachysterol: 0.2–1.2 mg daily.
Hypoparathyroidism Hydrochlorothiazide: If needed for hypercalciuria, 12.5–50 mg daily Second-line therapy by subcutaneous recombinant human PTH

Hypoparathyroidism Tetany
It is characterized by neuromuscular irritability manifested clinically by sensory, muscular dysfunction and autonomic manifestations.
Causes of Tetany:
Question 34. Write a short essay/note on causes of tetany/hypocalcemia.
Answer:
- Tetany is caused due to hypocalcemia or alkalosis or hypomagnesemia.
- Respiratory alkalosis alone (e.g., hyperventilation) can cause tetany, even in the absence of underlying hypocalcemia.
- Tetany is unusual among patients with chronic renal failure and hypocalcemia (occasionally severe) because of the protective effect of concurrent metabolic acidosis.
- Occurs when there is acute fall in serum ionized calcium to <4.3 mg/dL and total calcium concentration to 7‒7.5 mg/dL.
Clinical Features:
Question 35. Write a short essay/note on clinical features of tetany.
Answer:
- Symptoms begin with perioral and acral paresthesia leading to hyperventilation—respiratory alkalosis—exacerbation of paresthesia.
- Motor symptoms: Stiffness, clumsiness, myalgia, muscle spasms, cramps, carpal spasm, spasm of the respiratory muscles, and laryngismus stridulus.
- Autonomic manifestations are diaphoresis, bronchospasm, and biliary colic.


Question 36. Write a short note on Chvostek’s sign and Trousseau’s sign
Answer:
The classic physical findings in patients with neuromuscular irritability due to latent tetany are:
Chvostek’s sign: It is elicited by tapping the skin over the facial nerve in front of the external auditory meatus. It causes an ipsilateral contraction of the facial muscles, but up to 10% of normal population has a positive test.
Trousseau’s sign: Inflate BP cuff on arm to 20 mm Hg > systolic BP for 3 minutes and watch for carpopedal spasm [flexion at the wrist, flexion at the metatarsophalangeal (MP) joints, extension of the interphalangeal (IP) joints adduction thumbs/fingers]. It may also be induced by voluntary hyperventilation for 1‒2 minutes after release of the cuff.
Write a short essay/note on management of tetany.
Treatment:
Treatment Control of tetany:
Treatment Calcium gluconate: Slow intravenous (over 10 minutes) injection of 20 mL of 10% solution of calcium gluconate is rapidly effective in controlling the tetany.
Treatment Magnesium: If tetany is not relieved by above treatment, administration of magnesium may be necessary.
Treatment Correction of alkalosis
- If alkalosis is due to persistent vomiting, treat with intravenous isotonic saline and potassium.
- In alkalosis is due to alkali excess, it should be withdrawn. If needed, ammonium chloride 2 g 4 hourly orally will control tetany.
Treatment Hysterical hyperventilation: It may be controlled by rebreathing expired air from a suitable bag or inhalation of 5% carbon dioxide in oxygen.
Treatment Pseudohypoparathyroidism (PHP):
- PHP is an inherited disorder of target-organ unresponsiveness to PTH.
- It mimics hormone-deficient forms of hypoparathyroidism, with hypocalcemia and hyperphosphatemia, but the PTH level is elevated.
Treatment Clinical Features:
- PHP type IB is a disorder of isolated resistance to PTH, which presents with the biochemical features of hypocalcemia, hyperphosphatemia, and secondary hyperparathyroidism.
- PHP type IA has, in addition to these biochemical features, a characteristic somatic phenotype known as Albright’s hereditary osteodystrophy (AHO). This consists of short stature, round face, short neck, brachydactyly, shortened metatarsals, subcutaneous ossifications, and mental retardation. Due to shortening of the metacarpal bones most often the fourth and fifth metacarpals—affected digits have a dimple, instead of a knuckle, when a fist is made.
- Certain individuals in families with PHP inherit the somatic phenotype of AHO without any disorder of calcium metabolism; this state is called pseudopseudohypoparathyroidism or PPHP.

Adrenal Gland Disorders
Hormones Secreted by the Adrenal Gland:
Cushing’s Syndrome:
Question 37. Write a short essay on causes, clinical features, investigations, and management of Cushing’s syndrome.
Answer:
- Cushing’s syndrome is the term used to describe the clinical state of increased free circulating glucocorticoid.
- It occurs most often following the therapeutic administration of synthetic steroids or ACTH.
Question 38. Write a short essay on Cushing’s disease.
Answer:
- Cushing ’s disease results from corticosteroid excess due to pituitary dependent bilateral adrenal hyperplasia.
- The pituitary tumors producing Cushing’s disease are usually microadenomas (<10 mm in size) which usually do not cause symptoms by local mass effect.
- It usually develops sporadically but may be a component of multiple endocrine neoplasia type 1.
- All the spontaneous forms of the syndrome are rare.
Cushing’s Syndrome Causes of Cushing’s Syndrome:
Cushing’s Syndrome Common causes of ectopic ACTH secretion:
A Cushingoid appearance can be caused by excess alcohol consumption (pseudo-Cushing’s syndrome)—the pathophysiology is poorly understood.
Clinical Features:
Question 39. Write a short essay on major signs of Cushing’s syndrome.
Answer:
- Systemic fungal infections and tinea versicolor may develop in untreated patients.
- Higher risk of coronary artery disease and venous thrombosis.
- The predominant clinical features of Cushing’s syndrome are those of glucocorticoid excess.
- Skin pigmentation occurs only with ACTH-dependent causes.
- Impaired glucose tolerance or frank diabetes is common, especially in the ectopic ACTH syndrome.
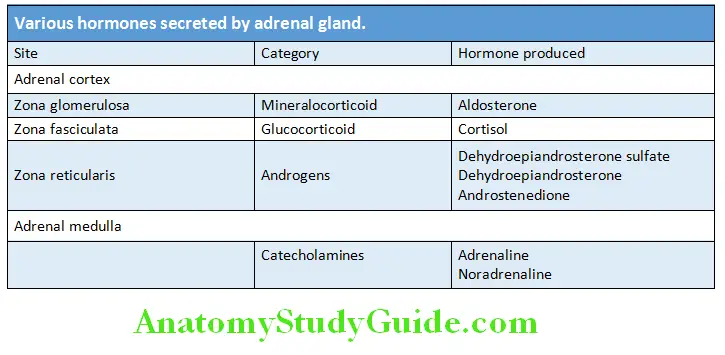

Clinical Features Mnemonic for Cushing’s syndrome:
C–Central obesity, Collagen fiber weakness, and Comedones (acne)
U–Urinary free cortisol increased along with glucose
S–Striae and Suppressed immunity
H–Hypercortisolism, Hypertension, Hyperglycemia, and Hypercholesterolemia
I-Iatrogenic due to administration of corticosteroids
N–Noniatrogenic and Neoplasms
G–Glucose intolerance and Growth retardation



- Hypokalemia due to the mineralocorticoid activity of cortisol is common with ectopic ACTH secretion
- Proximal muscle weakness, sleep apnea, osteoporosis, and hypertension are common features of the disease.
Clinical Features Features of Cushing’s syndrome due to ectopic ACTH secretion:
- Impaired glucose tolerance due to gluconeogenesis
- Hypokalemic alkalosis due to the mineralocorticoid activity of cortisol
- Skin pigmentation
Clinical Features Investigations in Cushing’s syndrome:
Clinical Features There are two phases of the investigation:
- Confirmation of the presence or absence of Cushing’s syndrome.
- Differential diagnosis of its cause (e.g., pituitary, adrenal or ectopic).
- Most obese, hirsute, hypertensive patients do not have Cushing’s syndrome.
- Some cases of mild Cushing’s have relatively subtle clinical signs.
- Confirmation rests on demonstrating inappropriate cortisol secretion, not suppressed by exogenous glucocorticoids.
- Random cortisol measurements are of no value.
Confirmatory tests to establish the presence of Cushing’s syndrome (2008 Endocrine Society Clinical Guidelines)
Clinical Features 48-hour low-dose dexamethasone test: Normal individuals suppress plasma cortisol to <50 nmol/L. Patients with Cushing’s syndrome fail to show complete suppression of plasma cortisol levels. This test is highly sensitive (>97%).
Clinical Features 24-hour urinary free cortisol measurements: This is simple, but less reliable—repeatedly normal values (corrected for body mass) render the diagnosis most unlikely, but some patients with Cushing’s have normal values on some collections (approximately 10%).
Clinical Features Plasma cortisol levels: In normal individuals, measurement of plasma cortisol levels at 8 AM and 12 midnight will show the lowest levels at midnight. This circadian rhythm is lost in Cushing’s syndrome and the cortisol levels remain the same throughout the day. A midnight level below 1.8 µg/dL is normal, and has a high sensitivity for excluding Cushing’s syndrome. However, it has a low specificity.
Clinical Features Overnight 1 mg dexamethasone suppression test:
Clinical Features Low dose dexamethasone suppression test:
- Late-night salivary cortisol (two measurements): It may be used as a screening test for Cushing’s syndrome.
- Concentration of cortisol in saliva is highly correlated with free plasma cortisol, irrespective of salivary flow rates and stable at room temperature for 1 week.
Clinical Features Tests that establish the cause of Cushing’s syndrome:
- The first step in the evaluation is to distinguish whether hypercortisolism is ACTH dependent or ACTH independent (primary adrenal disease) by measuring ACTH, best performed by two site immunoradiometric assay (IRMA). Plasma ACTH level at 8 AM
- Imaging for ACTH Independent conditions (primary adrenal disease): The next procedure after ruling out ACTH dependency is CT of the adrenal glands to look for adrenal mass.
- In patients with ACTH dependent Cushing’s, the final stage of the diagnostic evaluation is to determine the source of ACTH by

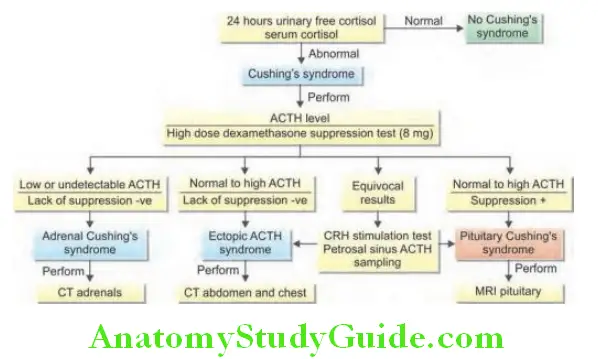
Clinical Features High-dose dexamethasone suppression test:
- 8 mg dexamethasone given orally at 11 PM, blood sample drawn at 8 AM the next day for serum cortisol
- Usually <5 µg/dL in most patients with Cushing’s disease.
Clinical Features CRH stimulation test: Patients with Cushing’s have ACTH and cortisol increases within 45 minutes after the IV administration of CRH.
- Increased in pituitary dependent disease
- Unchanged in ectopic ACTH syndrome and tumors of adrenal gland.

Clinical Features Vasopressin stimulation test: Stimulates ACTH in most patients with Cushing’s disease.
Clinical Features Petrosal venous sinus catheterization: ACTH is measured in petrosal and peripheral venous plasma before and within 10 minutes after administration of CRH.
Plasma potassium levels:
- Normal in pituitary dependant disease and tumors of adrenal gland.
- Low (3.5 mmol/L) in ectopic ACTH syndrome.
Clinical Features Other investigations:
Clinical Features Biochemical investigations: Blood glucose, cholesterol, and LDL may be raised.
Clinical Features Radiological investigations:
- Plain radiograph of the skull. Radiograph of chest to detect lung cancer.
- CT scan of anterior mediastinum, upper abdomen and pancreas to rule out tumors.
- CT/MRI head and MRI abdomen.
Clinical Features Management:
Clinical Features Exogenous Cushing’s syndrome: Taper and withdraw the glucocorticoid.
Clinical Features Cushing’s disease:
Clinical Features Treatment of choice: Trans-sphenoidal adenomectomy when there is clear circumscribed microadenoma. In the remaining patients subtotal resection of anterior pituitary.
Clinical Features Radiotherapy and radiosurgery: For recurrent or residual ACTH-secreting tumors. External pituitary irradiation alone is slow-acting and useful in only 50–60% of cases.
Clinical Features Medical therapy to reduce ACTH: Needed when surgery is delayed, contraindicated, or unsuccessful. “Block and replace strategy” with Ketoconazole 200 mg TID and increased to 400 mg TID. If ketoconazole is unsuccessful in controlling cortisol secretion, it should be maintained at a total dose of 1,200 mg/day and metyrapone or mitotane (adrenolytic) should be added.
Pituitary targeted therapies have been tried. Only the somatostatin analog, pasireotide, and the dopamine agonist, cabergoline (and the combination of the two), have shown potential benefit.
Clinical Features Bilateral adrenalectomy:
- Bilateral total adrenalectomy with lifelong daily glucocorticoid and mineralocorticoid replacement therapy is the final definitive cure.
- Done if the diagnosis is uncertain.
- Followed by pituitary irradiation with Yttrium-90 implantation to prevent the development of Nelson’s syndrome.
- Prednisolone and fludrocortisones should be given postoperatively for a variable length of time.
Clinical Features Adrenal tumors:
Clinical Features Surgical resection:
Adrenal adenomas: Surgical removal. Postoperatively, prednisolone is given as a replacement therapy till the contralateral adrenal, hypothalamus, and pituitary recovers.
Adrenal carcinomas: Surgical resection, irradiation of tumor bed, and administration of adrenolytic drug (mitotane).
Medical adrenalectomy: Medications that inhibit steroidogenesis include ketoconazole, metyrapone, mitotane, aminoglutethimide,
and octreotide.
Clinical Features Ectopic ACTH syndrome:
- Surgical excision of benign tumors (e.g., bronchial carcinoid).
- Radiotherapy and chemotherapy: For malignant tumors.
Recurrent tumors may be treated with ketoconazole, metyrapone, or aminoglutethimide.
Nelson’s Syndrome
Question 40. Write a short note on Nelson’s syndrome.
Answer:
- Nelson’s syndrome is increased pigmentation (high levels of ACTH) associated with an enlarging pituitary tumor postbilateral adrenalectomy.
- It occurs in about 20% of cases after bilateral adrenalectomy for Cushing’s disease.
- The syndrome is rare now that adrenalectomy is an uncommon primary treatment, and its incidence may be reduced by pituitary radiotherapy soon after adrenalectomy.
Treatment: Nelson’s adenoma may be treated by pituitary surgery and/or radiotherapy.
Hyperaldosteronism/Conn’s syndrome
Question 41. Write short essay on:
Answer:
Etiology, clinical features, diagnosis, and management of primary hyperaldosteronism Conn’s syndrome.
Excessive production of the aldosterone hormone is called hyperaldosteronism.
Hyperaldosteronism/Conn’s syndrome Classifiation:
Primary hyperaldosteronism: Develops due to an abnormality in the zona glomerulosa of the adrenal gland.
Secondary hyperaldosteronism: Develops due to the stimulation of aldosterone secretion by angiotensin II following activation of renin-angiotensin system.
Hyperaldosteronism/Conn’s syndrome Primary hyperaldosteronism:
Hyperaldosteronism/Conn’s syndrome Etiology:
- Adrenal adenoma (Conn’s syndrome)
- Bilateral hyperplasia of zona glomerulosa/bilateral adrenal hyperplasia
- Idiopathic
ACTH dependent (glucocorticoid-responsive or dexamethasone-suppressible): This is characterized by secretion of aldosterone under ACTH control. Therefore, treatment consists of administration of glucocorticoids to suppress release of ACTH.
Hyperaldosteronism/Conn’s syndrome Consequences:
Excess secretion of aldosterone results in sodium retention, potassium loss, and metabolic alkalosis.
Hyperaldosteronism/Conn’s syndrome Clinical features:
Hypertension: Most important clinical consequence of hyperaldosteronism.
- Tetany due to metabolic alkalosis
- Muscle weakness due to hypokalemia.
- Polyuria and polydipsia due to nephrogenic DI.
Hyperaldosteronism/Conn’s syndrome Investigations:
Hyperaldosteronism/Conn’s syndrome Investigation for diagnosis:
Hypokalemia: However, normal serum potassium does not exclude the diagnosis.
Urinary potassium loss: Levels > 30 mEq/day during hypokalemia are inappropriate.
Plasma aldosterone concentration (PAC): Elevated (>10 ng/dL) and is not suppressed with 0.9% saline infusion (2 L over 4 hours, 8 AM to 12 PM, ideally when the patient is seated) or fludrocortisone administration.
Suppressed plasma renin activity (PRA) or immunoreactivity.
PAC: PRA ratio [Plasma aldosterone: renin ratio (ARR)]: Used as the screening test for primary hyperaldosteronism.
A level above 20 is abnormal when plasma aldosterone is measured in ng/dL and PRA is measured in ng/mL/min.
Confirmatory aldosterone suppression test:
Oral salt loading test: After controlling hypertension and hypokalemia, a high sodium diet (5,000 g sodium diet) should be given for 3 days. On the third day of this diet, a 24-hour urine specimen is collected for measurement of aldosterone, sodium, and creatinine. The 24-hour sodium excretion should be >4,600 mg to indicated proper sodium loading. Urine aldosterone excretion > 12 µg/day indicates hyperaldosteronism.
Oral captopril test: In primary hyperaldosteronism, this test does not produce any significant decrease in PAC.
Investigation for differential diagnosis:
CT/MRI scanning: To detect adenoma and hyperplasia. Scanning of the adrenal with selenium-75 cholesterol to detect an adenoma.
Adrenal vein catheterization: To detect hypersecretion of aldosterone.
- Unilateral hypersecretion in adenoma.
- Bilateral hypersecretion in hyperplasia.
Dexamethasone suppression:
- Lowers plasma aldosterone transiently in adenoma.
- Prolonged suppression in glucocorticoid sensitive hyperaldosteronism.
Measurement of 18-OH-cortisollevels:
- Very high levels observed in adenomas and glucocorticoid-responsive hyperplasia.
- Slightly raised in idiopathic hyperplasia.
Management of primary hyperaldosteronism:
- Potassium (K+) supplementation
- Unilateral disease: Laparoscopic adrenalectomy, bilateral if multiple tumors are present. Surgery is the definitive treatment in unilateral disease.
- In patients with bilateral hyperaldosteronism, mineralocorticoid therapy is administered.
Aldosterone antagonists: Spironolactone and eplerenone are useful in patients where surgery cannot be performed. High dose of spironolactone (up to 400 mg/day) may be required. A few patients may develop gynecomastia with spironolactone and the incidence is lower with eplerenone. Amiloride (10–40 mg/day) may also be tried.
Secondary Hyperaldosteronism:
Question 42. Write a short note on secondary hyperaldosteronism.
Answer:
- Secondary hyperaldosteronism is secondary to an extra-adrenal cause.
- The aldosterone release occurs in response to activation of the renin-angiotensin system. It is characterized by increased levels of plasma renin that stimulates the zona glomerulosa.
Secondary Hyperaldosteronism Causes of secondary hyperaldosteronism:

Secondary Hyperaldosteronism Adrenocortical Insuffiency:
Classifiation and Causes:
Question 43. Write a short essay/note on classification and causes of adrenocortical insufficiency.
(or)
Write a short essay/note on etiology and clinical features of Addison’s disease.
Answer:
Adrenocortical insufficiency, or hypofunction, may be due to primary adrenal disease (primary hypoadrenalism) or decreased stimulation of the adrenals due to a deficiency of ACTH (secondary hypoadrenalism).
Classifiation and Causes Three-step process in the investigation of adrenocortical insufficiency:
Confirm adrenal insufficiency by demonstrating inappropriately low cortisol secretion.
- Determine whether the cortisol
- deficiency is primary or central AI.
- Determine the cause of the underlying disorder.

Classifiation and Causes Addison’s Disease:
Addison’s disease or chronic adrenocortical insufficiency is an uncommon disorder resulting from progressive destruction of the entire adrenal cortex.
Clinical features of Addison’s disease:
Question 44. Write a short essay/note on major clinical signs of Addison’s disease.
Answer:
- Clinical features are produced due to the deficiency of glucocorticoid, mineralocorticoid, and androgen as well as excess of ACTH.
- Fatigue and weight loss are the most prominent symptoms.
- Primary adrenal failure may present.
Acute: Hypotension and acute circulatory failure with shock out of proportion to severity of current illness, dehydration, an “acute abdomen”, unexplained hypoglycemia, and unexplained fever (Addisonian crisis)
Chronic: Vague features of ill health, sometimes including gastrointestinal symptoms, features suggestive of postural hypotension and salt craving.
- Skin pigmentation is nearly always present in primary adrenal insufficiency (but not in secondary).
- Cardinal features of Addison’s disease: Hypotension, pigmentation and previous history of acute adrenal crisis following stress, or slow recovery from illness.
Other autoimmune diseases associated with Addison’s disease:
-
- Hashimoto’s thyroiditis, primary atrophic hypothyroidism, pernicious anemia, type 1 diabetes mellitus, primary ovarian failure, and hypoparathyroidism.
- Type 1 polyglandular autoimmune syndrome is the combination of adrenal insufficiency, hypoparathyroidism, and chronic mucocutaneous candidiasis.
- Type 2 polyglandular autoimmune syndrome/Schmidt syndrome is the association of Addison’s disease and Hashimoto’s thyroiditis.

Investigations:
Write a short essay/note on biochemical abnormality in Addison’s disease.
Investigations Early morning (6 AM) cortisol level:
- Normally, levels are 10‒20 µg/dL
- Levels <3 µg/dL is suggestive of adrenal insufficiency.
- Levels >11 µg/dL exclude AI
- Random cortisol in ill patient 20 µg/dL reassuring.
Investigations Morning salivary cortisol concentration:
- Levels below 1.8 ng/mL is highly suggestive of adrenal insufficiency.
- Levels above 5.8 ng/mL rule out adrenal insufficiency.
Investigations Plasma ACTH level: Elevated in adrenal insufficiency.
Investigations ACTH stimulation test: There is failure of rise in plasma cortisol level following the administration of 0.25 mg (standard high dose test) of synthetic ACTH (cosyntropin)
- Low dose test (1 µg)
- Baseline and 30-minute cortisol levels
- More physiological ACTH level/stimulation
- Useful in central AI
- Useful for assessing recovery after chronic steroid treatment.
Investigations High dose (250 µg) test:
- Baseline, 30and 60-minute levels
- Stronger stimulation than 1 µg test.
Investigations Metyrapone test: Useful to detect partial ACTH deficiency and partial secondary adrenal insufficiency.
Investigations Insulin-induced hypoglycemia test: Insulin at a dose of 0.15 U/kg is administered to achieve hypoglycemia of 35 mg/dL or less. Cortisol levels are measured at 0, 30, and 45 minutes.
Investigations Corticotropin-releasing hormone test: To differentiate between secondary and tertiary adrenal insufficiency.
Investigations Other investigations: PRA is high and plasma aldosterone levels are low or normal.
Investigations Radiograph: In patients with tuberculous adrenalitis: Chest radiograph may show evidences of pulmonary tuberculosis and plain radiograph of abdomen, CT scan, and MRI scan may show calcification in the adrenal gland.
Investigations Adrenal autoantibodies: ACA—adrenal cortex antibody and anti-21-OH-hydroxylase antibody
Investigations Blood sugar: Low levels.
Investigations Peripheral blood: Mild anemia and mild eosinophilia.
Investigations Central AI: Evaluate for secretion of other pituitary hormones.
Investigations Management of primary adrenal insuffiency:
Investigations Acute treatment:
- Normal saline for volume resuscitation
- Look for/treat hypoglycemia by 25% dextrose
Investigations Steroids:
- Loading dose: 50–100 mg/m 2 hydrocortisone IV/IM
- Continue hydrocortisone with 50–100 mg/m 2/day, divided 6th or 8th hourly
Investigations Long-term treatment:
- Daily glucocorticoid replacement (hydrocortisone): 10–15 mg/m 2/day in divided doses.
- Daily mineralocorticoid replacement: Fludrocortisone 0.05–0.2 mg daily
- Relative steroid potencies.
Investigations Stress conditions:
- Primary goal is to avoid serious consequences of an adrenal crisis
- Always wear identification
Investigations Illness:
- Minor stress (e.g., sore throat, rhinorrhea, temperature < 38°C) may not require increase in dose.
- Moderate stress (e.g., severe URTI)—double the glucocorticoid (GC) replacement dose
- Major stress (e.g., T > 38°C and/or vomiting), three to four times the GC replacement dose.
Investigations Surgery:
- During general anesthesia, ± surgery, the GC requirement increases greatly.
- Dose equivalent to 100‒150 mg hydrocortisone for major surgical procedures in divided doses.
- Stress dosing is generally continued until the patient can tolerate oral intake, is afebrile, and is hemodynamically stable.
Tuberculous adrenalitis causing Addison’s disease is treated with anti-tuberculous chemotherapy
Acute Adrenal Crisis/Addisonian Crisis
Question 45. Write a short note on acute adrenal crisis.
Answer:
Acute adrenal crisis is a state ofacute adrenocortical insufficiency and occurs in patients with Addison’s disease who are exposed to the stress of infection, trauma, surgery, or dehydration due to salt deprivation, vomiting, or diarrhea.
Acute Adrenal Crisis/Addisonian Crisis Other causes: Acute adrenal crisis can also develop after:
- Bilateral adrenal infarction
Acute Adrenal Crisis/Addisonian Crisis Bilateral adrenal hemorrhage: Adrenal hemorrhage and often death may occur following meningococcemia (Waterhouse‒Friderichsen syndrome).
It is rare with secondary adrenal insufficiency.
Acute Adrenal Crisis/Addisonian Crisis Clinical Features:
Acute Adrenal Crisis/Addisonian Crisis Major manifestations:
- Shock (out of proportion to severity of illness)
- Nonspecific symptoms: Weakness, fatigue, lethargy, anorexia, nausea, vomiting, abdominal pain, confusion or coma.
- Abdominal tenderness “acute abdomen” and unexplained fever.
Crisis in patients with long-standing adrenal insufficiency: Shows features of chronic adrenal insufficiency and may show hyperpigmentation (due to chronic ACTH hypersecretion), weight loss, and serum electrolyte abnormalities.
Acute Adrenal Crisis/Addisonian Crisis Laboratory Findings:
- Hyponatremia
- Hyperkalemia
- Hypercalcemia
- Azotemia
- Lymphocytosis
- Eosinophilia
- Hypoglycemia.
Acute Adrenal Crisis/Addisonian Crisis Management:
- Establish IV access with a large bore needle.
- Withdraw blood for serum electrolytes, glucose and routine plasma cortisol and ACTH. Do not wait for results.
- IV infusion of 2–3 L normal saline bolus. Avoid iatrogenic fluid overload.
- IV hydrocortisone 100 mg, followed by 50 mg IV every 6 hours OR 200 mg/day IV infusion.
- After initial stabilization, continue IV NS at a slower rate for next 24–48 hours.
- Search and manage possible precipitating cause.
- In case patient is not a known case of AI, do short ACTH stimulation test.
- Determine the type of AI.
- Taper parenteral glucocorticoid over 1–3 days to oral maintenance dose.
- Start mineralocorticoid replacement with fludrocortisone 0.1 mg by mouth daily when saline infusion is stopped.
Acute Adrenal Crisis/Addisonian Crisis Equivalent doses of glucocorticoids:
- Compared to hydrocortisone, prednisolone has only 25% of mineralocorticoid activity
- Both dexamethasone and betamethasone have negligible mineralocorticoid activity.

Steroid Therapy:
Common Indication and Contraindications of Steroids:
Question 46. Write a short note on the use and abuse of steroids/the present role of steroid therapy.
Answer:

Steroid Therapy Common Contraindications for Steroids:
- Active tuberculosis
- Peptic ulcer
- Diabetes
- Uncontrolled hypertension
- Active infection
Side Effects of Corticosteroids Therapy:
Question 47. Write a short note on the complications of corticosteroid therapy.
Answer:

Measures to Reduce the Side Effcts of Corticosteroids Therapy:
Question 48. Write a short note on measures to minimize the side effects of corticosteroid.
Answer:
Side effcts of steroids can be reduced by administering them with the following rules:
- Lowest required dose
- Only under medical supervision
- On alternate days rather than daily
- Monitor intake of calories to prevent weight gain
- As a single dose than in divided doses
- Reduce intake of sodium
- In the morning
- Use H 2 receptor blockers or proton-pump inhibitors
- For the shortest required duration
- Provide high calcium intake and pump inhibitors
- For indications only
- Use bisphosphonates
Waterhouse–Friderichsen Syndrome:
Question 49. Write a short note on Waterhouse–Friderichsen syndrome.
Answer:
- It is characterized by an acute hemorrhagic infarction (destruction) of both the adrenal glands and is usually associated with fulminant meningococcal septicemia.
- It produces cutaneous petechiae, vasomotor collapse, and shock. Can occur at any age but is more common in children.
- Abrupt in onset and profound prostration occurs within a few hours.
- It produces petechiae, purpuric lesion, and hemorrhage into the skin.
Waterhouse–Friderichsen Syndrome Management:
Prompt recognition and appropriate therapy, intravenous fluids, high dose antibiotics (penicillin, cephalosporins, and sulfonamides), vasopressors, inotropes, plasma transfusion, steroids must be instituted immediately, or death follows within hours to a few days due to cardiac and/or respiratory failure.
Pheochromocytoma
Question 50. Write a short essay/note on clinical features, diagnosis, and treatment of pheochromocytoma.
Answer:
- Pheochromocytoma is a very rare tumor of the sympathetic nervous system composed of chromaffin cells that secretes catecholamines.
- Important because they are a rare cause of surgically correctable hypertension.
- Paraganglioma: Pheochromocytomas that develop in sympathetic ganglia are known as paragangliomas.
- It arises from both sympathetic and parasympathetic paraganglia, located anywhere from the base of the skull to the pelvis.
- Paragangliomas typically occur in the head and neck but are also found in the thorax, pelvis, and bladder.
- Traditionally, the features of pheochromocytomas have been summarized by the “rule of 10s”.
- Neurofibromatosis type 1 (Von Recklinghausen’s disease) is associated with an increased incidence of pheochromocytoma.
Rule of 10s’ for pheochromocytoma:
- 10% Extra-adrenal (closer to 15%)
- 10% Abdominal
- 10% Occur in children
- 10% Familial (now modified as closer to 25%)
- 10% Sporadic are bilateral or multiple (more if familial)
- 10% Not associated with hypertension
- 10% Malignant
- 10% Discovered incidentally
- 10% Recur (more if adrenal)
Pheochromocytoma Clinical Features:
The classic triad of symptoms in patients with a pheochromocytoma consists of episodic headache, sweating, and tachycardia
Pheochromocytoma 5 Ps: Pressure (90%), Pain (80%), Perspiration (70%), Palpitations (65%), and Pallor (42%)
Pheochromocytoma Paroxysmal hypertension:
- Characterized by episodes of pallor or flushing,
headache sweating, palpitations, and anxiety (fear of death). - Paroxysms last 10–60 minutes duration, daily to monthly
Pheochromocytoma Paroxysms are spontaneous or precipitated by:
- Diagnostic procedures, intra-arterial contrast
- Drugs (opioids, unopposed beta-blockade,
anesthesia induction, histamine, ACTH, glucagon, and metoclopramide) - Strenuous exercise, movement that increases
intra-abdominal pressure (lifting and straining) - Micturition (bladder paraganglioma)
Sustained hypertension is more common than paroxysmal hypertension.
Pheochromocytoma Complications of hypertension: Stroke, myocardial infarction, cardiomyopathy, and left ventricular failure.
Pheochromocytoma Gastrointestinal symptoms: Abdominal pain, vomiting, constipation, and weight loss.
Pheochromocytoma Hypercalcemia: Observed in associated MEN2 hyperparathyroidism
Pheochromocytoma Mild glucose intolerance:
- Lipolysis and weight-loss
Pheochromocytoma Investigations:
- Plasma metanephrine is the most sensitive test.
- Other investigations
Pheochromocytoma Chromogranin A: It is a major secretory protein present in the soluble matrix of chromaffin granules and is elevated.
Provocative (glucagon provocative test) and adrenolytic (clonidine, phentolamine test) tests: Rarely necessary.
CT/MRI scan: To localize tumor.
Scintigraphy: Useful for localization of tumor and includes
- MIBG (123I-labeled meta-iodobenzylguanidine) scintigraphy
- Somatostatin receptor scintigraphy using 111indium-labeled diethylenetriaminepentaacetic acid octreotide scan.
- (18F) fluro-dihydroxyphenylalanine (DOPA) PET scan.
Plasma noradrenaline: Selective venous sampling and estimation of plasma noradrenaline level may be useful in localizing the tumor in difficult cases.


Pheochromocytoma Management:
- Excision of the tumor is the main treatment.
- Special care has to be taken preoperatively, intraoperatively, and postoperatively.
Pheochromocytoma Preoperative preparation regimens:
- Combined α + β-blockade to be given at least 2 weeks preoperatively to control the hypertension. Antihypertensive agents used are: phenoxybenzamine (preferred drug for preoperative preparation). Initial dose is 10–20 mg in divided doses every 2–3 days as needed, final dose will be 20–100 mg/day. Other drugs used are selective α1-blocker (prazosin, terazosin, or doxazosin).
- Beta-blockers should never be started first. Drugs such as propranolol or metoprolol are used. Dose should be adjusted to control tachycardia. Typical maximum doses in this setting are 120 mg of propranolol or 200 mg of metoprolol.
- High sodium diet—patients are encouraged to start a diet high in sodium content (>5,000 mg daily) on the second or third day because of the catecholamine-induced volume contraction and the orthostasis associated with alpha-adrenergic blockade.
Pheochromocytoma If uncontrolled add:
- Metyrosine
Pheochromocytoma Calcium channel blockers: Nicardipine and amlodipine are most commonly used in this setting; starting dose of nicardipine is 30 mg twice daily of the sustained-release preparation and the starting dose of amlodipine is 2.5 or 5 mg administered once daily.
- Avoid diuretics as ECF volume is already contracted.
Intraoperative blood pressure needs to carefully monitored and controlled. Postoperative hypotension can be avoided by adequate fluid replacement and hypoglycemia (10–15% of patients) due to removal of catecholamine suppression of insulin secretion by glucose infusion. Postoperatively patients may become hypertension free.
Pheochromocytoma If tumor cannot be excised:
- Long-term treatment with αand β-adrenoreceptors blocking drugs (phenoxybenzamine and propranolol, or labetalol) is advocated.
- β-blockers should never be given alone.
- Patients can also be subjected to nuclear medicine treatment
Gonadal Disorders
Male Hypogonadism:
- Primary hypogonadism, if the serum testosterone and/or the sperm count are below normal and the serum LH and/or FSH concentrations are above normal.
- Secondary hypogonadism, if the serum testosterone concentration and/or the sperm count are below normal and the serum LH and/or FSH concentrations are normal or low.


Discuss the causes and approach to male hypogonadism.
Male Hypogonadism Classifiation and Causes of Male Hypogonadism:

Male Hypogonadism Physical Signs:
- Diminished muscle mass
- Loss of body hair
- Abdominal obesity
- Gynecomastia
- Testes frequently normal, occasionally small
Male Hypogonadism Prepubertal onset:
- Eunuchoidism
- Lack of adult male hair distribution
- Sparse axillary and pubic hair
- Lack of temporal hair recession
- High-pitched voice
- Infantile genitalia:
- Small penis, testes, and scrotum
- Fat deposition in pectoral, hip, thigh, and lower abdomen
- Eunuchoidal proportion
- Arm span > height > 5 cm
- Upper/lower segment ratio < 1
Male Hypogonadism Investigations:
Male Hypogonadism Serum testosterone: 8.00 AM
- Serum FSH and LH
Male Hypogonadism Semen analysis: Normally, >40% of the sperm ejaculated are motile, >32% have rapid forward progression, and >4% have normal morphology.
Male Hypogonadism Others: Peripheral leukocyte karyotype, other pituitary hormones, serum prolactin, iron saturation, and MRI brain.
Treatment:
Treatment of Primary Hypogonadism:
- Testosterone replacement for men who are hypogonadal only with signs and symptoms with subnormal morning serum testosterone.
- Treatment of the underlying disease
Write a short note on testosterone replacement.
Testosterone replacement
- Transdermal testosterone is started in gel form.
- Intramuscular preparations—testosterone enanthate (100–200 mg IM q 2 weeks)
- Oral agent—testosterone undecanoate (initial 120–160 mg/day in 2 divided doses for 2–3 weeks; usual maintenance dose 40‒120 mg/day), however parenteral formulations are preferred as adverse effects of oral preparation are hypertension and cardiovascular events.
Testosterone replacement Undesirable effects: BPH, prostatic cancer, dyslipidemia, polycythemia, and transaminitis.
Testosterone replacement Treatment of secondary hypogonadism:
- GnRH pulsatile infusion
- hCG
Impotence
Question 51. What is impotence? Discuss briefly.
Answer:
Impotence Definition: Male sexual dysfunction is termed “impotence”.
Impotence Manifestations:
- Loss of desire
- Inability to obtain and maintain erection
- Premature ejaculation
- Absence of emission
- Inability to achieve orgasm.
Impotence Causes of Male Infertility:
Erectile impotence may be due to various causes, but majority are of psychological origin. In each case of erectile impotence, it is necessary to rule out the organic causes.
Gynecomastia:
Write a short note on gynecomastia.
- It is clinically defined as the presence of a rubbery or firm mass concentrically extending from the nipples in a male secondary to proliferation of both stromal and epithelial component of the glands.
- It must be differentiated from pseudogynecomastia (lipomastia) seen in obese individuals, which is characterized by only fat deposition without glandular proliferation.
Causes of Gynecomastia:
Gynecomastia Clinical features:
Enlargement of breast: The principal complaint is unilateral or bilateral concentric enlargement of breast glandular tissue.
Breast pain: Present in one-fourth of patients and objective tenderness in about 40%.
- A complaint of nipple discharge can be elicited in 4% of cases.
- Patients with gynecomastia may have a slightly increased risk of development of breast carcinoma.
Gynecomastia Treatment:
Gynecomastia Medical treatment:
- The underlying disease should be corrected if possible, and offending drugs should be discontinued.
- Antiestrogens or selective estrogen receptor modulators, such as tamoxifen or raloxifene, have been found useful in relieving pain and reversing gynecomastia in some patients. Aromatase inhibitors have also been tried but are not as beneficial as tamoxifen.
Surgical treatment: Reduction mammoplasty should be considered for cosmetic reasons.
Gynecomastia Radiotherapy:
- Patients with prostatic carcinoma may receive low-dose radiation therapy (900c Gy or less) to the breasts before initiation of estrogen therapy. This may prevent or diminish the gynecomastia that usually results from such therapy.
- Radiotherapy should not be given to other patients with gynecomastia.
Gynecomastia Causes of male infertility:
Gynecomastia Endocrine:
-
- Hypothalamic-pituitary disorders
- Testicular disorders
- Defects of androgen action
- Hyperthyroidism
- Hypothyroidism
- Adrenal insufficiency
- Congenital adrenal hyperplasia
- Systemic illness
- Defects in spermatogenesis
- Immotile cilia syndrome
- Drug-induced
Gynecomastia Ductal obstruction:
-
- Congenital
- Acquired seminal vesicle disease
- Prostatic disease
- Varicocele
- Retrograde ejaculation
- Antibodies to sperm or seminal plasma
- Anatomic defects of the penis
- Poor coital technique
- Sexual dysfunction
- Idiopathic

Short Stature:
Question 52. Write a short note on short stature and its differential diagnosis.
Answer:
- Short stature is defined as a height that is below the 3rd percentile OR two or more standard deviations below the mean for chronological age and gender for a given population.


- A growth velocity that is below the 5th percentile for age and gender is called growth deceleration (e.g., <5 cm/year after the age of 5 years).
- Dwarfism is defined as short stature for the age of the patient.
- Most common causes of dwarfism are familial short stature and constitutional delay of growth and puberty.
Short Stature Proportionate Short Stature:
- Limbs proportionate to trunk
- Seen in most cases of familial short stature
Short Stature Disproportionate Short Stature:
- Limbs disproportionately short compared to trunk
- Seen mostly in cases of skeletal dysplasia
- Growth failure: Growth rate below the rate considered appropriate for sex and age.
Short Stature Tall Stature :
Tall stature is defined as height above 97th percentile for chronological age and sex or more than 2 SD above the mean for a defined population.


How do we calculate height of child based on parental height?
Estimated adult height of a child calculated on the basis of parental height using the following formulas:
- [(Paternal height + Maternal height)/2] + 2.5” (6.5 cm)
- [(Paternal height + Maternal height)/2] – 2.5” (6.5 cm)
Leave a Reply