Rheumatology and Connective Tissue Disorders
Introduction:
Question 1. Develop a systematic clinical approach to joint pain based on pathophysiology.
(or)
Prioritize based on clinical features for arriving at an etiological diagnosis in polyarthritis.
Answer:
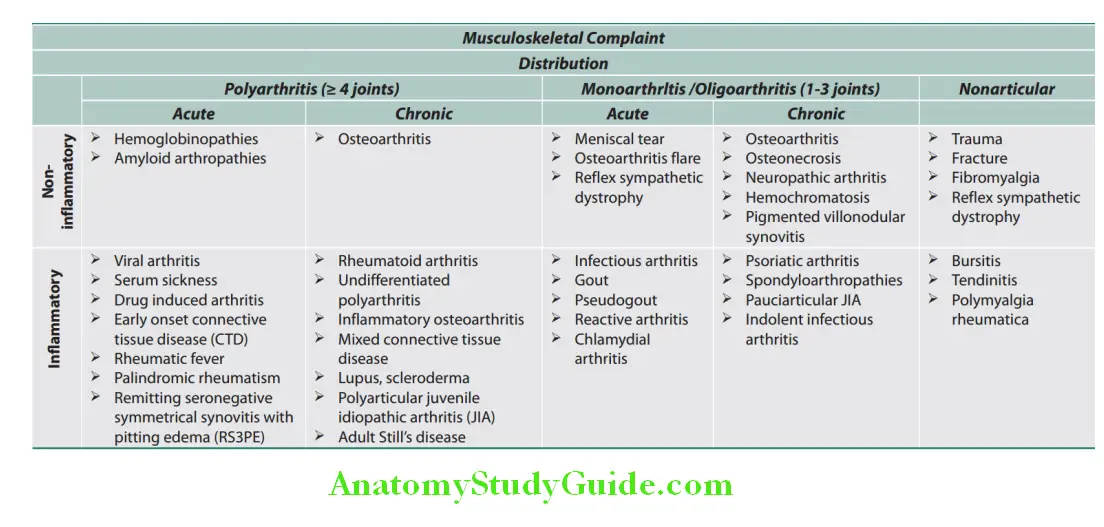
Clinical approach to polyarthritis is important and the diagnosis is often challenging due to the extensive differential diagnosis. Thorough history and physical examination is the cornerstone for diagnosis. The clinical factors helpful in narrowing the diagnosis are the chronology of symptoms, features of inflammation, joint distribution, extra-articular manifestations, disease course and patient demographics.
- Chronology:
- Acute polyarthritis may be due to a self-limited disease.
- It could be due to infectious, crystal induced or reactive causes.
- Rarely chronic polyarticular arthritides, like rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) can have acute presentation in the early course of illness.
- Chronic arthritis may be due to immunologic arthritides like RA, noninflammatory and nonarticular causes.
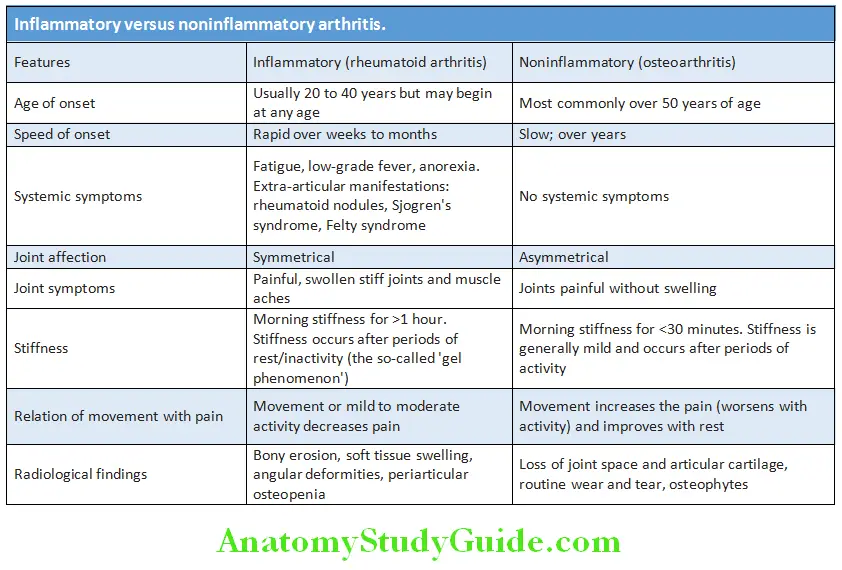
- Inflammation:
- Features of inflammation make it inflammatory arthritis and in the absence it is called athralgia.
- Erythema, warmth, pain and swelling are the cardinal features of inflammation. Fatigue, fever and weight loss can also be present.
- Morning stiffness lasting for longer than an hour is another clue.
- Palpate the joint capsule for synovial thickening, soft tissue swelling effusions.
- Presence of crepitus indicates underlying injury, osteoarthritis (OA) or previous inflammation. Heberden’s and
Bouchard’s nodes indicate osteoarthritis. - Subjective sense of swelling without objective sign of synovitis is seen in patients with fibromyalgia.
Question 2. Determine the potential causes of joint pain based on the presenting features of joint involvement.
Answer:
- Joint distribution:
- Crystal and infectios arthritides are often mono or oligoarticular whereas OA and RA are polyarticular.
- Joint involvement in RA tends to be symmetrical and polyarticular whereas spondyloarthropathy, gout and reactive arthritis involvement is asymmetric and oligoarticular.
- Psoriatic arthritis and OA can be either mono or polyarticular and symmetric or asymmetric.
- Upper extremities are commonly affected in RA and OA, whereas lower extremities are affected commonly in reactive arthritis, spondyloarthropathies and gout.
- Axial skeleton is involved in ankylosing spondylitis (Sp A) and OA. Osteoarthritis usually spares wrist, elbow and ankle unless there is a past history of trauma.
- Osteoarthritis involves distal interphalangeal (DIP) and proximal interphalangeal (PIP) joints with sparing of metacarpophalangeal joints. Rheumatoid arthritis spares DIP but affects PIP and metacarpophalangeal (MCP) joints.
- Psoriasis and crystal arthropathies affect all three joints.
- Extra-articular manifestations:
- SLE can present with malar rash and oral ulcers.
- Psoriatic skin and nail changes accompany psoriatic arthritis.
- Polymyositis is suggested by proximal muscle weakness.
- Reactive arthritis is suggested by a history of recent gastrointestinal or genitourinary infection, conjunctivitis and oral ulcers.
- Spondyloarthropathies can present with enthesitis or dactylitis causing sausage-shaped digits.
- Disease course:
- Intermittent arthritis is a feature of crystal arthropathies, Lyme arthritis and palindromic rheumatism that may progress to rheumatoid arthritis later.
- Migratory arthritis can be a feature of rheumatic fever, bacterial endocarditis, gonococcal infection and viral fever.
- Patient demographics:
- Incidence of RA, SLE, fibromyalgia are more among premenopausal women compared to men of same age group.
- Spondyloarthropathies occur equally in males and females.
- Gout usually occurs 20 years after puberty in men and 20 years after menopause in women. Rare among premenopausal women unless associated with renal failure.
- RA, SLE, reactive arthritis and sponyloarthropathies occur more among younger individuals whereas OA, polymyalgia rheumatic and giant cell arthritis occur more among elderly.
- Family history is important in spondyloarthropathies, rheumatoid arthritis and Heberden’s nodes of osteoarthritis.
Discusses the classification of arthritis with examples:
Differences between inflammatory versus noninflammatory arthritis is presented in Table:
Question 3. Describe the appropriate diagnostic work up based on the etiology.
Answer:
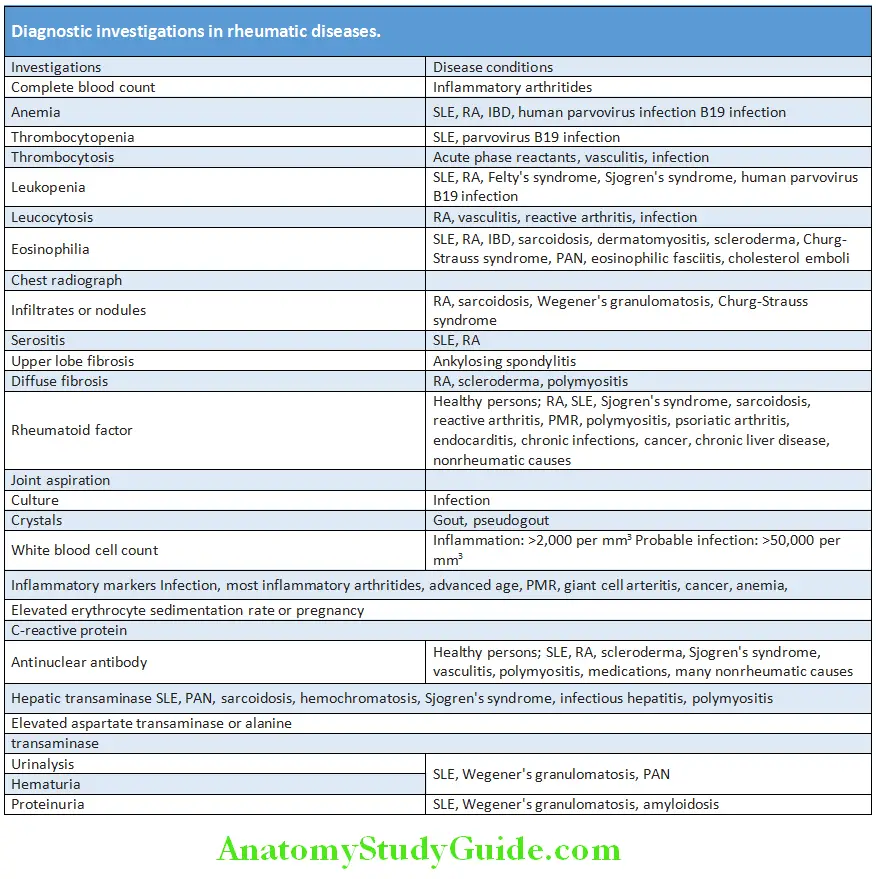
Rheumatological diagnostic tests should be interpreted in the appropriate clinical contex.
- A complete blood count, urine analysis, renal and liver functions test may provide dignostic clues.
- ANA can be positive in 5–10% of general population and may increase with age. Due to the high sensitivity a negative result rules out systemic lupus erythematosus but due to lack of specificity, positive result should be analyzed with caution.
- HLA-B27 positivity with a positive family history is highly suggestive of ankylosing spondylitis. However, relying only on HLA-B27 reports may lead to overdiagnosis as it can be seen in 8% of caucasians.
- Rheumatoid factor lacks sensitivity and specificity; hence both positive and negative results should be interpreted cautiously. Not useful if the patient lacks other criteria for diagnosis.
- Synovial fluid analysis helpful in diagnosing septic and crystal induced arthritis.
- Dignostic imaging is helpful in rheumatological disorders.
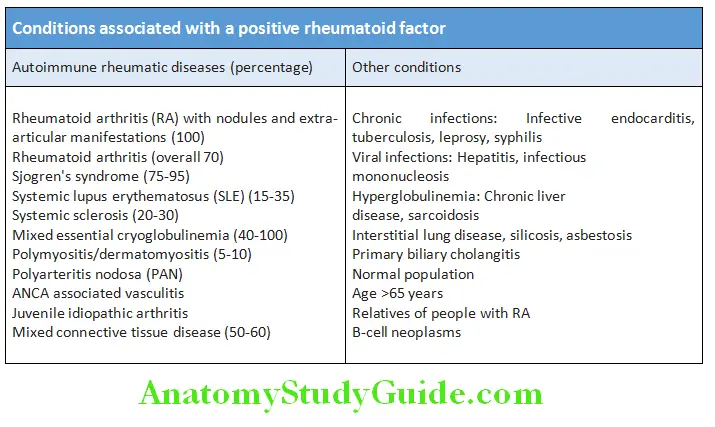
Rheumatoid Factor (RF):
Question 4. Write short note on rheumatoid factor and mention its significance.
Answer:
Rheumatoid factor is an autoantibody (IgM, IgG and IgA) directed against the Fc fragment of human immunoglobulin G (IgG).
Signifiance:
- RF has poor specificity and is positive in 70 to 80% of patients with rheumatoid arthritis (RA). It is appropriate and helpful only in patients suspected of having RA. It is not helpful in cases of low clinical suspicion.
- High RF titers are observed with more severe disease and extra-articular disease. The titers generally correlate with severity of disease. However, RF titers are not useful in assessing disease progression.
- It can also be positive in many other conditions.
- 10–30% of patients with long-standing RA are seronegative.
Anticyclic Citrullinated Peptide:
Antibodies (Anti-CCPs, ACPA):
Question 5. Enumerate the significance and indications for anticitrullinated peptide antibody (Anti-CCP).
Answer:
Citrullinated Peptide Antigens:
- These include fibrinogen, type II collagen, alpha enolase and vimentin.
- CCPs are derived from proteins in which arginine residues are converted to citrulline residues by an enzyme peptidyl arginine (which is abundant in inflamed synovium and in a variety of mucosal structures) during a variety of biological processes.
Anticyclic Citrullinated Peptide Antibodies (ACPA):
- These antibodies are useful for diagnosis of RF and may be involved in tissue injury. Many patients (about 70%) of RF have antibodies against cyclic citrullinated peptides (CCPs).
- These antibodies have much high specificity (93–98%) but less sensitive (60%) for diagnosis of RA.
- Anti-CCPs may be detected even in asymptomatic patients several years before the development of RA and are associated with severe disease. Thus, anti-CCP positive patients may require an aggressive treatment.
- Antibodies against CCPs (anti-CCPs) from immune complexes and deposit in various tissues mainly being the joints.
- Positive results can occur in several autoimmune rheumatic diseases (SLE, Sjogren), tuberculosis, hepatis C infection, and chronic lung diseases.
- Antibodies to mutated citrullinated vimentin (anti-MCV) is associated with more severe disease than anti-CCP antibodies.
- Serum 14-3-3e, which is an intracellular chaperonin protein, has been used diagnostically in RA.
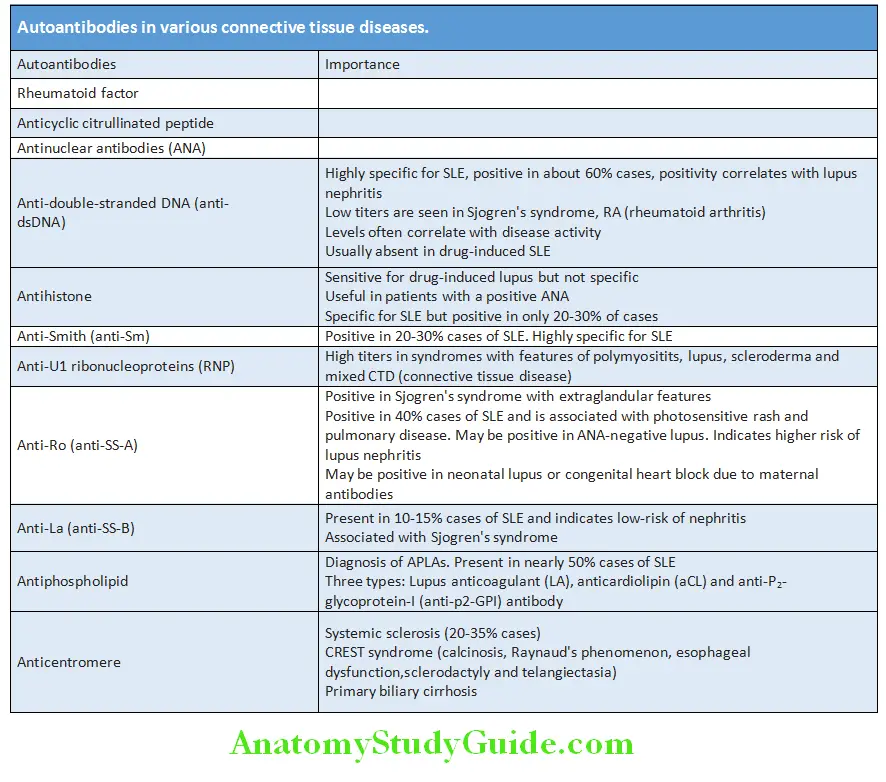
Autoantibodies in Various Connective Tissue Diseases:
Question 6. Enlist the various antibodies seen in patients with connective tissue diseases and mention its significance.
Answer:
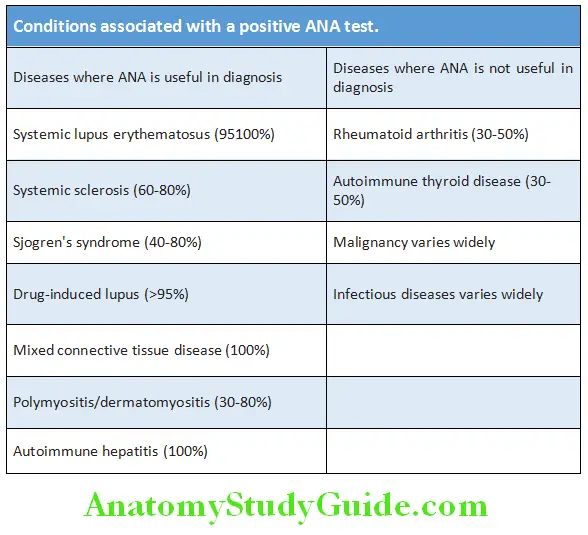
Antinuclear Antibodies:
Question 7. Enumerate the indications, significance and staining patterns of antinuclear antibodies.
Answer:
Antinuclear antibodies (ANA) are directed against one or more components of the cell nucleus. The component of the nucleus includes DNA, RNA, proteins as well as complexes of proteins with nucleic acid.
Significance:
- High titers of ANA (>1:160) are ofmore diagnostic significance than low titers.
- Circulating levels of ANA do not correlate with severity or activity of the disease.
- ANA is positive in several conditions.
- ANA is used as a screening test for systemic lupus erythematosus (SLE) and systemic sclerosis (SSc). ANA has high sensitivity for SLE (100%) but low specificity (10–40%). A negative ANA almost rules out SLE but a positive result does not confirm it. It should not be used to monitor the course of SLE or other diseases.
Read And Learn More: General Medicine Question And Answers
Tests for detection: ANAs are detected by indirect immunofluorescent staining of fresh-frozen sections of rat liver or kidney or Hep-2 cell lines (a line of human epithelial cells).

Extractable Nuclear Antibodies:
Question 8. Write short essay/note on extractable nuclear antibodies.
Answer:
These are antibodies to extractable nuclear antigens (ENA) and are directed against small ribonuclear proteins (RNA).
Their sensitivity and specificity are poor. Though used for diagnostic confirmation, they do not exclude a specific CTD.
They do not correlate with disease activity and may be found in patients without active disease

Antineutrophil Cytoplasmic Antibodies:
Question 9. Write short essay/note on antineutrophil cytoplasmic antibodies (ANCA), their significance/ANCA associated disorders.
Answer:
Antineutrophil cytoplasmic antibodies (ANCAs) are heterogeneous group of IgG autoantibodies directed against certain proteins (mainly enzymes) in the cytoplasmic granules of neutrophils and lysosomes of monocytes.
Patterns: On the basis of the target antigens and pattern of immunofluorescence staining, two common patterns of ANCA Have been distinguished.
- Anti-proteinase-3 (PR3-ANCA): It is also called cytoplasmic or cANCA. PR3 is a constituent of neutrophil azurophilic granule. It shares homology with many microbial peptides and antibodies against microbial peptides. Antibodies against proteinase-3 (PR3) form PR3-ANCAs. They produce cytoplasmic fluorescence (cANCA). They are associated with Wegener’s granulomatosis.
- Anti-myeloperoxidase (MPO-ANCA): It is also called perinuclear or pANCA. MPO is a lysosomal enzyme normally involved in producing oxygen-free radicals. Antibodies to myeloperoxidase (MPO) forms MPO-ANCAs which produces perinuclear fluorescence (pANCA). It can be induced by drugs (e.g. propylthiouracil). They are associated with microscopic polyangiitis and Churg-Strauss syndrome.
Signifiance:
- ANCA are strongly associated with small-vessel vasculitis and are useful in the diagnosis and monitoring of systemic vasculitis. For their significance in vasculitis, ANCA should be assayed both with indirect immunofluroscence (screening test) and direct ELISA for proteinase-3 or myeloperoxidase.
- ANCA are not specific for vasculitis. They may be positive in autoimmune liver disease, malignancy, infection [bacterial and human immunodeficiency virus (HIV)], inflammatory bowel disease, chronic hepatitis, primary sclerosing cholangitis, Felty’s syndrome, rheumatoid arthritis, SLE and pulmonary fibrosis.
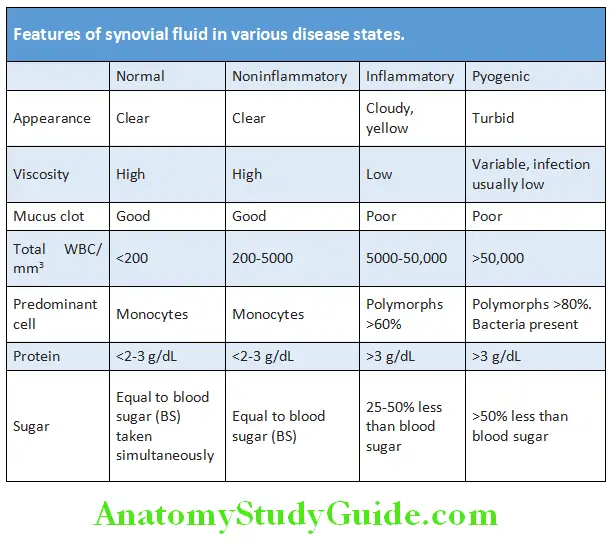
Synovial Fluid in Disease States:
Question 10. Differentiate the synovial fluid features in various arthritis.
(OR)
Enumerate the indications for arthrocentesis.
Answer:
- Arthrocentesis helps in the diagnosis of septic/ infective arthritis and crystal arthropathies and the indications are as below.
- Monoarthritis (acute or chronic)
- Monoarthritis in a patient with chronic polyarthritis
- Suspicion of joint infection
- Crystal-induced arthritis
- Hemarthrosis or trauma with joint effusion.
- Hemorrrhagic effusion may suggest trauma or mechanical derangement, coagulopathy or others.
- Counts less than 2,000 per mm3 may suggest noninflammatory causes like osteoarthritis or trauma.
- WBC count of at least 2,000 per mm3 in synovial fluid suggests inflammation, whereas a count higher than 50,000 per mm3 indicates infection. In later cases, synovial fluid needs to be cultured for ruling out infection.
- Crystal identification by polarized microscopy useful for diagnosis of gout and pseudogout.
Question 11. Describe the role of diagnostic imaging in rheumatological disorders.
Answer:
Currently available modalities include conventional radiography, computed tomography (CT), magnetic resonance imaging (MRI), arthrography, nuclear medicine scans (bone scintigraphy), and ultrasound.
- Conventional radiography:
- Presence of sacroiliitis indicates ankylosing spondylitis.
- Periarticular osteopenia with erosions, narrowing of the joint space and juxta-articular osteoporosis suggestive of rheumatoid arthritis.
- “Pencil-in-cup” deformities are suggestive of psoriatic arthritis.
- Chest radiographs may reveal underlying systemic disease. Radiographs showing infiltrates or nodules are suggestive of rheumatoid arthritis, sarcoidosis, Wegener’s granulomatosis or microscopic polyangitis. Serositis on radiograph suggests RA or SLE. Ankylosing spondylitis may present with upper lobe fibrosis whereas RA, scleroderma and polymyositis may present with diffuse fibrosis.
- Magnetic resonance imaging is sensitive for diagnosing soft Tissue injuries. MRI done in the early course of RA may reveal cartilage destruction that may not be picked up in routine radiographs. MRI can also be used to demonstrate tophi, synovial changes and erosions.
- Ultrasound is useful in the detection of soft tissue abnormalities such as tendinitis, tenosynovitis, bursitis, enthesitis and entrapment neuropathies.
- CT is useful in the diagnosis of low back pain syndromes. Also HRCT lung can help diagnosis of ILD.
- Nuclear medicine scans have great sensitivity for detecting many disease processes because they image and quantify physiologic biochemical processes, as well as provide anatomic information. The combination with single photon emission computer tomography (SPECT) increases anatomical resolution. Whole-body bone scintigraphy might also help differentiate inflammatory spondyloarthropathies from other inflammatory disorders such as SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis).
Rheumatoid Arthritis:
Question 12. Discuss the etiology, pathogenesis, clinical manifestations, diagnosis, investigations, complication, and management of rheumatoid arthritis.
Answer:
Rheumatoid arthritis (RA) is the most common form of chronic inflammatory, potentially crippling arthritis with multisystem involvement affecting approximately 1% of the adult population.
Etiology:
The cause is multifactorial and genetic, epigenetic and environmental factors play a part in the pathogenesis of RA.
- Genetic factors: Genetic susceptibility is a major factor in the pathogenesis of rheumatoid arthritis.
- HLA genes: RA is linked to specific HLA-DRB1 locus.
- Non-HLA genes: Polymorphism in PTPN22 gene, which encodes a tyrosine phosphatase.
- Abnormal DNA methylation, histone modifications, and microRNA regulation play a key role.
- Environmental arthritogenic agents: They are thought to initiate the disease process. Smoking and several microbial agents (e.g. virus, mycobacteria porphyromonas gingivalis, and Mycoplasma) have been suggested but not proved.
- Autoimmunity: The initial inflammatory synovitis, an autoimmune reaction with T cells is responsible for the chronic destructive nature of rheumatoid arthritis.
Pathogenesis:
The rheumatoid synovium (Pannus) behaves like a locally-invasive tumor. A cascade network of cytokines like granulocyte macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-2, IL-15, IL-13, IL-17, IL-18, interferon-gamma (IFNgamma), tumor necrosis factor (TNF) and transforming growth factor-beta (TGF-beta) are involved in the disease progression. B cells, CD4+ T cells, compliment mediated immune activation play a contributory role.
Clinical Features:
Question 13. Enumerate the clinical features of rheumatoid arthritis.
Answer:
- Gender: Female to male ratio is 3:1.
- Age: Most common age of onset is between 30 and 50 years, but the disease can occur at any age. In women in late childbearing years and in men during sixth to eighth decade.
Onset:
The presenting symptoms result from inflammation of the joints, tendons, and bursae. The onset varies:
- Slow and insidious in onset: Presents with malaise, fatigue, anorexia, weakness and generalized musculoskeletal pain.
- Acute onset: Sometimes RA may be very acute in onset, with morning stiffness, polyarthritis and pitting edema. This type occurs more commonly in old age.
- Palindromic onset: Occasionally, patient present with relapsing and remitting episodes of pain, stiffness and swelling that last for only a few hours or days alternating with symptom-free periods.
- Systemic onset/extra-articular involvement: Nonarticular symptoms, which will antedate the onset of arthritis
- Rarely, onset may be monoarticular.
Articular Manifestations:
Question 14. Describe the articular manifestations of rheumatoid arthritis.
Answer:
- Joints involved: RA can affect any of the synovial (diarthrodial) joints and involvement is usually in a symmetric distribution. Most commonly, RA starts in the small joints, namely metacarpophalangeal (MCP), proximal interphalangeal (PIP), and metatarsophalangeal (MTP) joints, followed by the involvement of large joints (wrists, knees, elbows, ankles, hips, and shoulders). Pitting edema over the dorsum gives rise to the “boxing glove” appearance. In RA, the hypertrophied synovium (also called pannus) invades and erodes contiguous cartilage, ligaments and bone.
- Symptoms: These include pain, swelling, and stiffness. Stiffness dominating in the mornings (‘morning stiffness’) lasting more than 1 hour is characteristic. Routine activities early in the morning like brushing teeth and combing hair may be very difficult.
Question 15. Write short note on rheumatoid hand.
Answer:
Hand and wrist: Hands are a major site of involvement and produces significant disability.
- The DIP joint is characteristically always spared unless the patient also has osteoarthritis (both coexist, particularly in elderly patients).
- Spindling of the fingers: It is the earliest finding characterized by swelling of the proximal, but not the distal interphalangeal joints.
- Deformities: Destruction of the joints and soft tissues may lead to chronic, irreversible deformities.
- Ulnar deviation: It results from subluxation of the metacarpophalangeal (MCP) joints, with subluxation of the proximal phalanx to the volar side of the hand.
- ‘Swan-neck’ deformity: It is due to hyperextension of the proximal interphalangeal joints (PIP) with flexion of the distal interphalangeal joints (DIP). At DIP joint there is elongation or rupture of attachment of the extensor tendon to the base of the distal phalanx; this results in mallet deformity of distal joint and in addition, an extensor tendon imbalance, leading to hyperextension deformity at PIP joint.
- ‘Boutonniere’ or ‘button-hole’ deformity: This deformity is due to flexion of the proximal interphalangeal (PIP) joints and extension of the distal interphalangeal (DIP) joints. Disruption of the central slip of the extensor tendon and the triangular ligament allows each of the conjoint lateral bands of the digit to slide volarly resulting in a pathologic flexion force and an extension lag; all tendons traversing the PIP joint in this setting elicit flexion of the joint.
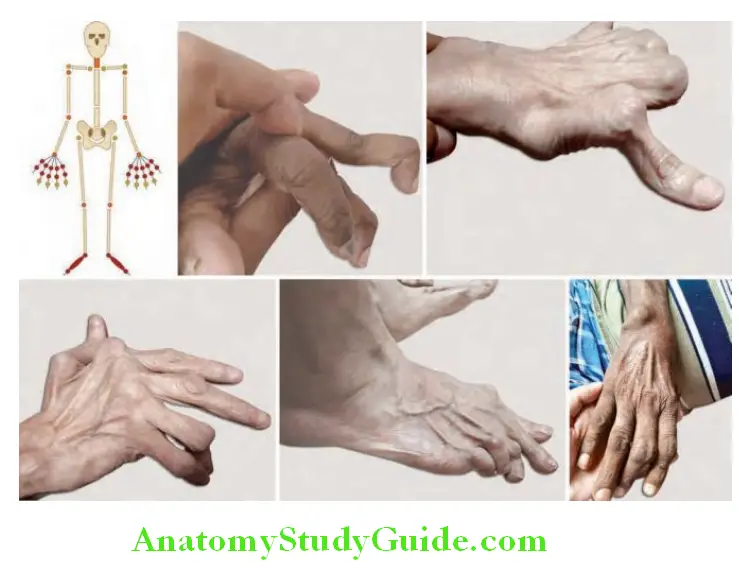
- ‘Gamekeepers thumb’: It is the result of hyperextension of the first interphalangeal joint and flexion of the first metacarpophalangeal joint. This leads to loss of thumb mobility and occurrence of pinch.
- Hitchhiker’s thumb (Z-shaped deformity) in which the thumb flexes at the metacarpophalangeal joint and hyperextends at the interphalangeal joint.
- Inflammation of the carpometacarpal joint leads to volar subluxation during contracture of the adductor hallucis.
- ‘Z’ deformity: It is due to radial deviation of the wrist, ulnar deviation of the digits with palmar subluxation of the first MCP joint with hyperextension of the first interphalangeal (IP) joint.
- Carpal tunnel syndrome: Due to synovial proliferation in and around the wrists producing compression of the median nerve.
- The “bow string” sign (prominence of the tendons in the extensor compartment of the hand).
Feet and ankle: In most patients, feet (particularly the MTP joints) are involved early:
- ‘Broadening’ of the forefoot: Due to swelling of the metatarsophalangeal joints.
- Subluxation of the toes at the MTP joints (“cock-up” deformities): Leads to skin ulceration on the top of the toes and painful ambulation because of loss of the cushioning pads which protect the metatarsals heads.
- Hallux valgus deformity and hammer toes.
Larger joints:
- Involvement of large joints (knees, ankles, elbows, hips and shoulders) is common but generally occurs somewhat later than small joint involvement.
- Shoulder joint involvement may present as glenohumeral arthritis, rotator cuff fraying and rupture.
- Synovial cysts: They present as fluctuant masses around involved joints (large or small). Best examples of synovial cyst is popliteal (‘Baker’s) cysts which develops in the knee joint. The knee synovitis produces excess synovial fluid that communicates posteriorly with the cyst (between the knee joint and the popliteal space) but is prevented from returning to the joint by a one-way valve-like mechanism. Ruptured Baker’s cysts can resemble a deep vein thrombosis.
Other joints:
- Axial skeleton: Although most of the spine is spared in RA, the cervical spine (atlantoaxial subluxation) can be involved producing quadriparesis.
- Cricoarytenoid joint involvement may present (30%) with hoarseness of voice and inspiratory stridor.
Extra-articular Manifestations:
Question 16. Describe the various systemic manifestations of rheumatoid arthritis.
Answer:
- Constitutional: RA is a systemic disease and features such as fatigue, weight loss, and low-grade fever (<38°C) occur frequently. Systemic features are more common in patients who have RF or ACPA antibodies, or both.
- Rheumatoid nodules:
- Rheumatoid nodules are usually subcutaneous and are typically benign.
- Significance: Nodules are seen in about one-fourth of patients with RA and, almost exclusively in those patients who are seropositive for rheumatoid factor. They are clinical predictors of more severe arthritis, sero-positivity, joint erosions and rheumatoid vasculitis.
- Sites: Typically occur on extensor surfaces, over joints or on areas subject to repeated trauma (particularly forearms sacral prominences, scalp and the Achilles tendon). Rarely, they develop in lungs, pleura, heart, pericardium or in the sclera of the eye.
- Characteristics: On examination, rheumatoid nodules are 2 mm to 5 cm in size, firm and nontender (unless traumatized). They have a characteristic histologic picture (central area of fibrinoid material surrounded by a palisade of proliferating mononuclear cells), and are thought to be triggered by small vessel vasculitis.
- Methotrexate therapy can trigger a syndrome of increased nodulosis despite good control of the joint disease of RA.
- Rheumatoid vasculitis:
- RA-associated small vessel vasculitis: It can manifest as digital infarcts, cutaneous ulcerations, palpable purpurae, distal gangrene and leukocytoclastic vasculitis. They require prompt more aggressive treatment with disease-modifying antirheumatic drugs (DMARDs).
- Other manifestations: Polyneuropathy, mononeuritis multiplex and visceral infarction (resulting in stroke and acute myocardial infarction) and mesenteric arteritis. Pyoderma gangrenosum occurs with increased frequency with RA.
- Dermatological manifestations:
- Rhematoid nodules
- Skin ulcers
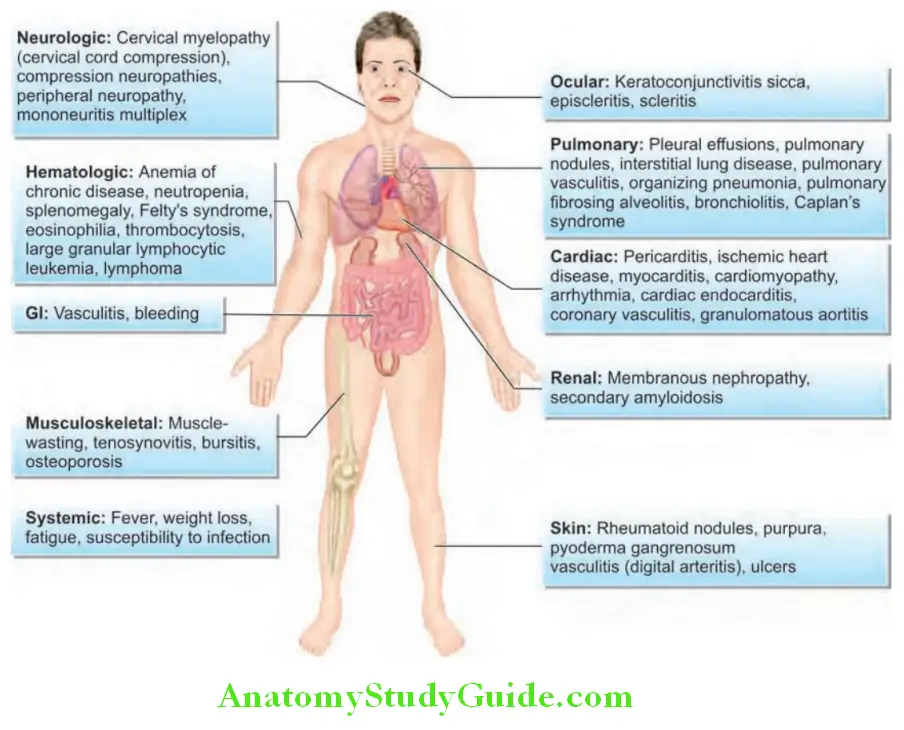
- Neutrophilic dermatosis (Sweet syndrome), pyoderma gangerenosum;
- Skin atrophy, petechiae, Raynaud’s phenomenon.
Pleuropulmonary Manifestations:
Question 17. Enlist the pleuropulmonary manifestations of rheumatoid arthritis.
Answer:
- Pleural lesions: Pleural effusions (exudative) occur more commonly in men and are usually small and asymptomatic.
- Pleural fluid is characterized by low glucose and pH.
- Pulmonary lesions:
- Rheumatoid nodules especially in men; usually solid, but may calcify, cavitate or become infected.
- Diffuse interstitial fibrosis causes dyspnea and may progress to cor pulmonale.
- Bronchiolitis obliterans with or without organizing pneumonia.
- Granulomatous pneumonitis.
- Pulmonary arteritis (vasculitis) may result in infarction.
- Caplan’s syndrome: It is characterized by the coexistence of seropositive RA and rounded fibrotic, peripherally located nodules of 0.5–5 cm diameter in the lung along with coal worker’s pneumoconiosis (CWP)/silicosis.
- Treatment induced: Methotrexate induced lung fibrosis, immunosuppression related infections like TB.
- Cardiac manifestations:
- Pericardial effusions are common. Rarely, long-standing pericardial disease results in a fibrinous pericarditis and constrictive pericarditis.
- Coronary artery disease due to premature atherosclerosis. Rarely, myocarditis, valvular involvement and conduction defects.
- Neurological manifestations:
- Peripheral nerve entrapment syndromes: For example, carpal tunnel syndrome (median nerve at the wrist), and tarsal tunnel syndrome (anterior tibial nerve at the ankle).
- Myelopathy: Due to spinal cord compression produced by acquired atlanto-axial dislocation.
- Mononeuritis multiplex: Due to vasculitis.
- Ophthalmological manifestations:
- Sjögren’s syndrome/Sicca complex: Comprising keratoconjunctivitis sicca, xerostomia and salivary (parotid) gland enlargement.
- Scleritis, episcleritis may progress to perforation of the orbit (scleromalacia perforans).
- Brown’s syndrome: Diplopia due to tendinitis of the superior oblique muscles.
- Osteoporosis: Osteoporosis secondary to rheumatoid involvements is very common especially with corticosteroid therapy and immobilization.
- Hematological manifestations:
- RBC: Normocytic normochromic anemia. Causes of anemia in RA include anemia of chronic disease, anemia due to gastrointestinal bleed secondary to NSAIDs/steroids, methotrexate induced folate deficiency, marrow suppression due to disease/drugs and part of hypersplenism if splenomegaly present.
- WBC: Eosinophilia and mild leukocytosis. Neutropenia in cases of Felty’s syndrome and large granular lymphocyte syndrome. Patients with RA are at an increased risk for infections, and this risk is further increased by some therapies.
- Platelets: Thrombocytosis.
- Felty’s syndrome: It is the triad of RA, splenomegaly, and neutropenia.
- Renal involvement: In the form of glomerulonephropathy, vasculitis, drug toxicities and secondary amyloidosis.
- Malignancies: RA patients have an increased risk of lymphomas.
Diagnosis:
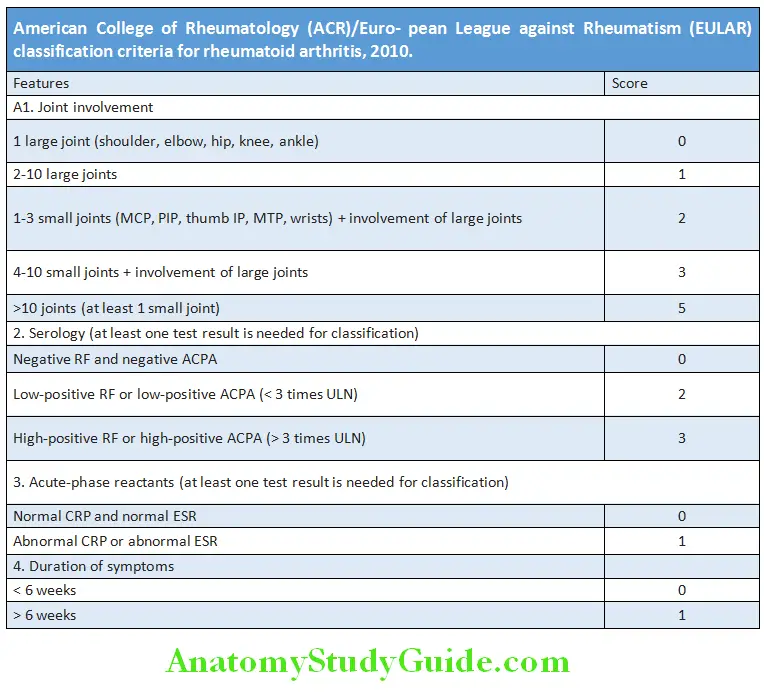
Question 18. Enumerate the ACR (American College of Rheumatology)/EULAR criteria for the diagnosis of rheumatoid arthritis.
Answer:
- Diagnostic criteria of rheumatoid arthritis: In 2010, a collaborative effort between the American College of Rheumatology and the ACR/European League against Rheumatism presented the criteria for the diagnosis of RA.
- The Disease Activity Score in 28 joints (DAS28) is used to grade severity of RA.
- Simplified Disease Activity Index (SDAI) and Clinical Disease Activity Index (CDAI) are also used in monitoring.
American College of Rheumatology (ACR) criteria for rheumatoid arthritis:
- Morning stiffness*
- Arthritis of 3 joint areas*
- Arthritis of the hands*
- Symmetric arthritis*
- Rheumatoid nodules
- Serum rheumatoid factor positive
- Radiographic changes
These criteria must be present for more than 6 weeks. Presence of 4 criteria favors definite diagnosis of RA.
Investigations:
Question 19. Discuss the diagnostic work up for rheumatoid arthritis.
Answer:
- Serology:
- Rheumatoid factor
- Anticyclic citrullinated protein antibodies (ACPA)
- Markers of acute inflammation:
- Anemia of chronic disease is seen in majority of patients, and the degree of anemia is proportional to the activity of the disease.
- Thrombocytosis is common, and platelet counts return to normal as the inflammation is controlled.
- Acute phase reactants, erythrocyte sedimentation rate and C-reactive protein levels parallel the activity of the disease, and their persistent elevation carries poor prognosis (joint destruction and mortality).
- WBC counts may be raised, normal, or profoundly depressed (Felty’s syndrome).
- Other autoantibodies : RA is associated with other autoantibodies, such as antinuclear antibodies (30% of patients) and antineutrophilic cytoplasmic antibodies, particularly of the perinuclear type (30% of patients).
- Radiographs of the affected joints: Radiological changes include symmetrical pattern of involvement, juxta-articular osteoporosis, soft tissue swelling, bone erosions and narrowing of the joint space.
- Ultrasonography and MRI: More sensitive than plain radiographs and detects soft-tissue synovitis before joint damage. Doppler ultrasound is useful for demonstration persistent synovitis when deciding on the need for DMARDs or assessing their efficacy.
- Synovial fluid analysis: White blood cell counts typically range from 5000–50,000 per microliter with approximately twothird of the cells being neutrophils with no crystals or organism. None of the synovial fluid findings are pathognomonic.
- Testing for latent tuberculosis: Before starting biological agents.
Question 20. Develop an appropriate treatment plan for patients with rheumatoid arthritis.
(or)
Describe the various treatment options for rheumatoid arthritis.
(or)
Describe the basis for biologics and DMARDs in rheumatologic diseases.
(or)
Describe the role and current status of various disease
Answer:
Management:
The early recognition of inflammatory arthritis, before irreversible injury has occurred, is the most important element in the effective management
General Measures:
The goal of therapy is disease remission and to maintain this remission by continuing therapy.
- Rest and splinting of the involved joints during the acute stage.
- Physiotherapy: Helps in mobilization and prevention of contractures.
- Smoking cessation, control of cardiovascular risks, screening for osteoporosis
Medical Therapy:
Three types of medical therapies are used in the treatment of RA:
- NSAIDs
- Glucocorticoids
- DMARDs (both conventional and biologic).
1. Nonsteroidal anti-inflammatory drugs (NSAIDs): NSAIDs produce symptomatic reliefbecause of its analgesic and anti- inflammatory properties. However, they do not alter the underlying disease process. Therefore, they are not used without the concomitant use of DMARDs. Their chronic use should be minimized because of its side effects such as gastritis/peptic ulcer disease and impairment of renal function.
2. Glucocorticoids: These are the most potent anti-inflammatory treatments available and cause dramatic and rapid improvement. Not only they produce symptomatic improvement, but also significantly decrease the radiographic progression of RA. However, the longterm therapy is associated with mulltisystem toxicities.
- Prednisolone is the most commonly used glucocorticoid and is administered orally.
- Bridge therapy: In ‘bridge therapy’, glucocorticoids are used first to shut off inflammation rapidly and then to taper them as the DMARD is taking effect.
- Pulse therapy: Remission is induced with IM (intramuscular) depot methylprednisolone 80–120 mg if synovitis persists beyond 6 weeks. Intra-articular long-acting glucocorticoids (e.g. triamcinolone hexacetonide) are used to reduce synovitis.
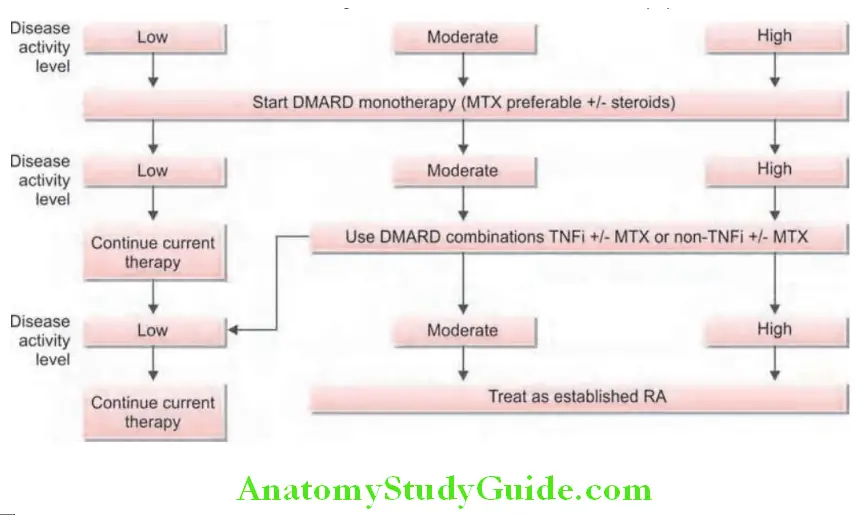
3. Disease modifying antirheumatic drugs (DMARDs):
Patients with RA must be started on DMARD therapy as soon as possible. DMARDs have the ability to inhibit greatly the disease process to modify or change the disabling potential of RA. DMARDs are so named because in most of the cases they are able to slow or prevent structural (radiographic) progression of RA.
Conventional DMARDs used in RA:
- Conventional DMARDs exhibit a delayed onset of action and take 2–6 months to exert their full effect.
- Start DMARD therapy early in the disease process. Early in the course of disease, most patients should be started on a combination of DMARDs and analgesics. Before using DMARDs, complete blood count, serum creatinine, aminotransferases, and screening for hepatitis C, hepatitis B, and latent tuberculosis infection. A chest radiograph should be obtained prior to initiating treatment with MTX.
Methotrexate: Currently, methotrexate is the DMARD of choice (considered as ‘gold standard’ drug) for RA and is the anchor drug for most combination therapies.
- Mechanism of action in RA: At the dosages used for RA, methotrexate stimulates extracellular release of adenosine from cells, which has anti-inflammatory and immunomodulatory properties. Enzymes inhibited by methotrexate in RA include thymidylate synthetase (TS) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase. It should not be prescribed in pregnancy.
- Dose: Usually given orally in the starting dose of 2.5–7.5 mg/week as a single dose. If there is nopositive response within 4–8 weeks, and there is no toxicity, the dose should be increased by 2.5–5 mg/week each month to 15–25 mg/week before considering, the treatment a failure. Oral absorption of methotrexate is variable. If oral treatment is not effective, it is given by subcutaneous injections. It should be monitored with full blood counts and liver biochemistry.
- Folic acid, 1 to 4 mg/day (or 5 mg once a week, on the day following methotrexate dose), reduces most methotrexate associated toxicities (e.g. gastrointestinal intolerance, stomatitis, hepatotoxicity, hyperhomocysteinemia, alopecia) without apparent loss of efficacy. If methotrexate alone does not sufficiently control RA, it is combined with other DMARDs.
Other DMARDs:
- Hydroxychloroquine is used usually in combination with other DMARDs, particularly methotrexate. It is given orally at a dose of 200–400 mg daily. It is the least toxic DMARD but also the least effective as monotherapy. Regular monitoring (every 6 months to a year) by ophthalmoscopy to detect any signs of retinopathy, bull’s eye maculopathy should be done.
- Sulfasalazine: It is effective when given in doses of 1–3 g daily. Monitoring of blood cell counts is recommended, particularly WBC counts, in the first 6 months. Combination of sulfasalazine + hydroxychloroquine + methotrexate is referred to as triple therapy.
- Leflunomide is a pyrimidine antagonist, also inhibits enzyme dihydroorotate dehydrogenase, interfering with cell signal transduction. It has a very long half-life and is given daily in a dose of 10–20 mg. The most common toxicity is diarrhea, which may respond to dose reduction. Leflunomide is teratogenic and hepatotoxic. It is used as monotherapy or in combination with methotrexate and other DMARDs.
- Others: These include minocycline, gold salts, penicillamine and cyclosporine are used sparingly now.
Conventional DMARDs used in RA:
- Methotrexate (MTX)
- Hydroxychloroquine
- Sulfasalazine
- Leflunomide
- Azathioprine
- Gold (auranofin)
- Minocycline
- D-penicillamine
Biologic DMARDs/Biologicals:
Question 21. List the biologicals used in the treatment of rheumatoid arthritis (RA).
Answer:
- They are usually given along with methotrexate or other conventional DMARDs.
- Disadvantages: High cost and long-term toxicities, notably infections (especially cellulitis, septic joints, tuberculosis, histoplas- mosis, coccidioidomycosis, and Listeria) and demyelinating syndromes.
TNF-α Blockers:
- Etanercept, a recombinant TNF receptor fusion protein. Administered subcutaneously in a dose of 25 mg twice weekly.
- Infliximab is a mouse/human chimeric monoclonal antibody against TNF-α. It is given intravenously (3–10 mg/kg) every 4–8 weeks.
- Adalimumab is a recombinant human IgG1 monoclonal antibody directed against TNF-α. It is administered subcutaneously in a dose of 40 mg every other week.
- Newer TNF inhibitors, certolizumab pegol and golimumab, are also effective.
IL-1 Receptor Blocker:
- Anakinra, a recombinant human interleukin-1 receptor antagonist. It is given subcutaneously in a dose of 100 mg daily.
- Tocilizumab and sarilumab are two newer agents.
Anti-CD20 Antibodies:
- Rituximab is a genetically engineered chimeric murine/human monoclonal antibody directed against the CD20 molecule found on the surface of B cells. It is given intravenously as two infusions 2 weeks apart for the treatment of patients with refractory RA.
T-cell Agent:
- Abatacept is a recombinant fusion protein of the extracellular domain of human CTLA4 and a fragment of the Fc domain of human IgGIt is administered by intravenous infusion every 4 weeks in a dose of approximately 10 mg/kg.
JAK inhibitors:
- Tofacitinib, baricitinib, peficitinib, and upadacitinib, can also be used in combination with MTX or as monotherapy.
Immunosuppressants: These are used as third-line drugs for RA that recurs or does not respond to second-line agents. These include
cyclophosphamide and azathioprine. However, in RA with acute vasculitis causing serious organ involvement, intravenous administration
of cyclophosphamide may be lifesaving.
Surgery:
Surgical therapy may improve pain and disability when there is failure of medical therapy. It is useful in maintenance of joint function,
and prevention and correction of deformities. Surgical procedures of the joint include
- Synovectomy of the inflammed joint before joint damage;
- Osteotomy to correct the deformity and relieve pain in the upper end of the tibia, and excision of the lower end of the ulna;
- Arthrodesis of the joint to relieve pain in the atlanto-axial joint, wrists, ankle and subtalar joints;
- Excision arthroplasty
- Joint replacement.
Physiotherapy, Rehabilitation and Assistive Devices:
Exercise and physical activity improve muscle strength. Judicious use of wrist splints can decrease pain.
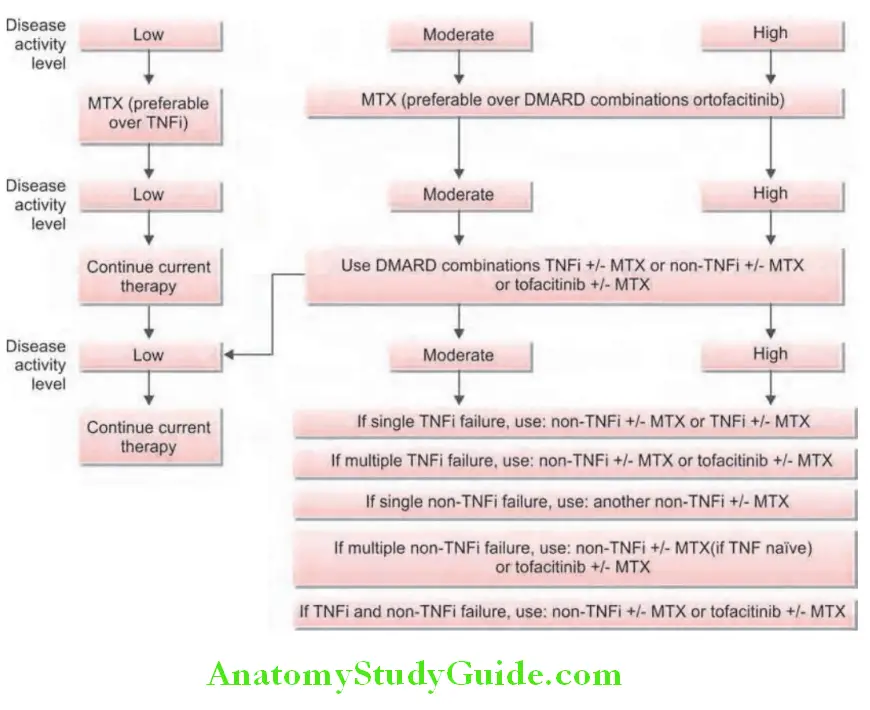
Treatment algorithm for rheumatoid arthritis is presented :
Felty’s Syndrome:
Question 22. Write short essay/note on Felty’s syndrome and its components.
Answer:
- Felty’s syndrome is the triad of RA, neutropenia and splenomegaly.
- It is seen in patients with long-standing chronic, severe and seropositive RA.
- Autoantibodies to deiminated histones (predominantly histone H3) and neutrophil extracellular chromatin traps (NETs) are postulate as the cause.
Clinical Features:
Clinical features of Felty’s syndrome:
- Features of RA (rheumatoid arthritis)
- Splenomegaly
- Lymphadenopathy
- Skin pigmentation
- Fever
- Weight loss
- Rarely recurrent infections and chronic leg ulcers
- Vasculitis
- Subcutaneous nodules
- Portal hypertension
- Higher risk of Hodgkin’s lymphoma
Laboratory Findings:
- Blood: Neutropenia 3 (absolute neutrophil count below 2000/microL), anemia and thrombocytopenia.
- Rheumatoid factor (RF): High titers.
- Circulating immune complexes (CIC): Increased levels.
Treatment:
- Do not require special specific therapy; instead treat underlying severe RA. Immunosuppressive agents particularly methotrexate and azathioprine are beneficial.
- G-CSF and GM-CSF can be tried.
- Splenectomy is indicated in: Severe and persistent neutropenia (< 500 cells/mm 3), recurrent or serious bacterial infections, chronic, nonhealing leg ulcers, hypersplenism producing severe anemia or thrombocytopenic hemorrhage.
Prognosis:
Poor with an increased mortality due to increased incidence of severe infection.
Nonsteroidal Anti-inflmmatory Drugs (NSAIDs):
Nonsteroidal anti-inflammatory drugs are the most widely used drugs, but they have an increased risk of cardiovascular disease. Oral NSAIDs are used for the management of pain due to inflammation.
Mechanism of NSAID action: Arachidonic acid (AA) is derived from membrane phospholipid and its metabolism occurs along two major enzymatic pathways namely; cyclooxygenase pathway (produces prostaglandins by the cyclooxygenase) (COX) and lipoxygenase pathway (produces leukotrienes by 5-lipoxygenase).
Question 23. Write briefly on COX enzymes and COX-2 inhibitors.
Answer:
Traditional NSAIDs versus COX-2 Inhibitors:
- Traditional NSAIDs (e.g. ibuprofen, diclofenac, and naproxen) exert their anti-inflammatory effect by inhibiting synthesis of prostaglandin from arachidonic acid by blocking both COX enzymes. They do not have a diseasemodifying effect in either osteoarthritis or inflammatory rheumatic diseases.
- Inhibition of COX-1 is required for anti-inflammatory and analgesic effects, but can damage the mucosa of stomach and duodenum and is associated with an increased risk of upper gastrointestinal ulceration, bleeding and perforation. Simultaneous administration of omeprazole (20 mg daily) or misoprostol (200 μg twice or 3 times daily) reduces the risk of NSAID-induced ulceration and bleeding. Other side-effects include fluid retention, renal impairment due to inhibition of renal prostaglandin production, and rashes.
- COX-2 (cyclooxygenase-2) selective NSAIDs (e.g. celecoxib, etoricoxib, etodolac, rofecoxib and valdecoxib) selectively inhibit COX-They have analgesic and anti-inflammatory properties similar to traditional NSAIDs. However, they are much less likely to cause gastrointestinal toxicity and have minimal antiplatelet effects. Similar to traditional NSAIDs, they can produce significant changes in renal function, and hence, should be cautiously used in patients with diabetes, dehydration and congestive heart failure.
- They play an important role in the management of inflammation and pain caused by arthritis. It has been observed that there is a higher risk of myocardial infarction and stroke (thromboembolic complications) in patients using COX-2 inhibitors compared to traditional NSAIDs. Hence, two COX-2 inhibitors namely rofecoxib and valdecoxib have been withdrawn. NSAIDs like diclofenac, nabumetone, meloxicam, and etodolac, are also relatively selective for COX-2 at lower doses.
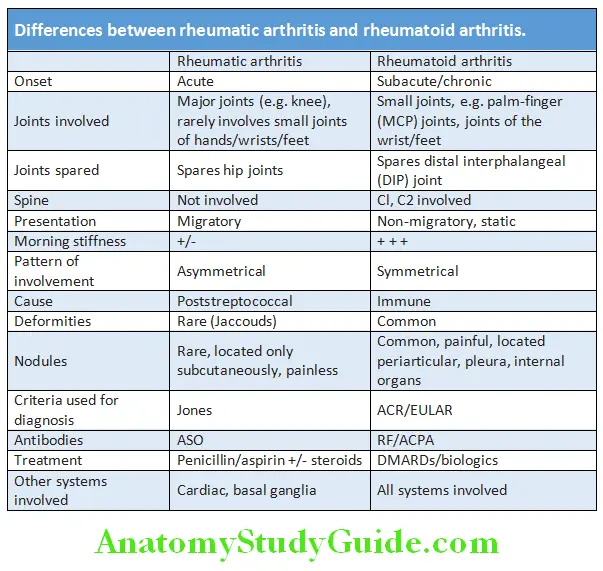
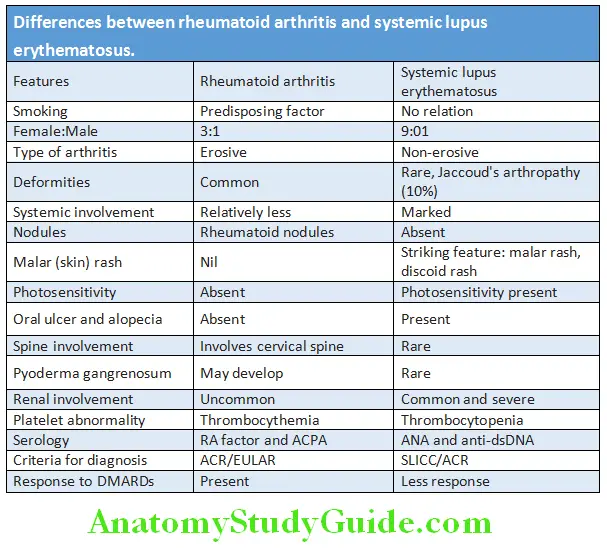
Differences between Rheumatic Arthritis and Rheumatoid Arthritis:
Question 24. Differentiate between rheumatic arthritis from rheumatoid arthritis.
Answer:
Systemic Lupus Erythematosus:
Question 25. Describe the clinical manifestations, diagnosis and management of systemic lupus erythematosus (SLE).
Answer:
- Systemic lupus erythematosus (SLE) is a chronic, multisystemic autoimmune disease that results from immune systemmediated tissue damage and is characterized by the presence of broad spectrum of autoantibodies.
- Arthralgia and rashes are the most common clinical features, whereas cerebral and renal diseases constitute most serious problems.
- Age: It usually occurs in young women between 20 to 30 years, but may manifest at any age.
- Sex: It predominantly affects women, with female-to-male ratio of 9:1.
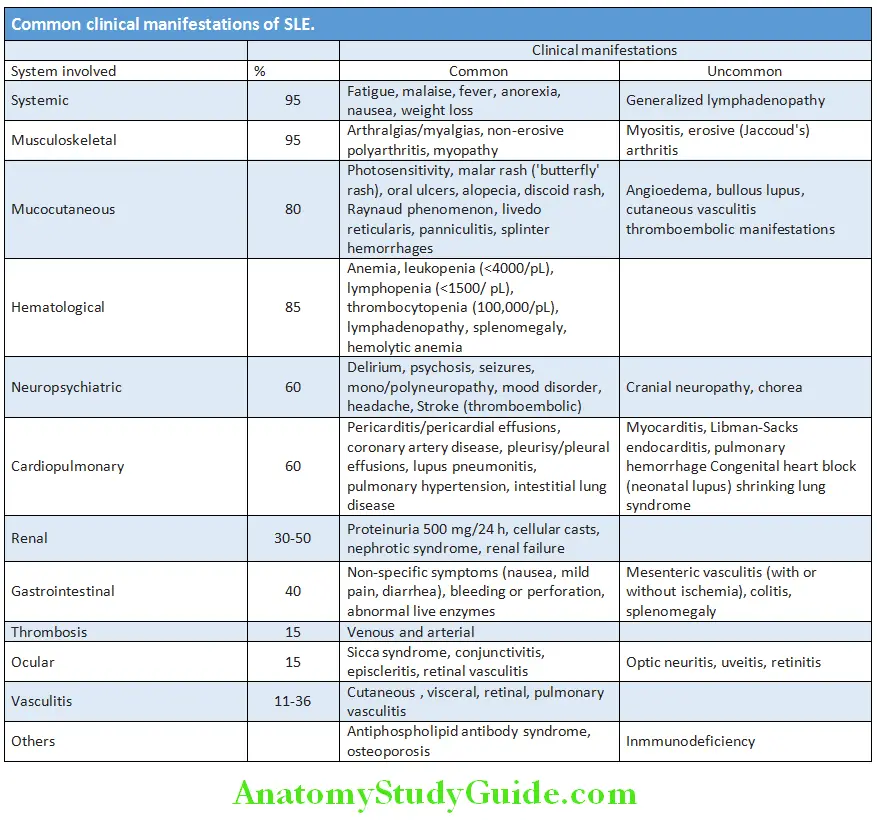
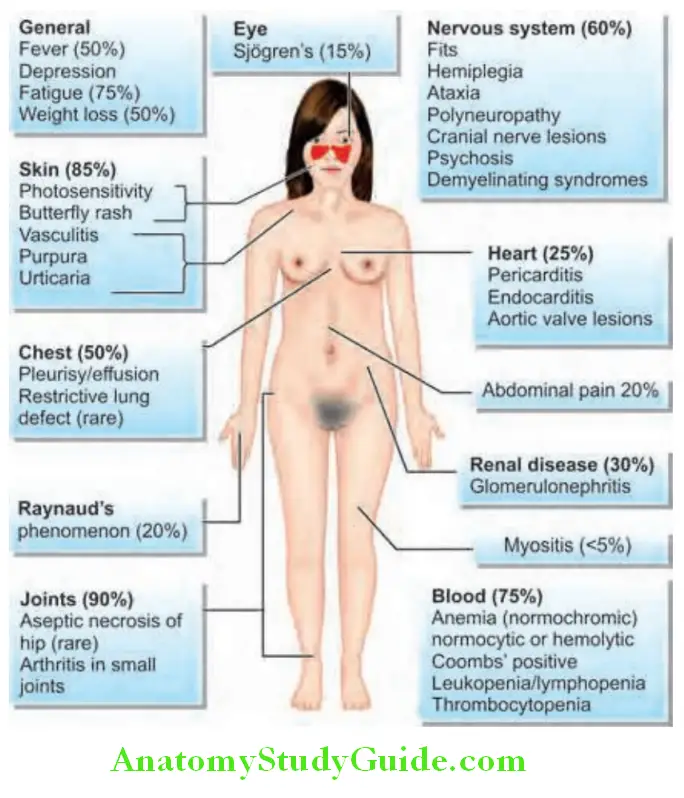
Clinical Features:
Question 26. Write short essay/note on clinical features of systemic lupus erythematosus
Answer:
Musculoskeletal Manifestations:
- Arthralgia and nonerosive arthritis are most common (>85%).
- Commonly involves proximal interphalangeal and metacarpophalangeal joints of the hand, along with the knees and wrists.
- In about 10% of patients, deformities result from damage to periarticular tissue, a condition termed Jaccoud’s arthropathy.
Cutaneous Manifestations:
- Classic malar rash consists of erythematous (flat or raised) facial rash with a butterfly distribution across the malar and nasal prominences and sparing of the nasolabial folds and is seen in 30–60% of patients. Butterfly rash is often triggered by sun exposure.
- Discoid lupus is a benign variant of lupus in which only the skin is involved. Discoid rash consists of erythematous, slightly raised patches with adherent keratotic scaling and follicular plugging. Discoid rash without any systemic features occurs in discoid lupus erythematosus (DLE). Only 5% of patients with DLE have SLE; however, as many as 20% SLE patients have DLE. This rash is primarily seen on the face and scalp.
- Generalized photosensitivity (skin rash on exposure to sunlight) and alopecia.
Mucous Membrane Manifestations:
Recurrent crops of small, painful ulcerations on the oral or nasal mucosa. Dryness secondary to Sjögren’s syndrome.
Hematologic Manifestations:
Antibodies that target each of the cellular blood elements are responsible for hematological changes.
- RBC: Normocytic normochromic anemia, reflecting chronic illness. Hemolytic anemia: Coombs’ test positive or microangiopathic hemolysis.
- WBC: Leukopenia, particularly lymphopenia.
- Platelets: Idiopathic/immune thrombocytopenic purpura induced by antiplatelet antibodies.
- Antibodies to clotting factors can contribute to impaired clot formation and hemorrhage.
Renal Manifestations:
Question 27. Enumerate the renal manifestations of SLE.
Answer:
- Kidney may be involved in 30–50% of SLE patients and is one of the most important organs involved.
- Often asymptomatic in most lupus patients, particularly initially. Hence, urinalysis should be done in any person suspected of having SLE followed by regular urinalysis and blood pressure monitoring.
- Characterized by proteinuria (>500 mg/24 hours) and/or cellular (red cell) casts.
- Histological classification of lupus nephritis.
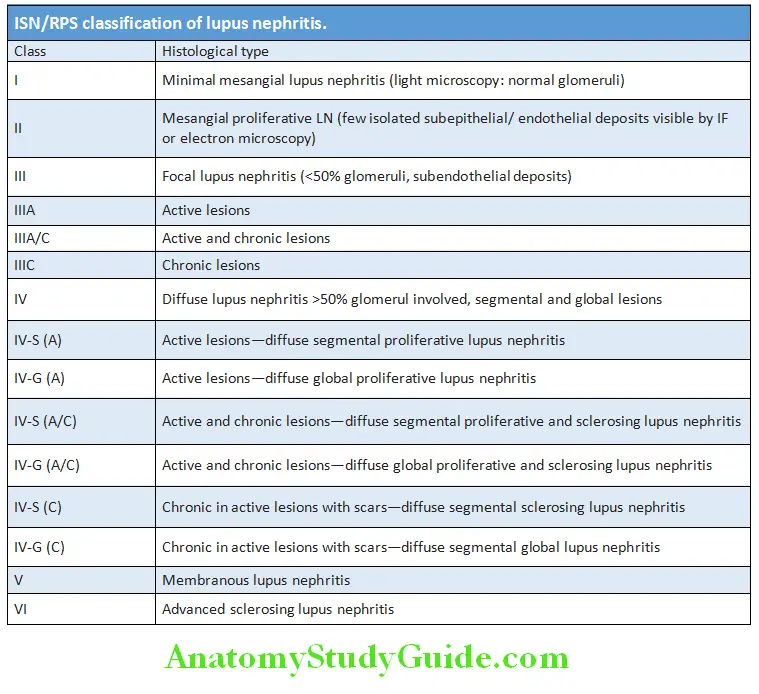
Neuropsychiatric Features:
- Nervous system involvement in SLE produces both
- neurologic and psychiatric manifestations.
- Clinical features include cognitive dysfunction (due to SLE cerebritis), headache and seizures, psychiatric features including depression and psychosis. Peripheral nervous system involvement can cause neuropathy. Cranial nerve and ocular involvement due to vasculopathy.
Cardiac Manifestations:
- Pericarditis most frequent; serious manifestations are myocarditis and Libman-Sack (noninfective endocarditis involving the mitral valve) endocarditis.
- Increased risk for myocardial infarction, usually due to accelerated atherosclerosis and vasculitis.
Pulmonary Manifestations:
- Pleural lesions: Pleurisy with or without pleural effusion—most common pulmonary manifestation. Pleural effusion is exudative with low C3 and ANA test positive in the pleural fluid.
- Lung lesions: Pneumonitis (exclude infection before ascribing a lung lesion to SLE), shrinking lung syndrome, interstitial inflammation leading to fibrosis and intra-alveolar hemorrhage.
Gastrointestinal Features:
Non-specific diffuse abdominal pain (caused by autoimmune peritonitis and/or intestinal vasculitis), nausea sometimes with vomiting and diarrhea. Increases in serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT). Mesenteric vasculitis can cause infarction or perforation of small intestine.
Ocular Manifestations:
- Sicca syndrome (Sjogren’s syndrome) and nonspecific conjunctivitis are common.
- Serious manifestations are retinal vasculitis (can cause infarcts and cytoid bodies) and optic neuritis. Complications of glucocorticosteroid therapy include cataracts (common) and glaucoma.
Other Manifestations:
Kikuchi-Fujimoto disease (splenomegaly, lymphadenopathy), antiphospholipid syndrome, osteoporosis, and complement deficiencies are associated with SLE. Increased risk of hematologic malignancies (particularly non-Hodgkin’s lymphoma) and possibly lung and hepatobiliary cancers.
Poor progmnostic factors in SLE include diffuse proliferative glomerulonephritis, male sex, older age at presentation, black race, hypertension and antiphospholipid antibody syndrome.
Clinical features are summarized diagrammatically Causes of death in SLE.
Causes of death in SLE:
- Infections and renal failure: Cause of death in the first decade of disease
- Thromboembolic events: Cause of death in the second and later decades
- Cardiovascular: Due to premature atherosclerosis
Laboratory Findings:
Purpose:
- To establish or rule out the diagnosis
- Follow The Course Of Disease
- To identify adverse effects of therapies.
Autoantibodies in SLE:
Question 28. Describe the diagnostic work up and the role of autoantibodies in systemic lupus erythematosus (SLE).
Answer:
- SLE is characterized by the production of several diverse autoantibodies. Some antibodies are against different nuclear and cytoplasmic components of the cell that are not organ specific. Other antibodies are directed against specific cell surface antigens of blood cells.
- Importance of autoantibodies:
- Diagnosis and management of patients with SLE
- Responsible for pathogenesis of tissue damage.
Types of Antibodies:
- Antinuclear antibody (ANA): Best screening test and >90% of the patients show a positive test. However, a positive test is not specific for SLE. It can be positive in some normal individual (especially elderly), other autoimmune diseases, acute viral infections, chronic inflammatory processes and with certain drugs.
- Anti-double-stranded DNA (anti-dsDNA) antibodies are common in SLE. Anti-Sm antibodies are highly specific for SLE. Rising levels of anti-dsDNA and low levels of complement (C3 and C4) usually reflect disease activity.
- Rheumatoid factor is positive in 30%.

Other Tests:
- Complete blood count: Essential and aids in diagnosis and management. Anemia (normocytic normochromic and Coombs’ positive), leucopenia, lymphopenia and thrombocytopenia may be present.
- Activated partial thromboplastin time (aPTT): Prolonged in the presence of pathogenic antiphospholipid antibodies. These antibodies give a false-positive result in the serologic test for syphilis.
- Erythrocyte sedimentation rate (ESR): Nonspecific indicator of systemic inflammation. It is often monitored in many patients to know the disease activity.
- C-reactive protein (CRP): It is an acute phase reactant which is relatively uninformative and normal in SLE. It does not usually rise with disease activity unless there is infection, arthritis or serositis.
- Urinalysis: When there is active nephritis, urinalysis shows proteinuria, hematuria and cellular (red and white blood cell casts in the urinary sediment suggest proliferative glomerulonephritis) or granular casts. Blood urea nitrogen and serum creatinine are elevated in acute kidney injury. If there is proteinuria, urine albumin/creatinine ratio should be measured. Renal biopsy confirms renal involvement.
- Complement levels: Low levels of C3 indicate active disease especially nephritis.
- LE cell: Phagocytic leukocyte (neutrophil or macrophage) that has engulfed the denatured nucleus of an injured cell is positive. Rarely done nowadays.
Imaging:
- Chest X-ray: To exclude other pathology, for pulmonary and cardiac pathology.
- High resolution CT: To demonstrate fibrotic lung.
- Magnetic resonance imaging (MRI) of brain or spinal cord in cases with central nervous system disease involvement.
- Echocardiography to diagnose pericardial and endocardial involvement.
Diagnostic Criteria for Systemic Lupus Erythematosus:
Question 29. List the diagnostic criteria/revised American Rheumatism Association criteria for systemic lupus erythematosus (SLE). The clinical presentation of SLE is so variable that the American College of Rheumatology has established a complex set of criteria
Answer:
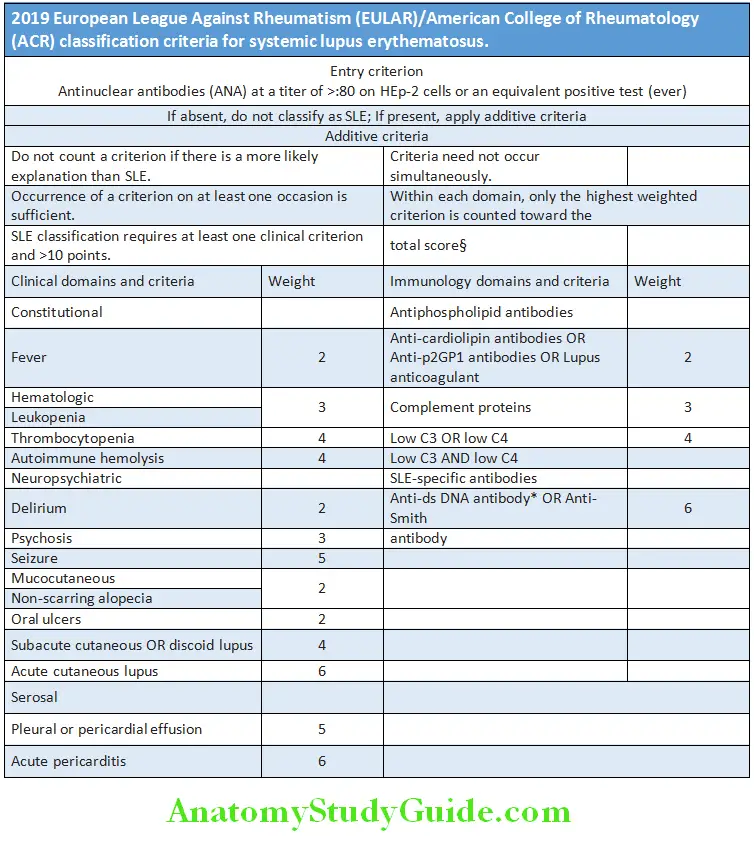
Systemic Lupus International Collaborating Clinics (SLICC) Classification 2012 criteria:
Differences between rheumatoid arthritis and systemic lupus erythematosus are presented in Table:
Management:
Question 30. Describe the management principles of systemic lupus erythematosus.
Answer:
General:
- To avoid known triggers of disease exacerbation, such as ultraviolet light, smoking cessation.
- Need for adequate rest.
Conservative therapies for management of non-life-threatening disease:
In patients with fatigue, pain, and autoantibodies of SLE, but without major organ involvement are managed by analgesics (NSAIDs), antimalarials (hydroxychloroquine, chloroquine, and quinacrine) and low-dose corticosteroids. Skin lesions and arthritis also respond to hydroxychloroquine. Photosensitive skin lesions need application of sun-screen lotions.
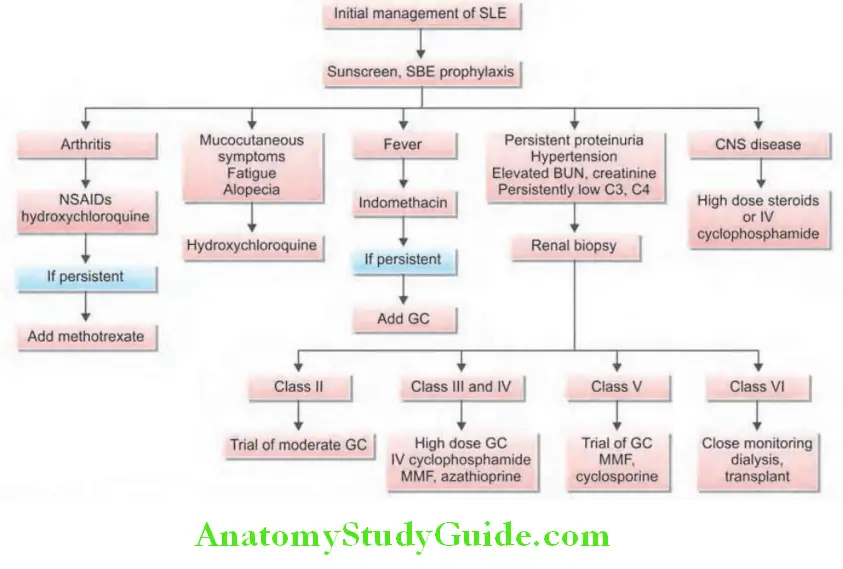
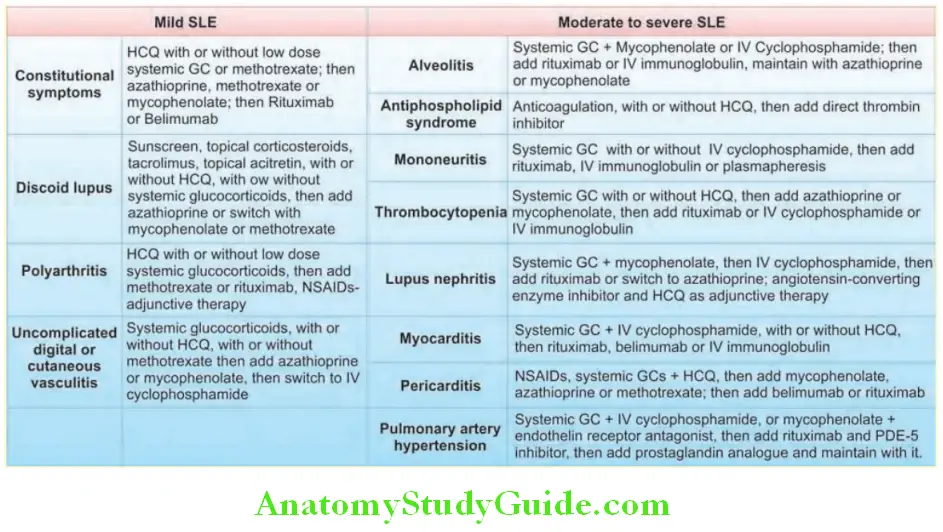
Life-threatening SLE Proliferative forms of lupus nephritis:
- Life-threatening SLE or patients with severe symptoms should receive corticosteroids.
- Acutely ill patients and patients with active proliferative glomerulonephritis may be treated with prednisone at 60 mg daily or 1 g of intravenous methylprednisolone administered daily for 3 days.
- Immunosuppressive agents: These include azathioprine, methotrexate, cyclophosphamide, my cophenolate mofetil and are also useful in controlling severe disease. They are particularly useful in patients with renal involvement. A combination of intravenous cyclophosphamide and steroids is the most effective regimen. Following intravenouscyclophosphamide, oral mycophenolate mofetil is an alternative to maintain remission.
- Drugs targeting B-cell pathways, such as belimumab and rituximab are used in refractory cases.
Drug-induced Lupus:
Question 31. Write short essay/note on drug-induced lupus.
Answer:
It is a syndrome consisting of positive ANA associated with symptoms such as fever, malaise, arthritis or intense arthralgias/
myalgias, serositis, and/or rash.
Etiology: The syndrome develops during therapy with certain medications and includes:
- Drugs: Antiarrhythmics (e.g. procainamide, diltiazem, disopyramide, and propafenone), antihypertensive (e.g. hydralazine), methyl dopa, angiotensin-converting enzyme inhibitors, beta blockers, antithyroid propylthiouracil, antipsychotics (e.g. chlorpromazine and lithium), anticonvulsants (e.g. carbamazepine and phenytoin), antibiotics (e.g. isoniazid, minocycline, and macrodantin), antirheumatic (e.g. sulfasalazine), diuretic (e.g. hydrochlorothiazide) and antihyperlipidemics (e.g. lovastatin and simvastatin).
- Biologic agents: Interferons and TNF inhibitors.
Symptoms: Usually resolve after discontinuation of the offending drug.
Features that differentiate drug-induced lupus from SLE.
Drug-induced lupus:
- Less female predilection than SLE
- Kidneys or brain involvement rare, less cutaneous involvement
- Antinuclear antibodies positive; antihistone antibodies
- Positive (95%); autoantibodies to dsDNA absent
- C3 levels normal
Seronegative Arthritis:
Question 32. Differentiate seropositive arthritis from seronegative arthritis.
Answer:
- Arthritis can be divided into seropositive and seronegative.
- Seropositive arthritis refers to the presence of rheumatoid factor and anti-CCP in the blood.
- Seronegative arthritis generally called ‘seronegative spondyloarthropathy’ and include any losing spondylitis, psoriatic arthritis and reactive arthritis.
Spondyloarthropathies (Spondyloarthritides):
Question 33. Write short essay/note on spondyloarthropathies.
(or)
Write short essay/note on seronegative spondyloarthropathies (SSA).
Answer:
Definition: The spondyloarthropathies (SpAs) are a group of related inflammatory joint diseases that share clinical features and genetic susceptibility.
Diseases included under spondyloarthropathies (SpAs):
- Ankylosing spondylitis (AnS)
- Reactive arthritis (ReA) (including Reiter’s syndrome)
- Psoriatic arthritis (PsA)
- Arthropathy associated with inflammatory bowel disease
- Undifferentiated spondyloarthritis
Common Features of SpAs:
- Common clinical features:
- Strong predilection for the spine, in particular the sacroiliac joints. Inflammatory back pain due to sacroiliitis and spondylitis.
- Tendency for new bone formation at sites of chronic inflammation, with joint ankylosis as a consequence.
- If peripheral arthritis occurs, it is usually in the lower extremity and asymmetrical.
- Predilection for sites of tendon insertion into bone (enthesis). Enthesitis is inflammation at sites where tendons, ligaments or joint capsules attach to bone and is one of the most specific clinical manifestations of the SpAs.
- RF negative; hence, known as ‘seronegative’.
- Common genetic predisposing factor: All subsets have an association with HLA-B27 allele.
Ankylosing Spondylitis:
Question 34. Discuss the clinical features, diagnosis and management ankylosing spondylitis, rheumatoid spondylitis or BekhterevStrümpell-Marie disease.
Answer:
Ankylosing spondylitis (AnS) is a chronic inflammatory disorder of unknown causes that primarily affects the axial skeleton (predominantly sacroiliac joints and spine). The peripheral joints and extra-articular structures are also frequently involved.
Age and gender: Usually begins in the second or third decade; male to female ratio is between 2:1 and 3:1.
Clinical Features:
Targets in ankylosing spondylitis: Mainly involves spine, sacroiliac joints and large peripheral joints (hip, shoulder) in an asymmetrical pattern.
Involvement of lumbar spine:
- Classical presentation is the insidious onset of inflammatory low back pain. The pain is dull and located in the lower lumbar regions or gluteal region and shows nocturnal exacerbation.
- Back pain persists for >3 months. It is accompanied by early-morning stiffness that typically improves with activity/exercise and exacerbated by inactivity.
- “Inflammatory back pain” typically exhibits at least four of the following five features:
- Age of onset <40 years
- Insidious onset
- Improvement with exercise
- No improvement with rest
- Pain at night
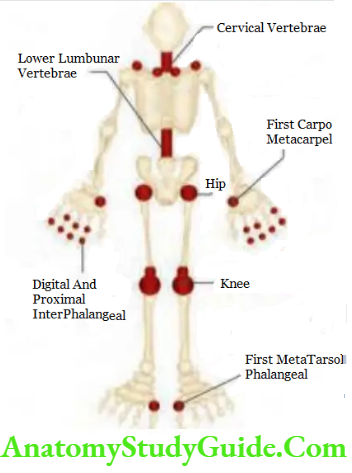
- Physical examination: Physical examination of the spine shows loss of spinal mobility in all directions and pain on sacroiliac stressing.
- Modified Schober test: Used to measure lumbar spine flexion. The patient stands upright with heels together. Two marks are made on the spine; one at the lumbosacral junction (identified by a horizontal line between the posterosuperior iliac spines) and another 10 cm above. The patient then bends forward maximally with knees fully extended. The distance between the two marks is measured. This distance increases by >5 cm in the case of normal mobility and by <4 cm in the case of decreased mobility.
Involvement of sacroiliac joint:
The tests mentioned below produce stress and pain in the sacroiliac joint.
- Patrick’s test: Pain in the sacroiliac joints may be elicited either by direct pressure or by maneuvers that stress the joint, namely, Patrick’s test. One leg is guided into ‘figure of 4’ position with the ipsilateral ankle resting across the contralateral thigh. The ipsilateral knee is then pressed downwards with one hand while providing counter pressure with the other hand on the contralateral anterior superior iliac spine.
- FABER (flexion abduction external rotation) maneuver: Patient lies supine while the examiner flexes and externally rotates the hip.
- Gaenslen’s maneuver: The examiner extends the hip by letting the leg dangle off the side of the examining table.
- Occiput to wall test (Flesch test): Normally zero, in AnS it is increased.
Enthesitis:
- Enthesitis is inflammation at the sites of tendon/ligament insertions.
- Heel pain due to inflammation at the Achilles tendon (Achilles tendinitis) and plantar fascia (plantar fasciitis) at calcaneal insertions is common. Like arthritis, enthesitis is also aggravated by rest and improved with activity.
- Other sites of enthesitis include superior and inferior aspects of patella, metatarsal heads and spinal ligament insertions on vertebral bodies.
Other sites:
- Involvement of the thoracic spine, costovertebral joints and costosternal joints leads to chest pain, reduced expansion of chest (<5 cm) and thoracic kyphosis.
- Involvement of the cervical spine produces neck pain and a forward stoop of the neck.
- Peripheral arthritis usually occurs late and asymmetric.
- Involvement of hips and shoulders result in pain and limitation of movement.
- Dactylitis (sausage digits) is characterized by diffuse swelling of toes or fingers.
Extra-articular manifestations:
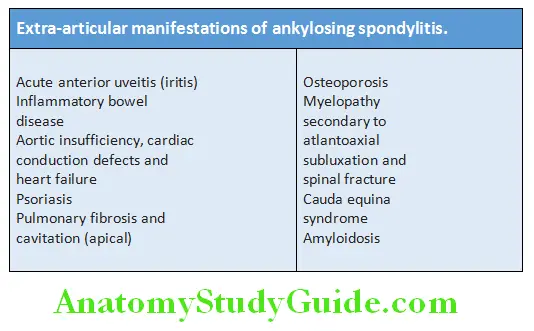
Investigations:
Describe the diagnostic work up and management of ankylosing spondylitis:
- Erythrocyte sedimentation rate (ESR) and C-reactive protein: Raised.
- Tests for rheumatoid factor (RF): Negative.
- Anti-cyclic citrullinated peptide (CCP) and antinuclear antibodies (ANAs): Negative.
- HLA-B27: Present in >90% cases. High sensitivity and specificity.
- MRI and bone scan: They can detect early sacroiliitis.
Radiographic Findings:
X-ray:
- Sacroiliac joint: The medial and lateral cortical margins of both sacroiliac joints lose definition owing to erosions. They show irregularity and loss of cortical margins (blurring of the cortical margins of the subchondral bone), erosions in the joint line, and ‘pseudowidening’ of the joint space. Subsequently, it shows subchondral sclerosis, joint space narrowing and fusion, bony ankylosis reflecting complete bony replacement of the sacroiliac joints.
- Lumbar spine: Radiographs of the spine may show anterior ‘squaring’ (loss of the normal anterior concavity of the lumbar vertebra), ‘shiny corners’ (subchondral sclerosis at the upper edge of the vertebral body), or even ‘barreling’ of one or more vertebral bodies due to erosion and sclerosis of the anterior corners of vertebrae with subsequent erosion. These are manifestations of enthesitis.
- Bridging syndesmophytes are areas of calcification that follow the outermost fibers of the annulus and may also be seen in AS. Progressive ossification may lead to formation of marginal syndesmophytes make the diagnosis clear. They are observed as bony bridges connecting successive vertebral bodies anteriorly and laterally. In advanced disease, ossification of the anterior longitudinal ligament and facet joint fusion may also be seen. Eventually, the changes may result in a ‘bamboo spine’, so called because the multiple bridging syndesmophytes (bridging the intervertebral spaces) can mimic the appearance of bamboo.
- Diffuse osteoporosis of spine and atlantoaxial dislocation can be seen as late feature. Erosive changes may be observed in the symphysis pubis, the ischial tuberosities and peripheral joints.
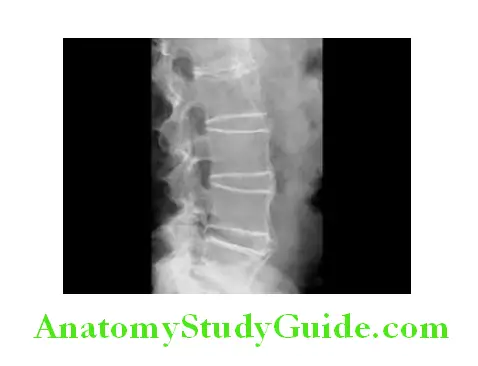
Magnetic resonance imaging (MRI):
MRI is much more sensitive for detection of early sacroiliitis than X-ray and can also detect inflammatory changes in the lumbar spine.
Diagnostic Criteria:
Modified New York Criteria (1984) for ankylosing spondylitis:
Clinical criteria:
- Low back pain and stiffness for >3 months that improve with exercise but are not relieved by rest
- Limitation of motion of the lumbar spine in both sagittal and frontal planes
- Limitation of chest expansion
Radiologic criteria:
- Sacroiliitis: Grade ≥2 bilateral or grade 3 or 4 unilateral
Grading:
- Definite AS (ankylosing spondylitis) if the radiologic criterion is associated with at least one clinical variable.
- Probable ankylosing spondylitis (AS) if:
- The three clinical criteria are present
- The radiologic criterion is present without the clinical criteria

Management:
- Early diagnosis is important to maintain posture and range of motion.
- Regular exercise is started with active and passive physiotherapy before syndesmophytes have formed.
- Symptomatic relief of pain and stiffness can be obtained with long-acting or slow-release NSAID or an NSAID suppository improves sleep, peripheral arthritis and enthesitis are managed with NSAIDs or local steroid injections. Indomethacin is the most effective drug and may be given up to a maximum dose of 100 mg/day. Systemic glucocorticoids have limited role.
- Sulfasalazine and methotrexate are useful to control effective for peripheral arthritis but not for spinal disease.
- Peripheral arthritis and enthesitis are managed with NSAIDs or local steroid injections. Rarely systemic corticosteroids may be necessary.
Anti-tumor necrosis factor therapy:
Biologic agents, such as monoclonal antibodies to TNF-α (infliximab, adalimumab, certolizumab, golimumab) or the soluble TNF receptor (etanercept) are options for patients with active ankylosing spondylitis who are not satisfactorily responded to NSAIDs. Duration of treatment is 2–3 years.
Dosage:
- Infliximab: 3–5 mg/kg body weight intravenously every 6–8 weeks.
- Etanercept: 25 mg subcutaneously twice a week initially/50 mg once weekly.
- Adalimumab: 40 mg biweekly by subcutaneous injection.
- Golimumab: 50 or 100 mg every 4 weeks, given by subcutaneous injection.
- Secukinumab is an interleukin-17 monoclonal antibody that has been approved for treatment of ankylosing spondylitis.
Others:
- Reconstructive surgery
- Pamidronate
- Control of uveitis by local steroids
Reactive Arthritis:
Question 35. Describe the etiology, clinical features, diagnosis and management of reactive arthritis.
(or)
Write a short note on Reiter’s syndrome.
Answer:
Reactive arthritis (ReA) refers to acute nonpurulent arthritis (aseptic arthritis) complicating an extra-articular infection elsewhere in the body, most typically enteric (GIT) or urogenital (GU tract) infections.
Classical triad of Reiter’s syndrome is:
- Nonspecific Urethritis
- Conjunctivitis
- Arthritis.
However the term is no longer in common use.
Etiology:
- GIT infections: Common enteric organisms that trigger include Salmonella typhimurium, Shigella flexneri, Yersinia enterocolitica, Escherichia coli, Clostridum difficile, and Chlamydia.
- GUT infection: Common urogential organisms are Chlamydia trachomatis or Ureaplasma urealyticum.
- More than 75% of ReA patients have the histocompatibility antigen HLA-B27.
Clinical Features:
- Age and gender: Predominantly a disease of young (18–40 years) men, with a male preponderance of 15:1.
- Constitutional symptoms are common and include fatigue, malaise, fever, and weight loss.
- Arthritis: Arthritis is typically an acute, asymmetrical, lower limb (e.g. knees, ankles, midtarsal and MTP joints) arthritis and occurs 1 to 3 weeks after the GIT or GUT infection, arthritis usually persists for 3–5 months.
- Enthesitis is common.
- Dactylitis or ‘sausage digit,’ is a diffuse swelling of a solitary finger or toe may also be seen. It is the result of inflammatory changes affecting the joint capsule, entheses, periarticular structures, and periosteal bone.
- Sacroiliitis may be seen in 25% patients.
- Extra-articular features
- Urogenital lesions: Urethritis and prostatitis in males, cervicitis or salpingitis in females.
- Mucocutaneous lesions: Circinate balanitis (a painless erythematous lesion of the glans penis), keratoderma blennorrhagica (hyperkeratotic lesions on the palms of the hands or the soles of the feet) and nail dystrophy.
- Ocular disease: Common and includes conjunctivitis, anterior uveitis and acute iritis.
- Others: Cardiac conduction defects, aortic regurgitation, central or peripheral nervous system lesions, and pleuropulmonary infiltrates.
- Usually a self-limiting course of 3–12 months. About 50% of patients develop recurrent bouts of arthritis and 15–30% develop chronic arthritis or sacroiliitis.
Investigations:
- Erythrocyte sedimentation rate (ESR) and acute-phase reactants: Usually raised.
- Peripheral blood: Anemia and polymorphonuclear leukocytosis.
- Synovial fluid: Characteristic synovial fluid is leukocyte-rich (>2000 white blood cells/mL with a predominance of neutrophils) and may contain multinucleated macrophages (Reiter’s cells).
- HLA-B27: Positive in more than 75% of cases.
- Serum tests for rheumatoid factor and antinuclear antibodies: Negative.
- Urethritis: Confirmed in the ‘two-glass test’ by demonstration of mucoid threads in the first-void specimen that clears in the second specimen.
Radiological features:
- Shows periarticular osteoporosis, joint space narrowing and proliferative erosions in chronic or recurrent disease.
- Periostitis, especially of metatarsals, phalanges and pelvis, and large, fluffy calcaneal spurs at the insertion of the plantar fascia. Sacroiliitis (less common than ankylosing spondylitis) is often asymmetrical and sometimes unilateral.
Syndesmophytes are predominantly coarse and asymmetrical, often extending beyond the contours of the annulus
(‘non-marginal’).
Management:
- Acute attack:
- Treated with rest, oral NSAIDs and analgesics. Indomethacin, 75–150 mg/d in divided doses, is the initial treatment of choice.
- Intra-articular or local steroid injections and rarely systemic steroids in severe cases.
- Non-specific chlamydial urethritis: Treated with doxycycline or ciprofloxacin or a single dose of azithromycin course.
- No role of antibiotics in reactive spondyloarthropathy secondary to enteric infections.
- Sulfasalazine, azathioprine and methotrexate may be useful in patients with disabling relapsing and remitting arthritis.
- DMARDs: For patients with persistent marked symptoms, recurrent arthritis or severe keratoderma blennorrhagica.
- Anterior uveitis is a medical emergency and treated by topical, subconjunctival or systemic corticosteroids.
- TNF-α blocking agents are the drugs of next choice in severe and persistent disease.
Psoriatic Arthritis:
Question 36. Write short note on psoriatic arthritis.
Answer:
- Psoriatic arthritis (PsA) is a seronegative inflammatory arthritis that characteristically occurs in patient with psoriasis, a past or family history of psoriasis or with characteristic nail changes.
- Occurs in about 7–20% of patients of psoriasis.
Types: Wright and Moll described five patterns of psoriatic arthritis.
Newer pattern of psoriatic arthritis is described in HIV patients.
Patterns of psoriatic arthritis:
- Asymmetric inflammatory oligoarthritis
- Symmetric polyarthritis similar to RA (rheumatoid arthritis)
- Arthritis of the DIP (distal interphalangeal) joints
- Spondyloarthropathy ( sacroiliitis and spondylitis)
- Arthritis mutilans
Clinical Features:
It presents with pain and swelling affected joints and enthesitis.
- Asymmetrical inflammatory oligoarthritis: Most characteristically occur in the hands and feet, when synovitis is associated with tenosynovitis, enthesitis and inflammation of intervening tissue resulting in a ‘sausage-shaped digit’ or dactylitis. Usually, less than four joints are involved (oligoarthritis).
- Symmetrical polyarthritis: It is more common in women and may strongly resemble RA.
- Arthritis of the DIP joints: Uncommon and affects men more than women. It involves finger DIP joints and surrounding periarticular tissues, and is accompanied by nail dystrophy.
- Psoriatic spondylitis: Presents similar to AS but with less severe involvement. It is typically unilateral or asymmetric in severity.
- Arthritis mutilans is a severe, deforming erosive arthritis accompanied by prominent cartilage and bone destruction results in marked instability of fingers and toes. The encasing skin appears invaginated and ‘telescoped’ and the finger can be pulled back to its original length.
- Enthesitis and tenosynovitis is common.
- Skin lesions: Characteristic skin lesion of psoriasis may be present.
- Nail changes: Psoriatic nail dystrophy, onycholysis, pitting, and hyperkeratosis observed in nearly 80% patients with arthritis.
- Uveitis: Unilateral or bilateral and is generally chronic.
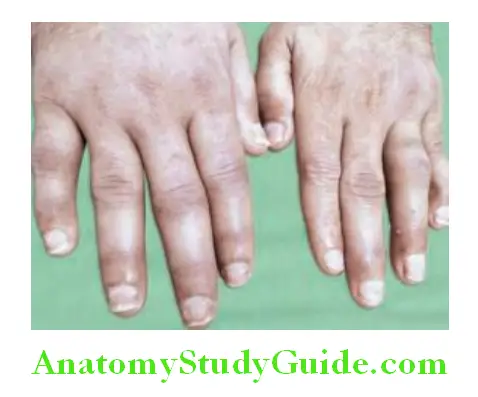
Question 37. Differentiate psoriatic arthritis from rheumatoid arthritis:
Answer:
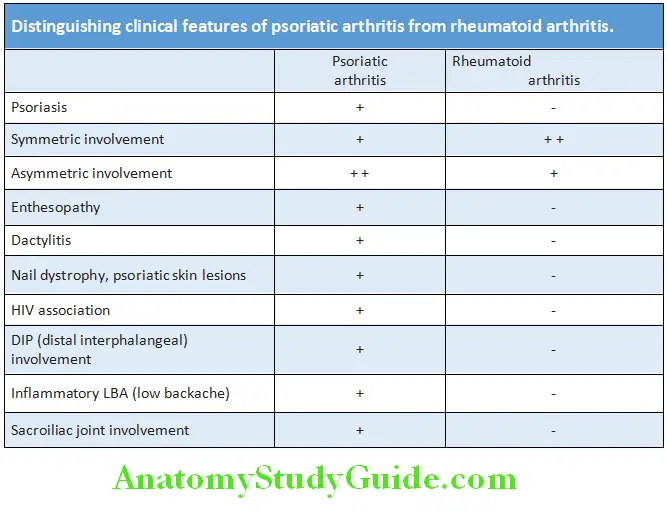
Diagnosis:
- Acute phase reactants (ESR and CRP): Raised in active disease.
- Rheumatoid factor and antinuclear antibodies: Negative.
- Radiological findings: May be normal or show erosive change with joint space narrowing similar to those of RA, but osteoporosis is relatively less common. Distal interphalangeal joints may show ‘pencil-in-cup’ changes because of marked resorption of bone. Other findings include enthesitis with periosteal reaction, sacroiliitis and spondylitis.
- MRI and ultrasound with power Doppler are used to detect synovial inflammation and inflammation at the enthuses (enthesitis). Classification of Psoriatic Arthritis (CASPAR) criteria is used for diagnosis which include skin lesions, nail changes, negative RF, dactylitis and juxta-articular bone formation—3 out of 5 need to be present.
Management:
- NSAIDs and analgesics may be sufficient to control symptoms in mild disease.
- Local synovitis may be treated by intra-articular corticosteroid injections.
- DMARDs for persistent synovitis unresponsive to conservative treatment. Sulfasalazine or methotrexate slows the development of joint damage.
- Anti-TNF-α agents (e.g. etanercept, golimumab, infliximab and adalimumab) should be considered for patients with active synovitis who respond inadequately to standard DMARDs.
- Hydroxychloroquine must be avoided as it can cause exfoliative skin reactions.
- Alefacept is a fusion protein of soluble lymphocyte function antigen 3 with Fc fragments of IgGIt is used for moderate-to-severe psoriasis and associated arthritis.
Enteropathic Arthritis:
Question 38. Write short note on spondyloarthropathy associated with inflammatory bowel disease.
Answer:
Also called as enteropathic arthritis associated with inflammatory bowel disease (ulcerative colitis and Crohn’s disease).
An acute inflammatory oligoarthritis occurs in 10–20% of patients with inflammatory bowel disease (IBD). More often in patients with Crohn’s disease than in those with ulcerative colitis.
Clinical Features:
Peripheral arthritis:
- The arthritis is asymmetrical, inflammatory, nonerosive polyarthritis and mainly involves lower-limb joints (knees, ankles, hips).
- The clinical activity of the peripheral arthritis parallels the activity of the gut inflammation. Effective treatment of the GI disease usually controls the joint disease as well.
- It is not associated with HLA-B27 and follows a transient course.
Sacroiliitis:
- It is clinically and radiologically similar to classic AS.
- It can manifest before or after the onset of IBD and there is no correlation between activity of the spondylitis and bowel disease.
- HLA-B27 is found in 50% of patients and it follows a chronic course.
Others: Enthesitis, dactylitis and extra-articular manifestations of IBD.
Treatment:
- NSAIDs to be used cautiously because they may worsen diarrhea.
- Monoarthritis is treated by intra-articular corticosteroids.
- Sulfasalazine more effective as it may help both bowel and joint disease. Azathioprine and methotrexate may also be used.
- TNF-α blocking drug infliximab used for IBD can help the arthritis.
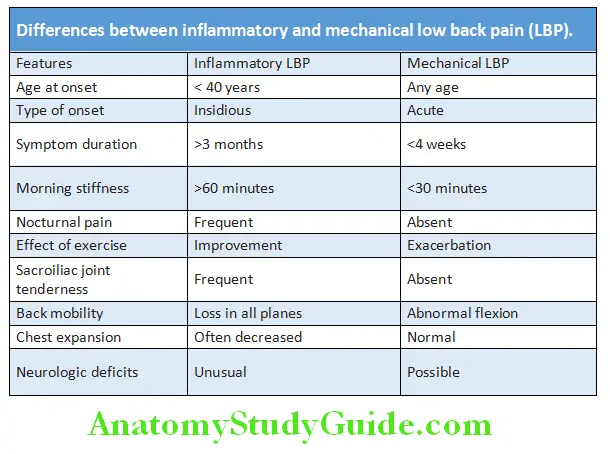
Differences between inflammatory and mechanical low back pain (LBP)
Question 39. Describe the clinical clues to differentiate inflammatory low back pain of ankylosing spondylitis from mechanical low back pain?
(or)
Differentiate between inflammatory versus mechanical back pain
Answer:
Adult Onset Still’S Disease (Aosd):
Question 40. Discuss the clinical features of adult onset still’s disease.
Answer:
AOSD is a rare inflammatory multisystem disease occurring in adults presenting as fever (quotidian/double quotidian), rash, arthritis, sore throat, hepatosplenomegaly and serositis (pleural and pericardial effusion).
The rash is classically evanescent, salmon-colored, macular or maculopapular which is usually nonpruritic and occurs with the fever. Arthritis and arthralgia mostly involve the knee, wrist and the ankle. Myalgia is seen in more than 50 percent of the cases. Macrophage activation syndrome, a rare and fatal complication of Adult Onset Still’s disease characterized by leukopenia, hypertriglyceridemia, transaminitis and intavascular coagulation.
Diagnosis:
Adult Onset Still’s disease (ASOD) is primarily a diagnosis of exclusion.
- Neutrophilia, thrombocytosis, raised ESR, CRP are present. Increased LDH and liver enzymes are seen
- Marked elevation of ferritin (>3000 mg/ml) found in more than 70 % of patients. Reduced level (<20% ) of glycosylated fraction of ferritin is a more useful marker.
- RF and ANA are negative.
- Yamaguchi criteria is the accepted diagnostic criteria.
Treatment:
- Often a self limited disease, rarely can have an intermittent flares and chronic articular form.
- NSAID’s and glucocorticoids can be given to reduce systemic inflammation.
- DMARD’s like methotrexate may be used as steroid sparing agents.
- Anti-TNF alpha antagonist and IL-1 antagonist (Anakinra) and cyclosporine can be used for patients presenting with macrophage activation syndrome.
Juvenile Idiopathic Arthritis/Juvenile Rheumatoid Arthritis:
- Formerly called Still’s disease
- Onset is before 16 years of age, manifestations persist for at least 6 weeks, and the etiology is unknown.
- JIA can manifest with following clinical types
- Systemic arthritis associated with fever, rash, lymphadenopathy and organomegaly
- Polyarthritis, involvement of 4 or more joint areas. RF may be positive or negative
- Oligoarthritis, involving 4 or less joint areas
- Enthesitis related arthritis
- Psoriatic arthritis
- Undifferentiated arthritis
- Other features include polyserositis and macrophage activation syndrome
- Mild to disease responds to NSAIDs and steroids
- Biologic agents, such as anakinra, canakinumab, or tocilizumab are useful in severe disease and systemic complications.
Vasculitis:
Question 41. Define and classify vasculitis.
Answer:
Definition: Vasculitis refers to a heterogeneous group of disorders that is characterized by destructive inflammation of blood vessel walls. The lumen of the inflamed blood vessels is liable to occlude or rupture or develop a thrombus, and this is associated with ischemia of the tissues supplied by the involved vessel.
Classifiation:
Vasculitis may involve a single organ (e.g. skin), or it may involve several organ systems. One of the basis for classifying the vasculitides is the size of the predominant blood vessels involved
Classification of the primary vasculitides according to size of predominant blood vessels involved (Chapel Hill Consensus Conference).
Predominantly large-vessel (aorta and its major tributaries) vasculitis:
- Giant cell arteritis
- Takayasu arteritis
- Cogan syndrome
Predominantly medium-vessel (medium and small-sized arteries and arterioles) vasculitis:
- Polyarteritis nodosa
- Primary central nervous system disease
- Buerger disease
Predominantly small-vessel (small arteries, arterioles, venules and capillaries) vasculitis:
- ANCA-associated small-vessel vasculitis
- Granulomatosis with polyangiitis (Wegener granulomatosis)
- Microscopic polyangiitis
- Churg-Strauss syndrome (allergic angiitis and granulomatosis)
- Drug-induced ANCA-associated vasculitis
- Immune complex mediated
- Cutaneous leukocytoclastic angiitis (‘hypersensitivity’ vasculitis)
- Henoch-Schönlein purpura
- Urticarial vasculitis
Clinical Manifestations:
Question 42. Write short essay/note on clinical features of vasculitis.
Answer:
Constitutional symptoms: Fever, weight loss, malaise, arthralgias/arthritis (common to vasculitides of all vessel sizes). Typical clinical manifestations of large, medium, and small vessel vasculitis are presented and vasculitis associated with granulomatous inflammation is listed in Table.
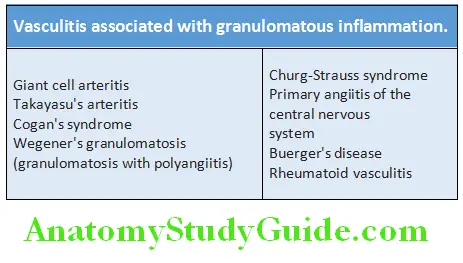
Giant Cell Arteritis (GCA):
Question 43. Write short essay/note on temporal arteritis, cranial arteritis or giant cell arteritis.
Answer:
Giant cell arteritis (temporal arteritis, Horton disease or cranial arteritis) is inflammatory granulomatous arteritis of large and medium sized arteries in the head and neck. It can involve aorta and its primary and secondary branches.
Age and gender: It is a disease of the elderly and average age at onset is 70. It is extremely rare below 60 years of age. Female to male ratio is about 3:1.
Vessels involved: One or more branches of the carotid artery (e.g. temporal artery, occipital, ophthalmic, posterior ciliary arteries and vertebral arteries). It can involve multiple arteries, aorta and its branches.
Clinical Features:
- Onset may be gradual or sudden.
- Systemic manifestations: Severe malaise, tiredness, fever, anemia, fatigue and weight loss.
- Severe headache: Most severe and often localized along the course of the superficial temporal artery or occipital region. Arteritis often occurs in the carotid/ temporal arteries. On palpation, the involved artery is tender, thickened and cord-like or nodular. It may be accompanied by tenderness of the scalp or of the temple.
- Jaw claudication: When chewing, eating or talking, due to ischemia of the masseter muscles.
- Visual symptoms: These include loss of visual acuity, reduced color perception and pupillary defects. It can even present as sudden blindness in one eye due to occlusion of the posterior ciliary artery.
- Neurological manifestations: It occurs in about 30% of patients and includes transient ischemic attacks, brainstem infarcts and hemiparesis.
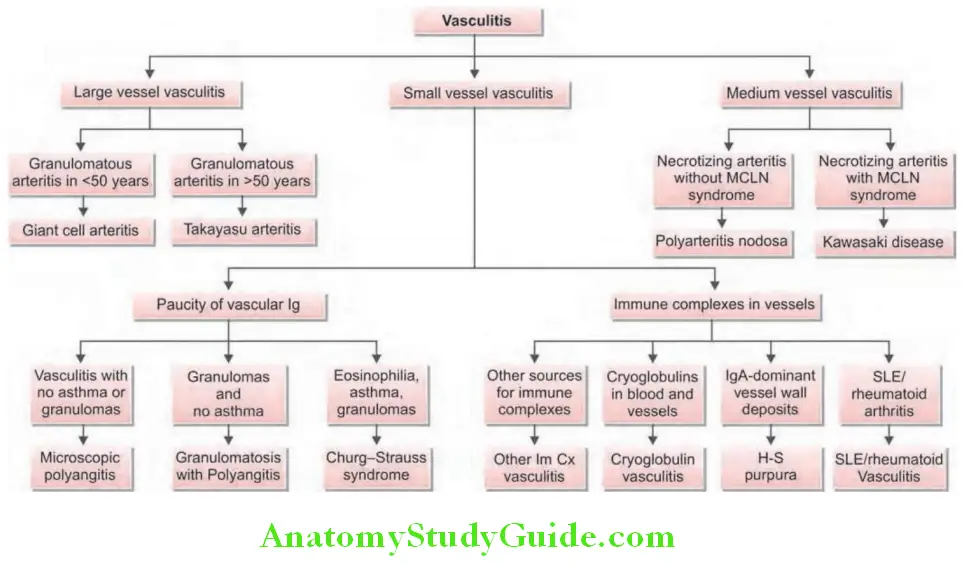
Investigation:
- Normochromic, normocytic anemia.
- ESR is raised (50–120 mm/h) and the CRP very high.
- Temporal artery biopsy from the affected side is the definitive diagnostic test. Characteristic biopsy findings are fragmentation of the internal elastic lamina with necrosis of the media, mixed inflammatory cell infiltrate and granulomatous inflammation of the intima and media with giant cells.
- Color Doppler Ultrasound or arteriography: It may be used for guiding the biopsy. Duplex ultrasonography can detect the characteristic appearance.
Treatment:
Corticosteroids are the treatment of choice and should be commenced urgently in suspected giant cell arteritis to prevent the risk of visual loss. It responds dramatically within 24–48 hours of starting corticosteroids. Prednisolone is given at a doseof 40–60 mg/day. NSAIDs should not be used.Tociluzumab and methotrexate have been used as steroid sparing agents.
Polymyalgia Rheumatica (PMR):
Question 44. Write short note on polymyalgia rheumatica.
Answer:
PMR is characterized by pain, aching, and stiffness in the muscles of the neck, shoulders, and hip-girdle area that are usually much worse in the morning. PMR is closely associated with GCA (giant cell arteritis).
Criteria for the Diagnosis of PMR:
Treatment:
Corticosteroids: Prednisolone (10–15 mg/day) is rapidly effective. Symptoms are relieved within 48–72 hours and the ESR normalizes after 7–10 days.
Criteria for the diagnosis of PMR (polymyalgia rheumatica):
- Age >50 years
- Aching and stiffness for at least 1 month, affecting at least two of the three areas (i.e. shoulders, neck, and pelvic girdle)
- Morning stiffness lasting at least 1 hour
- ESR >40 mm/hour
- Exclusion of other diseases except GCA (giant cell arteritis)
- Rapid response to prednisone (20 mg/day)
- All of the above criteria must be met to diagnose PMR
- Classic Polyarteritis Nodosa (PAN):
Question 45. Describe the clinical features, diagnosis and management of classic polyarteritis nodosa (PAN).
Answer:
Definition: Polyarteritis nodosa is also called classic polyarteritis nodosa is a systemic, necrotizing vasculitis of small and medium-sized muscular arteries that spares the smallest blood vessels (e.g. arterioles, venules or capillaries). It characteristically involves the renal and visceral arteries and is not associated with glomerulonephritis.
Two additional features that distinguish PAN from other forms of systemic vasculitis are:
- Confinement of the disease to the arterial rather than the venous circulation.
- Absence of granulomatous inflammation.
Organ systems involved: Commonly involves skin, kidneys, heart, liver, and gastrointestinal tract. PAN does not involve pulmonary arteries. About 30% of patients with PAN have chronic hepatitis B with HBsAg-HbsAb complexes in affected vessels.
Clinical Features:
- Age and gender: Peak incidence between 40 to 50 years of age. Predominantly affects middle aged males (M:F=2:1).
- Subacute onset of constitutional vague symptoms such as fever, weight loss, weakness, malaise and arthralgias are present in >50% of cases. Others include testicular pain or tenderness, mononeuritis multiplex, and intestinal angina (postprandial pain caused by the involvement of mesenteric vessels). Renal involvement produces renal insufficiency and hypertension. Cutaneous manifestations include erythema nodosum, purpura, livedo reticularis, and ulcers.
- Absence of pulmonary artery involvement helps distinguish PAN from most cases of vasculitis. However bronchial arteries may be involved.
Diagnosis:
- Anemia, thrombocytosis, and raised acute phase reactants.
- Hepatitis B surface antigen (HBsAg): It may be positive.
- p-ANCA: Present in about 20% of cases.
- Arteriograms: Demonstrates aneurysms of small and medium-sized arteries in the kidney, liver, and visceral vasculature.
- Biopsy: Presence of characteristic findings of vasculitis of involved organ confirms the diagnosis. Biopsy of symptomatic organs such as nodular skin lesions, painful testes, and nerve/muscle reveals vasculitis. Microscopy shows destruction of the blood vessel wall by inflammatory cells, accompanied by fibrinoid necrosis.
Treatment:
- Glucocorticoids and cytotoxic agents: In idiopathic PAN, remissions or cures is achieved by high doses of glucocorticoids alone. Cyclophosphamide, mycofenolate, rituximab and azathioprine is indicated when PAN is refractory to corticosteroids or when there is serious involvement of major organs.
- PAN with hepatitis B: Antiviral therapy used in combination with glucocorticoids and plasma exchange.
Microscopic Polyangiitis:
Question 46. Write short note on microscopic polyangiitis.
Answer:
Definition: Microscopic polyangiitis (MPA) is the most common ANCA-associated necrotizing vasculitis with few or no immune complexes in the involved vessels. It affects small vessels (capillaries, venules or arterioles).
Clinical Features:
- Age and gender: Most common age of onset is 40–60 years. More common in males than females.
- Microscopic polyangiitis is the most common cause of the pulmonary-renal syndrome, i.e. alveolar hemorrhage and glomerulonephritis. Alveolar hemorrhage associated with hemoptysis and respiratory compromise.
- The five most common clinical manifestations of MPA are:
- Glomerulonephritis (~80% Of Patients)
- Weight Loss (>70%)
- Mononeuritis Multiplex (60%)
- Fever (55%)
- A variety of cutaneous findings-cutaneous vasculitis (>60%).
- Other features: Migratory arthralgias or arthritis (either pauciarticular or polyarticular), palpable purpura, sometimes with skin ulcerations, nodules, livedo reticularis, and digital gangrene may be seen.
Diagnosis:
p-ANCA is positive in most patients, although c-ANCA may also be present in 40% cases.
Treatment:
- Glucocorticoids in combination with either rituximab or cyclophosphamide: It is the cornerstone of treatment regimens, because most patients with MPA have major organ involvement such as glomerulonephritis, alveolar hemorrhage, or vasculitic neuropathy.
- Cyclophosphamide may be administered on either a daily or intermittent basis.
- ‘Pulse’ methylprednisolone (1 g/d for 3 days) may be considered for patients with severe organ involvement at diagnosis.
- In severe cases (e.g. renal failure or diffuse alveolar hemorrhage), additional use of plasmapheresis is beneficial.
- Other treatment regimens includes mycofenolate, methotrexate, azathioprine and intravenous immunoglobulin.
Churg-Strauss Syndrome:
Question 47. Write short essay/note on Churg-Strauss syndrome/Eosinophilic granulamatosis with polyangitis (EGPA).
Answer:
Churg-Strauss syndrome (CSS) or eosinophilic granulamatosis with polyangitis.is a rare syndrome that affects small to medium-sized arteries and veins of multiple organ systems.
Hallmarks: It is characterized by asthma, peripheral and tissue eosinophilia, extravascular granuloma formation, and systemic vasculitis.
Classic Clinical Features:
Classic clinical features of Churg-Strauss syndrome:
- Allergic rhinitis and nasal polyposis (75%)
- Asthma (90%)
- Peripheral eosinophilia (10–60% of all circulating leukocytes)
- Fleeting pulmonary infiltrates and occasional alveolar hemorrhage
- Cutaneous nodules, GI bleed, eosinophilic gastroenteritis
- Acute renal insufficiency, acute glomerulonephritis, mononeuritis multiplex, venous thromboembolism, and stroke
- Coronary arteritis pericarditis, rhythm abnormalities myocarditis and heart failure
Investigations:
- Striking eosinophilia >10% (>1000 cells/µL) in peripheral blood.
- Raised ESR fibrinogen, or a 2-globulins and CRP.
- Increased IgE levels.
- Urine: RBC casts and proteinuria.
- Positive antinuclear cytoplasmic antibodies against myeloperoxidase (p-ANCA) in ~48% of cases.
- Chest X-ray: May show infiltrates and pleural effusion.
- Biopsy: Three histologic criteria for the diagnosis:
- The presence of necrotizing vasculitis
- Tissue infiltration by eosinophils, and
- Extravascular granuloma.
Treatment:
- High-dose steroids and cyclophosphamide, followed by maintenance therapy with low-dose steroids. Some cases may require addition of cyclophosphamide, mycofenolate and leflunomide
- In severe cases, anti-TNF-α agents like infliximab and etanercept are used
- Anti-interleukin (IL)-5 monoclonal antibodies (mepolizumab, reslizumab) and an anti-IL-5 receptor antibody (benralizumab) are approved for use in severe asthma
Granulomatosis with Polyangiitis (Wegener’s Granulomatosis):
Question 48. Write short essay on clinical features, diagnosis and treatment of granulomatosis with polyangiitis (Wegener’s granulomatosis).
Answer:
Definition: Granulomatosis with polyangiitis is a distinct clinicopathologic entity characterized by necrotizing vasculitis, which involves the upper respiratory tract, the lungs, and the kidneys.
It is one of the most common forms of systemic vasculitis. It involves small to medium-sized blood vessels. It affects both the arterial and venous circulations.
Clinical Features:
- Age and gender: It usually presents during fourth to fifth decade. Males are affected more often than females.
- Nasal involvement: Found in about 90% of patients. These include nasal crusting, epistaxis (bleeding), paranasal sinus pain, sinusitis, nasal mucosal ulceration and obstruction. Inflammation of nasal cartilage may lead to nasal septal perforation and collapse of the nasal bridge (‘saddle nose’ deformity). Erosive sinus disease and subglottic stenosis (narrowing of the trachea due to inflammation just below the vocal cords) are highly characteristic of granulomatosis with polyangiitis.
- Lesions in the mouth: Two classic mouth lesions are gum inflammation (‘strawberry gums’) and tongue ulcers.
- Deafness both conductive and sensorineural hearing loss are typical of the disease and proptosis due to inflammation of the retro-orbital tissue, diplopia due to entrapment of the extra-ocular muscles, and loss of vision due to optic nerve compression may occur.
- Lung involvement may produce cough, hemoptysis (due to alveolar hemorrhage) and dyspnea.
- Renal involvement occurs in the form of rapidly progressive glomerulonephritis, resulting in proteinuria, hematuria, red blood cell casts in the urine and renal failure.
- Other manifestations: These include palpable purpura, conjunctivitis, episcleritis, scleritis, cranial neuritis and mononeuritis multiplex.
Diagnosis:
- ESR: Markedly elevated.
- Non-specific abnormalities: These include mild anemia, leukocytosis and mild hypergammaglobulinemia (particularly of the IgA class). Complement levels will be normal or elevated.
- Serum antiproteinase-3 ANCA (c-ANCA): Positive in about 90% of patients.
- Chest radiograph: It may show pulmonary infiltrates and nodules that may cavitate.
- Imaging of the upper airways or chest with MRI: It can be useful in localizing abnormalities.
- Lung biopsy: Used for confirmation of the diagnosis. It shows the characteristic necrotizing granulomatous vasculitis.
- Renal biopsy: It may show a segmental necrotizing crescentic glomerulonephritis, with no immunoglobulin deposition (‘Pauci-immune’).
Treatment: Treatment is similar to that for microscopic polyangiitis.
Behçet’s Disease/Syndrome:
Question 49. Describe the symptoms, diagnosis and treatment of Behçet’s disease.
Answer:
Behçet’s syndrome is characterized by triad of:
- Recurrent Episodes Of Oral/Mouth Ulcers
- Genital Ulcers
- Eye inflammation (i.e. iritis).
Pathologic process is a vasculitis (leukocytoclastic) that may affect small, medium, and large vessels in either the venous or the arterial circulation in any organ.
Clinical Features:
Diagnostic criteria: Diagnostic criteria of International Study Group for Behçet’s syndrome is presented. However, the spectrum of Behçet’s syndrome consists of many manifestations (e.g. neurological involvement seen in 5% of patients, recurrent vascular thrombosis) that are not included in these criteria. Patients can have non erosive arthritis and cutaneous nodules too.
- Positive pathergy test: Highly specific to Behçet’s disease but may be positive in only 50% of Behçet’s patients. It involves pricking the skin with a needle and looking for evidence of pustule development within 48 hours.
- ESR and CRP raised but not autoantibodies.
Diagnostic criteria for Behçet’s syndrome:
One required manifestation:
Recurrent oral ulceration: Aphthous or herpetiform, at least three times in 12 months, usually deep and multiple, and last for 10–30 days. This is the cardinal clinical feature.
plus
At least two of the following:
- Recurrent genital ulceration occurs in 60–80% of cases.
- Characteristic eye lesions: Anterior or posterior uveitis or retinal vasculitis or cells in vitreous on slit-lamp examination.
- Characteristic skin lesions: Erythema nodosum, pseudofolliculitis, papulopustular lesions and acneiform nodules.
- Positive pathergy test.
Treatment:
- Oral and genital ulceration: Topical steroid preparations (soluble prednisolone mouthwashes, steroid pastes). Thalidomide (100–300 mg/ day for 28 days initially) is very effective for resistant oral and genital ulceration but is teratogenic and neurotoxic.
- Colchicine can be effective for erythema nodosum and joint pain. Apremilast is an alternative to colchicine
- Steroids and immunosuppressants and ciclosporin are indicated for chronic uveitis and rare neurological complications. Anti-TNF agents can be used to control severe uveitis and serious neurological manifestations and gastrointestinal Behçet’s disease.
Henoch-Schönlein Purpura/IgA Vasculitis:
Question 50. Write short note on Henoch-Schönlein purpura and anaphylactoid purpura.
Answer:
- Henoch-Schönlein purpura (also referred to as IgA vasculitis or anaphylactoid purpura), is the commonest systemic small-vessel vasculitis in children. It is caused by IgA dominant immune complex deposition following an infectious trigger. It often occurs after upper respiratory tract infections.
- Usually occurs in children (>90% of cases) and young adults.
Diagnostic Criteria:
Usual presentation Often present with the tetrad of:
- Purpura (vasculitic/non-thrombocytopenic/palpable purpura): Palpable and found over the buttocks and lower legs.
- Arthralgia or arthritis: Most patients develop a transient nonmigratory polyarthralgias in the absence of frank arthritis and resolves without permanent damage to joints.
- Gastrointestinal involvement: Seen in about 70% of pediatric patients. It is characterized by recurrent colicky abdominal pain associated with nausea, vomiting, diarrhea, or constipation, and the passage of blood and mucus per rectum. In some cases, there may be intussusceptions.
- Renal involvement: Seen in 10–50% of patients. It usually produces mild glomerulonephritis causing proteinuria, microscopic hematuria, with red blood cell casts in the urine. It usually resolves spontaneously without treatment.
Other manifestations include scrotal pain keratitis, uveitis, respiratory and neurological involvement.
Prognosis: HSP is self-limited most of the time. The vast majority of cases resolve within 6–8 weeks. Adult cases are sometimes more recalcitrant (not responsive to treatment).
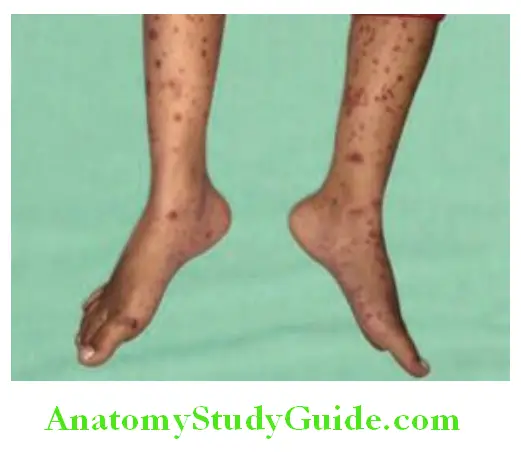
Investigations:
- Acute phase reactants are elevated. Serum IgA levels are elevated.
- Skin biopsy: Findings are pathognomonic. It demonstrates leukocytoclastic vasculitis in capillary venules and direct immunofluorescence of the skin biopsy shows IgA immune complexes deposit in the small vessels.
Treatment:
- Usually a self-limiting disorder that settles spontaneously without specific treatment.
- Patients respond to bed rest and NSAIDs.
- Corticosteroids and immunosuppressive therapy may be indicated when HSP is severe disease, particularly in the presence of nephritis.
Cogan’s Syndrome (CS):
An immune-mediated condition that primarily affects young adults, is associated with ocular inflammation (usually interstitial keratitis) and audiovestibular dysfunction. Maybe associated with large- or medium- to small-sized vessel vasculitis or an aortitis.
Buerger’s Disease (Thromboangiitis Obliterans):
- Nonatherosclerotic, segmental, inflammatory disease that affects the small to medium-sized arteries and veins of the extremities.
- Classic patient is a young male smoker.
- Identification of the typical pattern of vascular involvement by angiography.
- Major risk factor is tobacco exposure.
- Absence of fibrinoid necrosis (it is a hallmark of most systemic vasculitides).
- Initially, nonspecific pains in the calf, foot, or toes.
- Later, the progression of thrombosis and vasculitis can lead to horrific pain in the digits and limbs and ultimately to gangrene and tissue loss, through either autoamputation or elective amputation.
- The only effective intervention in Buerger disease is complete smoking cessation.
- Intermittent pneumatic compressions, calcium channel blockers, phosphodiesterase inhibitors may help in pain relief.
Takayasu Arteritis:
Question 51. Write short essay/note on Takayasu arteritis.
Answer:
- Takayasu arteritis, named for the Japanese ophthalmologist who first described the ocular manifestations in 1908.
- It is a large-vessel vasculitis of unknown cause that chiefly affects women during their reproductive years.
- Sites: Most common sites are the aorta (65%) and the left subclavian arteries (93%).
- Presentation:
- Vascular manifestations include: Bruit, claudication, hypertension, light-headedness (associated with vertebral or carotid artery disease), unequal blood pressures in the extremities, carotidynia, aortic regurgitation, and loss of a pulse. Absence pulses in upper limbs (called reverse coarctation).
- Bruits: Most frequent over the carotid arteries, but also often develop in the supraclavicular or infraclavicular space (reflecting subclavian disease), along the flexor surface of the upper arm (from axillary artery disease), or in the abdomen (from renal or mesenteric artery vasculitis). Many patients have multiple bruits.
- A widened pulse pressure and diastolic murmur along the right sternal border may signal the aortic regurgitation that develops in 20% of patients. Stroke, angina, and congestive heart failure affect a significant minority of patients.
- Cardiac complications: Hypertension, congestive heart failure, angina, and aortic regurgitation.
- About 50% of patients develop constitutional or musculoskeletal symptoms. Asthenia, weight loss, fever, myalgia, and arthralgia occur commonly.
- Criteria for the diagnosis of Takayasu arteritis.
American College of Rheumatology classification criteria for Takayasu arteritis:
- Onset at age <40 years
- Limb claudication
- Decreased brachial artery pulse
- Unequal arm blood pressures (>10 mm Hg)
- Subclavian or aortic bruit
- Angiographic evidence of narrowing or occlusion of the aorta or its primary branches, or large limb arteriti
Investigations:
- Elevated erythrocyte sedimentation rates or C-reactive protein values during phases of active disease.
- Anemia and thrombocytosis
- Vascular abnormalities can be imaged by conventional angiography, MRI, MRA, CT angiography, or ultrasonography. Conventional angiography is the ‘gold standard’ for precisely delineating the stenosis, occlusions, and aneurysms.
Treatment:
- High-dose glucocorticoids in combination with a glucocorticoid-sparing agent (methotrexate, mucophenolate, leflunomide)
- Vascular intervention—for treatment of stenosed arteries leading to organ ischemia or hypertension and for aneurysmal disease.
Question 52. Differentiate between giant cell arteritis and Takayasu’s arteritis.
(or)
Discuss the pathogenesis and clinical features and management of osteoarthritis/degenerative joint diseases.
Answer:
Differences between giant cell arteritis and Takayasu’s arteritis.
Osteoarthritis:
Osteoarthritis (OA) is the most common form of arthritis. OA refers to a clinical syndrome of joint pain accompanied by varying degrees of functional limitation and reduced quality of life.
OA is a noninflammatory, slowly progressive joint disease, mainly involving the cartilage. It shows progressive destruction of articular cartilage of weight-bearing joints of genetically susceptible older persons. It leads to narrowing of joint, subchondral bone thickening, and finally nonfunctioning, painful joints.
Joints Affcted:
- Weight bearing joints (knee, hips and cervical and lumbar segments of the spine).
- Non-weight bearing distal interphalangeal (DIP) joints, thumb bases (first CMC joints and trapezioscaphoid joints), first MTP joints, lower cervical and lumbar facet joints, knees, and hips.
Etiology:
Osteoarthritis (OA) is a multifactorial disease having both genetic and environmental components
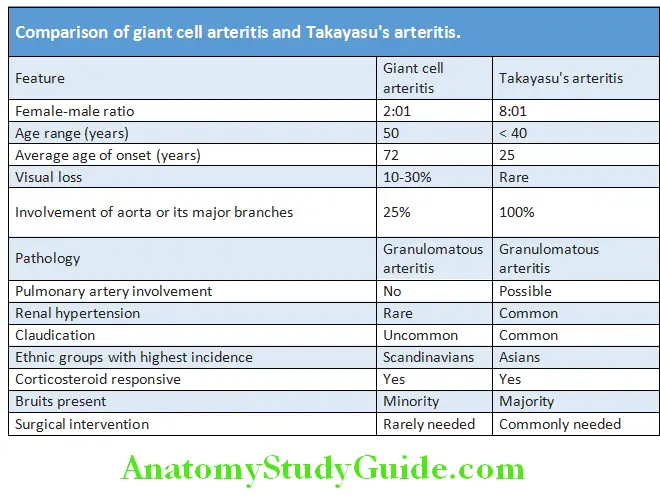

Types of Osteoarthritis:
Idiopathic or primary osteoarthritis: It develops as aging process and may affect few (oligoarticular) or many joints.
- Localized: Most commonly affects the hands, feet, knee, hip, and spine.
- Generalized: Involves three or more joint sites
Secondary osteoarthritis: It appears in younger individuals with predisposing condition.
- Previous injuries/trauma to a joint
- Congenital or developmental deformity of a joint(s)
- Secondary to systemic disease [e.g. diabetes, marked obesity, acromegaly, hypothyroidism, neuropathic (Charcot) arthropathy]
- Calcium pyrophosphate dihydrate deposition disease (CPPD)
- Bone and joint disorders including rheumatoid arthritis, gouty arthritis, septic arthritis, and Paget disease of bone and osteonecrosis
Identifible Causes of Osteoarthritis:

Clinical Manifestations:
Age of onset: Usually after age 40.
Joints affected in osteoarthritis:

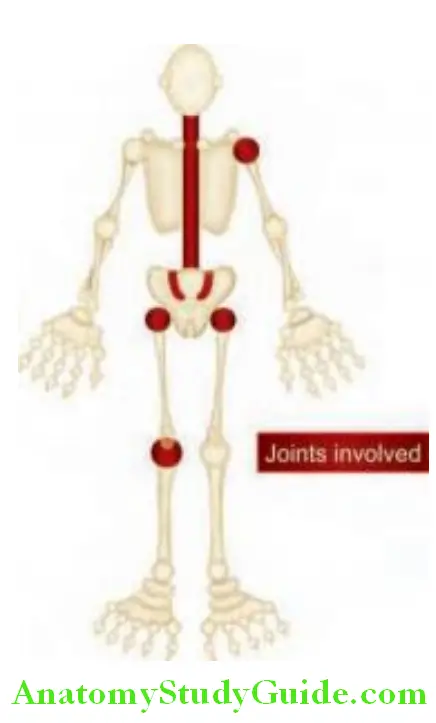
Signs, Symptoms and Diagnostic Features of Osteoarthritis:
Symptoms:
- Joint pain that increases with activity
- Stiffness: Morning stiffness relatively brief (for <30 minutes) and self-limited
- Gelling
Physical fidings:
- Crepitus (a grating sensation) on active motion
- Bony enlargement at the joint margin, hard bony enlargements, called Heberden’s nodes (on DIP) and/or Bouchard’s nodes (on PIP).
- Decreased range of motion
- Malalignment
- Tenderness to palpation over the joint
- Absence of warmth
Synovial flid analysis:
- Clear fluid
- Noninflammatory synovial fluid (<1000 WBC/mm 3)
- Normal viscosity
Erythrocyte sedimentation rate normal for age Negative serologic tests for antinuclear antibody and rheumatoid factor
Radiographic features:
Nonuniform joint space narrowing. Subchondral eburnation (bony
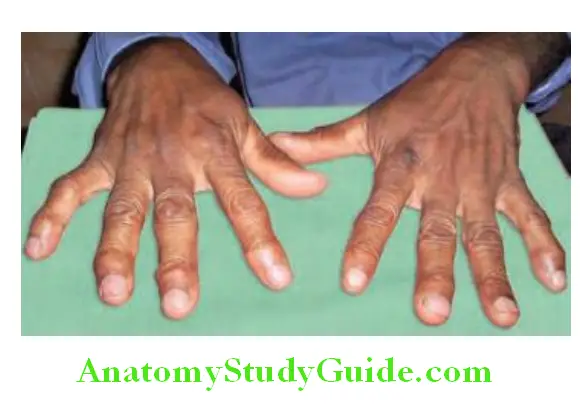
Diagrammatic representation of joint changes in osteoarthritis:
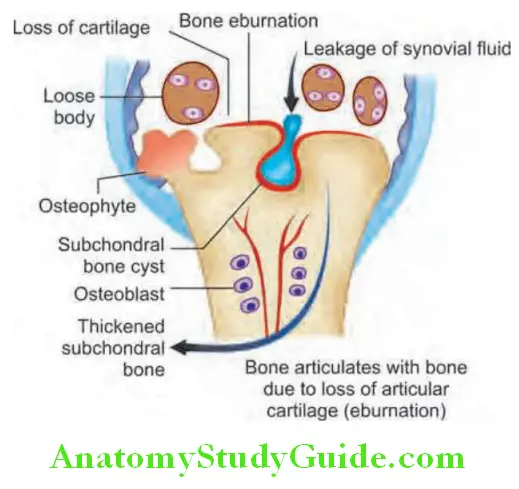
Prevention:
Preventing the onset of OA requires lifestyle changes.
- Primary prevention: These include:
- Weight control: Maintaining or reducing weight.
- Occupational injury prevention: Avoiding repetitive joint use and its injuries.
- Sports injury prevention: Precautions to prevent injury such as warming up and using proper equipment.
- Secondary prevention: Early diagnosis so that appropriate early intervention can be utilized. However, this is difficult in OA since no effective biomarkers are available to determine the progression of the disease.
- Tertiary prevention: Reduce, delay the onset of complications and disability. The strategies include: self-management (weight control, physical activity, education); home help programs; cognitive behavioral interventions; rehabilitation services and medical surgical treatments.
Therapeutic Options in Osteoarthritis:
Question 53. Write short essay/note on management of osteoarthritis.
Answer:

Systemic Sclerosis (Scleroderma):
Question 54. Describe the clinical manifestations, diagnosis and management of systemic sclerosis.
Answer:
Systemic sclerosis (SSc) is a chronic multisystem disease of connective tissue affecting the skin, musculoskeletal system, internal organs (e.g. gastrointestinal tract, lungs, heart and kidneys) and vasculature. Scleroderma literally means ‘hard skin’ and the hallmark of SSc is thickening and hardening of the skin (scleroderma) due to skin fibrosis.
Classification:
Systemic sclerosis is classified and subdivided into two principal subsets defined largely by the anatomic skin distribution and associated with distinct clinical and laboratory manifestations.
- Diffuse cutaneous systemic sclerosis (DCSS: 30% of cases): It is associated with progressive skin thickening, starting in the fingers and ascending from distal to proximal extremities, the face, and the trunk. These patients have a risk of early pulmonary fibrosis and involvement of kidney and other systems.
Question 55. Write short note on CREST syndrome and its clinical features.
Answer:
- Limited cutaneous systemic sclerosis (LCSS: 70% of cases): Usually these patients have long-standing Raynaud’s phenomenon before other manifestations. Skin involvement progresses slowly and remains limited to the fingers (sclerodactyly), distal extremities, and face, without involvement of trunk. Few of these patients have prominent Calcinosis cutis, Raynaud’s phenomenon, Esophageal dysmotility, Sclerodactyly (scleroderma of the fingers), and Telangiectasia, which is termed CREST syndrome. CREST syndrome generally show slow progression.
- Other two types are:
- Systemic sclerosis sine scleroderma: A small group of patients have no detectable skin involvement but have clinical features such as Raynauds, digital ulcers, and autoantibodies for SSc.
- Systemic sclerosis with overlap syndrome: SSc with overlap or features of another systemic rheumatic disease such as systemic lupus erythematosus (SLE), rheumatoid arthritis, polymyositis, or Sjögren’s syndrome.
Age and gender: Peak incidence is between 30 and 50 years of age with female to male ratio of 3:1.
Etiology: The cause is poorly understood. There is associations with alleles at the HLA locus have been found. There is immunological dysfunction of T lymphocytes, especially Th1 and Th17 subtypes.
Clinical Features:
Question 56. Write short note on symptoms and signs of systemic sclerosis.
Answer:
Skin Manifestations:
- Limited disease or CREST syndrome: Skin involvement restricted to sites distal to the elbow or knee (apart from the face).
- Diffuse disease: Skin involvement proximal to the knee and elbow and on the trunk.
Changes in skin:
- Symmetrical and bilateral skin thickening is the hallmark of SSc.
- Early stage: Shows non-pitting edema of fingers and flexor tendon sheaths.
- Later stages: Skin becomes shiny, firm, thickened, and distal skin creases disappear. After many years, the skin may become thin, atrophic and tightly bound to underlying subcutaneous tissue.
- Hyperpigmentation: Skin generally becomes hyperpigmented. In dark-skinned individuals, vitiligo-like areas of hypopigmentation may occur. Because pigment loss spares the perifollicular areas, the skin may develop a ‘salt-and-pepper’ appearance most prominently on the upper back and chest.
- Changes in facial skin: Produces a beak-like nose, ‘masklike’ face, and decreased oral aperture (microstomia).
- Other manifestations: These include flexion contractures, ulcers over fingertips and bony prominences (due to breakdown of atrophic skin), telangiectasia, calcinosis cutis and dry coarse skin.
Musculoskeletal Manifestations:
- Range from mild arthralgias to frank nonerosive arthritis with synovitis resembling rheumatoid arthritis.
- Generalized arthralgia, morning stiffness and flexor tenosynovitis are common.
- Muscle weakness and wasting due to disuse atrophy, myopathy and myositis.
- Restricted hand/limb function associated with contractures of the joints is due to sclerosis of skin rather than joint disease.
- Bone resorption is most common in the terminal phalanges, where it causes loss of the distal tufts (acro-osteolysis) (pseudoclubbing).
Vascular Manifestations:
- Involvement of the vasculature is universal feature of SSc.
- Raynaud’s phenomenon is an episodic reversible vasoconstriction of the vessels of the digits (fingers and toes) that can result in ischemia of the digits. It occurs in all patients with SSc and can precede other features by many years. This is the first manifestation of the disease in almost every patient.
- Triggering factors: Factors which trigger attack include exposure to cold, a decrease in temperature, emotional stress, and vibration. Typical attacks start with pallor (vasoconstriction), followed by cyanosis (ischemia) of variable duration and erythema (reperfusion) develops spontaneously or with rewarming of the digit.
- A diffuse vasculopathy of peripheral arteries (in the extremities) may cause narrowing or occlusion of the vessel lumen leading to tissue ischemia. This may produce skin ulceration over pressure areas, localized areas of infarction and pulp atrophy at the fingertips.
- Vascular disease is fundamental to organ damage and subsequent malfunction of the heart (cardiomyopathy), lung (pulmonary hypertension), kidney [scleroderma renal crisis (SRC)], and other organs in SSc.
Gastrointestinal Manifestations:
Gastrointestinal disease in scleroderma usually involves both the upper and lower gastrointestinal tract but clinical expression is highly variable.
- Esophageal involvement: It can manifest as reflux with erosive esophagitis, dysphagia and odynophagia, strictures, and dilatation and atony of lower esophagus.
- Stomach involvement: It results in dilatation, atony and delayed gastric emptying. Recurrent occult upper gastrointestinal bleeding may occur due to gastric antral vascular ectasia (GAVE) (‘watermelon’ stomach).
- Small and large bowel involvement: It results in intermittent abdominal distension, abdominal pain, constipation, intestinal obstruction, malabsorption (due to bacterial overgrowth) and steatorrhea. Pseudoobstruction is a known complication.
Pulmonary Manifestations:
It is a major cause of morbidity and mortality.
Main forms of lung disease:
Inflammatory alveolitis leading to interstitial fibrosis and pulmonary arterial hypertension.
Other Manifestations:
Renal involvement: Occurs in only a minority of patients and can be acute or chronic. Acute hypertensive renal crisis (scleroderma renal crisis-SRC) characterized by sudden onset of malignant hypertension and renal failure was used to be the most common cause of death. At present, this is not the case because ACE inhibitors with dialysis and renal transplantation have improved the outlook.
Cardiac involvement: Myocardial fibrosis may cause arrhythmias, conduction defects and cardiomyopathy. Occasionally, pericarditis with or without effusion may develop. Localized scleroderma: Localized form without systemic involvement (morphea).
Diagnosis:
Criteria for diagnosis of scleroderma:
- Thickened (sclerodermatous) skin changes proximal to the metacarpophalangeal joints
OR
- At least two of the following:
- Sclerodactyly
- Digital pitting (residual loss of tissue on the finger pads due to ischemia)
- Bibasilar pulmonary fibrosis
Investigations:
- ESR: Elevated.
- IgG levels: Raised.
- Anemia: It may be due to:
- Chronic Disease
- Iron Deficiency Due To Gastrointestinal Bleed,
- Folate And B12 Deficiency Due To Blind Loop Syndrome
- Microangiopathic hemolytic anemia due to renal involvement.
- Urea and creatinine: Raised in acute kidney injury.
- Urine microscopy and if there is proteinuria, measurement of urine albumin/creatinine ratio.
- Autoantibodies associated with scleroderma
- Rheumatoid factor: Positive in 30%.
- Nail-fold capillary changes: Visualization of capillaries of the skin at the nail-fold by diascopy gives insight into a patient’s microvasculature status. Nail-fold capillary dropout and dilated capillary loops are seen in nearly every patient with scleroderma but are not specific for scleroderma.

- Imaging:
- Chest X-ray: For changes in cardiac size and evidence of lung disease.
- X-ray of hands: For deposits of calcium around fingers. In severe cases, erosion and absorption of the distal phalanges (acro-osteolysis).
- Barium swallow: To demonstrate impaired esophageal mo- tility. Scintigraphy, manometry, impedance, and upper GI endoscopy are also useful.
- High-resolution CT: To demonstrate interstitial lung disease.
- Other investigations: Echocardiography for cardiac involvement and pulmonary hypertension.
ACR/EULAR critera for classification of systemic sclerosis provided in Table:
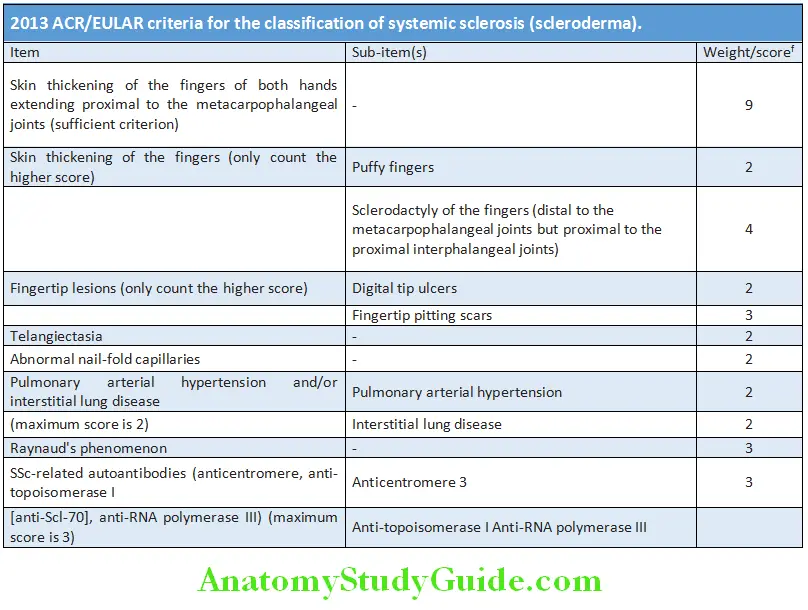
Treatment:
There is no cure and no treatment can halt or reverse the fibrotic changes produced in systemic sclerosis. Treatment should be organ-based to ameliorate the effects of the disease on target organs.
- General measures:
- Education, counseling and family support.
- Regular exercises and skin lubricants.
- Monitoring of blood counts, renal functions and analysis of urine on regular basis.
- Control of Raynaud’s syndrome and digital ulcers
- Raynaud’s phenomenon: Avoid triggering factors. Use hand warmers, and oral vasodilators (calcium-channel blockers, ACE inhibitors, angiotensin receptor blockers). Parenteral vasodilators (prostacyclin analogues and calcitonin gene-related peptide) may be beneficial in severe digital ischemia.
- Digital ulcers: Endothelin 1 antagonist bosentan promotes healing of digital ulcers. If ulcers become infected, antibiotics may be required.
- Surgical management: Lumbar sympathectomy, radical micro-arteriolysis (digital sympathectomy), and thoracic sympathectomy may be beneficial.
- Hypertension: Treated aggressively with ACE inhibitors, even if renal impairment is present.
- Esophageal reflux: Treated with proton pump inhibitors and anti-reflux agents, but prokinetic drugs are rarely useful.
- Intestinal involvement: Symptomatic malabsorption requires nutritional supplements. Antibiotics may be necessary for small intestinal bacterial overgrowth syndromes, and metoclopramide or domperidone may be useful for symptoms of pseudo-obstruction.
- Joint involvement: Treated with analgesics and/or NSAID. If synovitis is present, immunosuppressants such as methotrexate can be used.
- Pulmonary hypertension: Treated with oral vasodilators, oxygen and warfarin. Advanced cases require prostacyclin therapy (inhaled, subcutaneous or intravenous) or the oral endothelin-receptor antagonists (bosentan and sitaxsentan). Low-dose oral corticosteroids and cytotoxic drugs (e.g. cyclophosphamide or azathioprine) are indicated when myositis or pulmonary fibrosis is co-exist.
- D-Penicillamine: May reduce skin thickening and also systemic involvement.
- Progressive and diffuse skin involvement without visceral involvement, methotrexate (MTX) or mycophenolate mofetil (MMF).
Prognosis:
Prognosis is highly dependent on the extent of major organ disease. Can be predicted to some extent by the degree of skin involvement:
- Limited scleroderma has a normal life expectancy, about 90% 5-year survival rate.
- Diffuse skin disease have only about a 70–80% 5-year survival rate.
Sjögren’S Syndrome:
Question 57. Discuss the clinical manifestations, diagnosis and management of Sjögren’s syndrome (SS).
Answer:
It mainly affects the exocrine (salivary and lachrymal) glands and is characterized by the immune-mediated destruction of exocrine glands with secondary development of keratoconjunctivitis sicca (dry eyes) and xerostomia (dryness of the mouth).
Classifiation:
- Primary: Sjögren’s syndrome (sicca syndrome): It occurs in the absence of any underlying connective tissue disorder.
- Secondary: It occurs in association with other autoimmune disorders. These include rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis (scleroderma), mixed connective tissue disease, primary biliary cirrhosis, vasculitis and chronic active hepatitis.
Age and gender: Usual age of onset is 40 to 50 years, with female to male ratio of 9:Mechanisms of tissue destruction: Lymphocytic infiltration and immune-complex deposition.
Clinical Features:
Question 58. Write short note on keratoconjunctivitis sicca.
Answer:
Laboratory Investigations:
- Schirmer tear test: This is the most commonly used test for the diagnosis of SS. It measures wetting of standardized tear test strip by tear flow over 5 minutes. A standard tear test (absorbent) paper strips is placed between the eyeball and inside of the lower eyelid. Normal result is more than 10 mm of wetting in 5 minutes and if wetting is <10 mm in 5 minutes it indicates defective tear production.
- Rose Bengal staining: Staining of the eyes with rose Bengal may show punctate or filamentary keratitis over the area not covered by the open eyelid.
- Biopsy: If the diagnosis remains in doubt, it can be confirmed by biopsies of the salivary gland or of the lip (labial minor salivary gland). They show a focal infiltration by lymphocytes and plasma cells. Minor salivary gland biopsy remains a highly specific test for the diagnosis of SS.
- Tests for salivary gland involvement: Other diagnostic tests include sialometry (to measure salivary flow), sialography, and scintigraphy. Newer imaging techniques such as ultrasound, MRI or MR sialography of the major salivary glands are also useful.
- Common laboratory abnormalities:
- Most patients have an elevated ESR, raised immunoglobulin levels (hypergammaglobulinemia), and circulating immune complexes.
- Autoantibodies
- Rheumatoid factor: Usually positive (75–90%).
- Antinuclear antibodies: Positive in 80% of cases.
- Anti-mitochondrial antibodies: Positive in 10%.
- Anti-Ro (SSA) antibodies: Positive in 60–90% (in 10% of cases of RA and secondary Sjögren’s syndrome). This antibody can cross the placenta and cause congenital heart block.
- Anti-La (SSB) antibodies: Present in 40%.
- Others: Leukopenia, thrombocytosis.
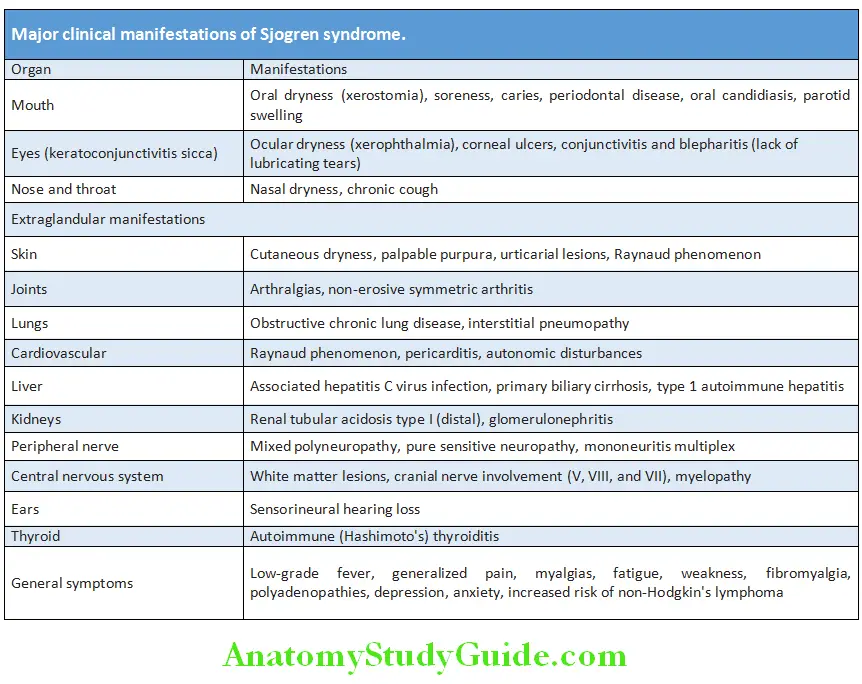
Treatment:
- Management is symptomatic.
- Ocular symptoms: Symptomatic treatment is with artificial tears. Lachrymal substitutes (e.g. hypromellose) during the day and more viscous ophthalmologic lubricating ointment at night. Cyclosporine emulsion may be useful in ocular dryness.
- Oral symptoms: Best replacement for xerostomia is water, lubricating agents and saliva-replacement solutions. Stimulation of saliva flow by sugar-free chewing gum or lozenges may be helpful. Vaginal dryness is treated with lubricants such as K-Y jelly.
- Muscarinic agonists (pilocarpine and cevimeline) have been recently used for the treatment of sicca symptoms in SS. They stimulate the M1 and M3 receptors present on salivary glands and tear glands leading to increased secretory function.
- Hydroxychloroquine is helpful for fatigue and arthralgia.
- Glucocorticoids and immunosuppresive agents are reserved for potentially severe disease. Corticosteroids are used to treat persistent salivary gland swelling or neuropathy. Other agents that can be used are hydroxychloroquine, methotrexate, leflunomide, azathioprine, sulfasalazine, mycophenolic acid, and cyclosporine.
- Biological agents, including infliximab, interferon-α and anti-CD20 antibodies (rituximab) are tried.
Overlap Syndromes and Mixed Connective Tissue Disease (MCTD):
Question 59. Define mixed connective tissue disease/overlap syndromes.
Answer:
Overlap syndromes are diseases in which clinical or laboratory signs and symptoms of another defined connective tissue disease occur, such as systemic lupus erythematosus, scleroderma, rheumatoid arthritis, Sjogren’s syndrome, Churg–Strauss arteritis, thrombotic thrombocytopenic purpura, antiphospholipid antibody syndrome and autoimmune thyroid disease.
Mixed connective-tissue disease (MCTD) is a disorder with overlapping clinical features of systemic lupus erythematosus, scleroderma, and myositis, with the presence of a distinctive antibody against U1-ribonucleoprotein (RNP). U1 RNP consists of ribonucleic acid (RNA) plus three proteins (A’, C, and a 68-70 kD protein).
Crystal Arthropathies:
Gout:
Question 60. Describe the etiology, clinical manifestations, diagnosis and management of gout.
(or)
Discuss the causes, clinical manifestations, investigations and management of hyperuricemia.
Answer:
Gout is a heterogeneous group of inflammatory diseases characterized by hyperuricemia (plasma urate level above 6.8 mg/dL) and monosodium urate crystal deposition in and around synovial joints and kidneys.
Etiology of Gout and Hyperuricemia:
Question 61. Write short note on causes of hyperuricemia.
Answer:
Uric acid is the end product of purine metabolism, which is eliminated only in the urine. Humans do not have uricase, an enzyme which degrades uric acid. Usually, there is a balance between uric acid production and tissue deposition of urates.
Classification of hyperuricemia:
Urate overproduction:
- Primary hyperuricemia:
- Idiopathic
- Complete or partial deficiency of HGPRT
- Superactivity of PRPP synthetase
- Secondary hyperuricemia:
- Excessive purine consumption
- Myeloproliferative or lymphoproliferative disorders
- Hemolytic diseases
- Psoriasis
- Glycogen storage diseases: types 1, 3, 5, and 7
Uric acid under excretion:
- Primary hyperuricemia:
- Idiopathic
- Secondary hyperuricemia:
- Decreased renal function
- Metabolic acidosis (ketoacidosis or lactic acidosis)
- Dehydration
- Diuretics
- Hypertension
- Hyperparathyroidism
- Drugs including cyclosporine, pyrazinamide, ethambutol and low-dose salicylates
- Lead nephropathy
Overproduction and under excretion:
- Alcohol use
- Glucose-6-phosphatase deficiency
- Fructose-1-phosphate-aldolase deficiency
Clinical Features:
The natural history of gout has four stages:
- Asymptomatic hyperuricemia: It is a state in which serum urate exceeds the level of solubility but symptoms of crystalline deposition have not occurred. Appears at puberty in males and after menopause in females.
- Acute gouty arthritis:
- Acute gouty arthritis usually follows decades of asymptom- atic hyperuricemia. Usually the first attack occurs in a mid- dleaged male.
- Presentation: It appears as sudden excruciating (agonizing) joint pain, swelling, intense redness and tenderness of the first
metatarsophalangeal (MTP) joint (termed podagra). Other joints that can be involved are tarsal joints, ankles, knees and wrists. Gout can also cause bursitis and tenosynovitis. 20% patients can present with a polyarticular pattern is the initial manifestation. - Systemic symptoms: The acute attack may be accompanied by fever, chills, and malaise, leukocytosis and raised ESR.
- Factors which precipitate acute attack are those that cause fluctuations (rapidly raising or lowering) in serum urate levels. These include overindulgence in certain high-purine foods, alcohol ingestion, starvation, dehydration, certain drugs (e.g. diuretic, salicylates, urate-lowering drugs allopurinol and radiographic contrast agents), trauma and surgery.
- Termination of attack: Even if not treated, acute attacks of gout are usually self-limited, subside spontaneously and first several acute attacks last for 5 to 8 days. Recovery is usually associated with desquamation of the overlying skin. There is no damage to any organ system during the acute attack.
- Intercritical gout: With resolution of the attack, patients enter an interval termed the ‘intercritical period’. It is the asymptomatic period between the first acute attack and subsequent attacks. During this period, the previously affected joints are free of symptoms. Despite this, monosodium urate crystal deposition continues.
- Chronic tophaceous gout (tophi and chronic gouty arthritis):
- Eventually the untreated patient progresses to chronic polyarticular gout (advanced gout). This stage usually develops after several years (10 or more years) of acute gouty arthritis, which may lead to severe crippling disease.
- The intensity of pain in chronic is not severe compared to that of acute attacks.
- Most characteristic lesion of advanced gout is subcutaneous tophus. It results from deposition of crystals in cartilage, synovial membranes, tendons and soft tissues. It can occur anywhere in the body but is most common in the fingers, wrists, ears (helix and antihelix), knees, and olecranon bursa.
Nephropathy:
Occurs in 90% of patients with gouty arthritis. Two types of parenchymal renal damage can occur.
- Urate nephropathy: It is characterized by deposition of urate crystals in the interstitial tissue. It may lead to albuminuria, isosthenuria or renal failure.
- Obstructive uropathy (nephrolithiasis): Chronic hyperuricemia may be complicated by formation of uric acid renal stone with blockage of urine flow. The factors favoring stone formation are hyperuricosuria, overproduction of purine, increased ingestion of purine, uricosuric drugs and acidic urine.
- Other features: Hypertension is a risk factor for development of gout and gout may be associated with increased incidence of hypertension and cardiovascular disorders.
Investigations:
- Joint fluid microscopy: Most specific and diagnostic test but is technically difficult. Synovial fluid examined by compensated polarized microscopy shows urate crystals. These crystals are bright yellow in color and appear as slender, needle shaped, negatively birefringent structures.
- Acute gout: Synovial fluid is turbid due to the greatly elevated cell count (>90% neutrophils).
- Chronic gout: More variable but occasionally it appears white due to the high crystal load. Crystals may be seen between attacks.
- Serum uric acid: Raised hyperuricemia (>7.0 mg/dL in males and >6.0 mg/dL in females) does not confirm the diagnosis. However, levels may be normal (in 50% cases) during an acute attack.
- Serum urea, creatinine: Monitoring is needed to detect any signs of renal impairment.
- Raised ESR and CRP and neutrophilia are observed during acute gout, and they return to normal as the attack subsides.
Plain radiographs: Usually normal in acute gout. In chronic or tophaceous gout, well-demarcated rounded or oval punched out erosions with hypertrophic calcified ‘overhanging edge’ (Martel sign) are characteristically observed. RA causes marginal erosions within the limits of the joint capsule.
Dual-energy computed tomography (DECT): Identifies urate deposits in articular and periarticular locations and can distinguish urate from calcium deposition.
Question 62. Write short note on management of acute attack of gout.
Answer:
Management:
Treatment of acute attack:
- NSAIDs: They rapidly relieve pain and swelling during the acute attack and are the agents of choice. Commonly used drugs include indomethacin (75 mg immediately, then 50 mg 6–8 hours), naproxen (750 mg immediately, then 500 mg every 8–12 hours), diclofenac (75–100 mg immediately, then 50 mg every 6–8 hours) and fenoprofen. NSAIDs may cause renal impairment.
- Local icepacks can produce symptomatic relief.
- Oral colchicine: It is the second drug of choice which acts by inhibiting microtubule assembly in neutrophils. It is very effective at a dose of 1.2 mg immediately, then 0.6 mg every 6–12 hours. Side effects include diarrhea or colicky abdominal pain.
- Corticosteroids: If patient cannot tolerate NSAIDs or colchicine, oral prednisolone (20–30 mg/day) or intramuscular or intra-articular depot methylprednisolone may be tried.
- ACTH gel: A single injection of intramuscular ACTH gel (25 to 80 IU) is useful for terminating an acute attack of gout.
- Interleukin 1 inhibitors: Anakinra, canakinumab have been tried.
Prophylaxis:
Indicated in patients with reccurent flares, tophi with joint damage and patients with renal insufficiency.
Dietary advice:
- Avoid alcohol intake, especially beer, which is high in purines and fructose. Carbonated soft drinks are also rich in fructose.
- Avoid meat and shellfish.
- Controlled weight reduction if patient is obese.
- Avoid use of thiazides or loop diuretics.
Drugs for Prophylaxis:
Urate-lowering therapy:
It is indicated in patients who develop more than one acute attack within one year and those with complications. Xanthine oxidase inhibitors are used in urate overproducers while uricosuric agents are used in urate underexcretors.
- Allopurinol is the drug of first choice. It is an xanthine oxidase inhibitor that blocks the conversion of xanthine into uric acid. Thus it reduces uric acid production and serum uric acid levels rapidly.
- Indication: Should be used when the attacks are frequent and severe (despite dietary changes), associated with renal impairment or tophi, or when NSAIDs or colchicine are difficult to tolerate.
- Dosage: 300–900 mg daily.
- Common side effects: These include gastrointestinal intolerance and skin rashes. Allopurinol hypersensitivity syndrome is the most severe reaction characterized by fever, skin rash, eosinophilia, hepatitis, progressive renal insufficiency, and death.
- Febuxostat is a non-purine selective inhibitor of xanthine oxidase, is a new urate lowering drug that is possibly well-tolerated, safer in renal impairment and superior to allopurinol. Dose is 40–120 mg/day.
- Uricosuric agents like probenecid, benzbromarone, lesinurad, and sulfinpyrazone are indicated in selected cases (when uric acid excretion in the urine is below 600 mg/day).
- Uricosuric drugs are risky if urinary urate excretion is already >800 mg/24 hours. These are contraindicated in those with urate calculi. Uricosurics are ineffective in renal impairment (creatinine clearance < 50 mL/minute).
- Uricase, analogs as pegloticase and rasburicase are used to promote depletion of body urate pools.
Indications for urate-lowering drugs:
Hyperuricemia associated with increased uric acid production
- Urinary uric acid excretion of 1000 mg or more in 24 hours
- Very high levels of serum uric acid
- Hyperuricemia associated with HGPRT deficiency or PRPP synthetase overactivity
- Uric acid nephropathy
- Nephrolithiasis
- Prophylaxis before cytolytic therapy
- Evidence of bone or joint damage
Question 63. Write short note on Lesch–Nyhan syndrome.
Answer:
- Lesch–Nyhan syndrome is an X-linked recessive disorder characterized by three major clinical elements—overproduction of uric acid, neurological disability and behavioral problems.
- Hyperuricemia may lead to nephrolithiasis, renal failure, gouty arthritis and tophi.
- Neurological disability is characterized by dystonia or other movement disorders like choreoathetosis, ballismus, spasticity and hyperreflexia.
- Behavioral abnormalities include intellectual disability and aggressive and impulsive behaviors.
- Hypoxanthine guanine phosphoribosyltransferase (HPRT) that plays a key role in recycling purine bases is deficient in these individuals due to mutation of HPRT gene on X chromosome.
- Prognosis is bad as the treatment options are limited.
- Allopurinol is useful for treating hyperuricemia.
- Motor disabilities managed by baclofen and benzodiazepines and behavioral abnormalities managed by behavioral therapy and medications.
Calcium Pyrophosphate Dihydrate Deposition Disease/Pseudo-gout:
Question 64. Write short essay/note on pseudo-gout; calcium pyrophosphate dihydrate deposition disease; pyrophosphate arthropathy.
Answer:
Calcium pyrophosphate dihydrate (CPPD) deposition disease is a condition associated with deposition of calcium pyrophosphate dihydrate (CPPD) crystals within articular and hyaline cartilage.
Common Causes:
Common causes of calcium pyrophosphate:
- Idioydrate crystal deposition disease.
- Idiopathic (most frequent): Occurs in association with aging
- Complication of primary osteoarthritis
- Long-term consequence of mechanical joint trauma or knee meniscectomy
Familial:
- Associated with systemic metabolic disease, e.g. hyperparathyroidism, dialysis dependent renal failure, hemochromatosis, hypomagnesemia.
Clinical Features:
- May be asymptomatic or may result in a variety of clinical presentations.
- A common presentation is acute inflammatory arthritis that resembles acute gout. Hence, also known as ‘pseudogout’. However, in contrast to gout it is more common in elderly women and usually affects the knee followed by the wrist, shoulder, ankle and elbow. Examination of joint reveals a warm, tender erythematous joint with signs of a large effusion. Fever is common.
- Trigger factors: These include trauma, intercurrent illness and surgery.
- Chronic arthropathy: It may also occur in association with CPPD crystal deposition disease.
Investigations:
- Examination of synovial fluid: CPPD crystals can be demonstrated using compensated polarized microscopy and helps in differentiation from gout. CPPD crystals are generally rhomboid-shaped and positively birefringent. They appear blue when parallel to the long axis of the compensator and yellow when perpendicular. The synovial fluid is turbid and may be uniformly blood-stained.
- X-rays of the affected joint: It may show calcification in hyaline cartilage and/or fibrocartilage (chondrocalcinosis).
Basic Calcium Phosphate Crystal Deposition:
- Basic calcium phosphate (BCP) deposition disease is due to the deposition of hydroxyapatite or apatite crystals and other basic calcium phosphate salts (e.g. octacalcium phosphate, tricalcium phosphate) in soft tissues.
- Main sites of deposits: BCP crystal deposits are mainly seen in tendons, ligaments and hyaline cartilage. In the shoulder, it may manifest as calcific tendinitis of the rotator cuff.
- Fluids for crystal detection: BCP crystals may be detected as nonbirefringent globular clumps within leukocytes in synovial and bursal fluids. Under light microscopy BCP crystal clumps stain positive with the calcium-binding dye such as alizarin red S.
Therapeutics for Crystal Deposition Disease:
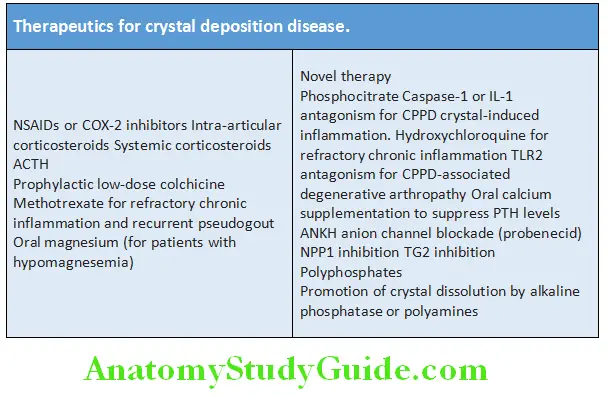
Miscellaneous:
Relapsing Polychondritis (RP):
- Immune-mediated condition associated with inflammation in cartilaginous structures and other connective tissues throughout the body.
- Sites: These include ears, nose, joints, respiratory tract, and others. Unilateral or bilateral external ear inflammation that characteristically spares the earlobe. episcleritis, scleritis, nasal bridge collapse, larynx and tracheal involvement with respiratory distress is seen.
- The McAdam’s criteria require at least three criteria out of six of the following: 1) bilateral auricular chondritis, 2) non-erosive seronegative polyarthritis, 3) nasal chondritis, 4) ocular inflammation, 5) respiratory tract chondritis, and 6) cochlear and/or vestibular dysfunction.
- Associated with diseases: Thirty percent occur in association with disease such as systemic vasculitis (particularly Wegener granulomatosis), connective tissue disorder (e.g. rheumatoid arthritis or systemic lupus erythematosus), or a myelodysplastic syndrome.
- Presentation: Patients can present with respiratory obstruction, aortitis and mitral regurgitation.
Treatment: Steroids and in severe disease immunosuppresive agents (Dapsone, Methrotrexate) are used.
Nonarticular Rheumatism:
Question 65. Differentiate articular from nonarticular disorders.
Answer:
- Nonarticular rheumatism is not actually a true arthritis. It refers to aches and pains that arise from structures outside of joints.
- Forms: Four forms namely tendinitis, bursitis, FMS (fibromyalgia syndrome), and the myofascial pain syndrome.
- Common types of tendinitis and bursitis:
- Tennis elbow: Pain over the lateral epicondyle of the elbow due to inflmmation of the tendons of the wrist extensor muscles that insert at this location.
- Golfer’s elbow: Pain over the medial epicondyle due to inflmmation of the wrist flxor tendons that insert at this location.
- Shoulder impingement syndrome: It is due to impingement of the tendons of the rotator cuff with shoulder abduction or flxion. It can be associated with supraspinatus tendinitis, subacromial bursitis, or rotator cuf tears.
- Housemaid’s knee: It is prepatellar bursitis produced due to repetitive trauma or overuse such as kneeling. Another common region for bursitis is over the greater trochanter of the lateral hip.
Fibromyalgia Syndrome:
Question 66. Write short note on the criteria for diagnosis of fibromyalgia syndrome (FMS).
Answer:
Fibromyalgia syndrome is characterized by chronic widespread pain, and is defined as pain for more than three months both above and below the waist.
- Diagnostic criteria for FMS:
- At least 3 months of widespread pain that is bilateral, above and below the waist.
- It includes axial skeletal pain and pain to palpation at a minimum of 11 of 18 predefined tender points.
- The diagnosis of other diseases does not exclude the diagnosis of FMS.
- Physical examination: Normal except for tender points in precise locations. Usually these points are tender bilaterally.
- Tender points: Typically located at the occiput, at the midportion of the trapezius, the origin of the supraspinatus, low anterior cervical region, second costochondral junction, lateral epicondyle, outer upper quadrant of the buttocks, greater trochanter region, and medial knee area.
- Differential diagnosis: Medical illnesses that may exhibit symptoms similar to those of FMS include: Celiac sprue,
hepatitis C, hyperparathyroidism, hypothyroidism, and polymyalgia rheumatica (PMR). - Nonpharmacological treatment includes patient education and initiation of exercises
- Amitriptyline, duloxetine, milnacipran, or pregabalin can be used as mono or combination therapy
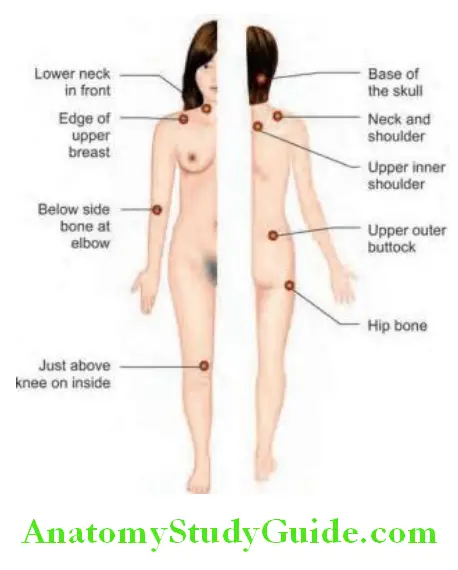
Osteoporosis:
Question 67. Describe the risk factors, prevention and treatment of osteoporosis.
Answer:
Osteoporosis is the most common bone disorder and is a component of the fragility syndrome.
Defiition:
- It is a bone disease characterized by low bone mass and micro-architectural disruption of bone tissue that leads to increased bone fragility and fracture risk.
- WHO definition: Osteoporosis is defined as a bone mineral density (BMD) of 2.5 standard deviations (SDs) below the young healthy adult mean value (T-score ≤−2.5) or lower as measured by DXA (dual-energy X-ray absorptiometry). The term ‘established osteoporosis’ includes the presence of a fragility fracture. There is inverse relationship between bone mineral density and fracture risk in postmenopausal women and older men. However, this definition should not be applied to young women, men or children.
- National Institute of Health (NIH) definition: Osteoporosis is defined as a skeletal disorder characterized by compromised bone strength, which predisposes an individual to an increased risk of fracture.
- Osteopenia: It is the term used when the BMD values lies between −1 and −2.5 SDs below the young adult mean.
Epidemiology:
Most common bone disorder. More common in women (4:1). More common in Caucasian population than in other races.
Pathogenesis:
- In osteoporosis, there is loss of bone mass, despite normal mineralization. In contrast, osteomalacia occurs when bone is not being properly mineralized, despite the normal production of bone matrix.
- Osteoporosis develops due to an imbalance in bone remodeling. It can be due to increased breakdown of bone (absorption of bone) by osteoclasts and/ or decreased bone formation by osteoblasts. The process of mineralization of new bone matrix remains normal.
- Osteoporosis affects both trabecular (long thick bones, e.g. femur) and cortical bone (high surface area, think bones, e.g. spine). When it affects trabecular bones, reabsorption of bone is the main mechanism.
- Genetic factors play important role and multiple genes are involved, including collagen type 1A, vitamin D receptor and estrogen receptor genes.
- Nutritional factors, sex hormone status and physical activity also play a role.
- Cigarette smoking is an independent risk factor.
Classifiation of Osteoporosis:
- Generalized osteoporosis: It involves the entire skeleton.
- It may be:
- Primary or
- Secondary to variety of conditions
- Localized: Limited to certain bone or region (e.g. disuse osteoporosis of a limb).
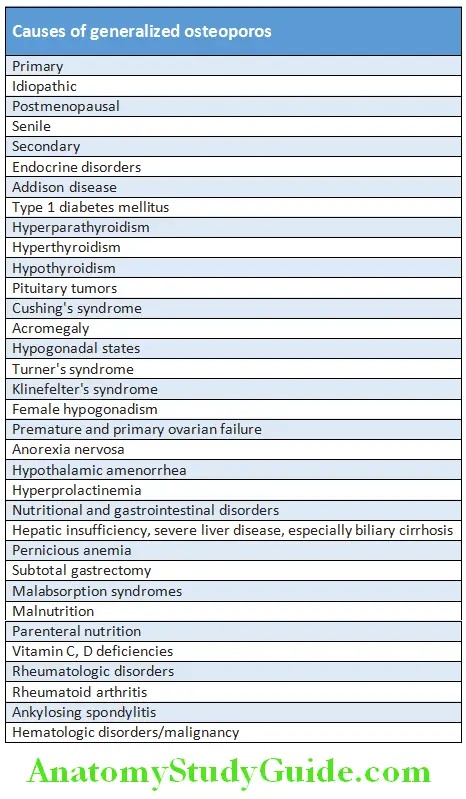
Clinical Features:
- Risk of fracture: Osteoporosis can be clinically silent and does not have specific symptoms. However, it increases the risk of bone fractures and fracture is the only cause of symptoms and thereby increases the morbidity and mortality in elderly.
- Fragility fracture: It is a low-trauma fracture, i.e. mechanical forces (situations) that would not ordinarily result in fracture in healthy people. They typically occur in trabecular bone. The most common sites are:
- Spine (vertebrae): Sudden severe back pain (in the spine) that radiates around to the front suggests vertebral crush fracture (compression fracture).
- Hip (proximal femur): Fractures of the proximal femur usually occur in elderly persons falling on their side or back.
- Usually requires prompt surgery.
- Wrist (distal radius): Colles’ fractures follow a fall on an outstretched arm.
- Other sites include arm (humerus), pelvis, ribs, and other bones.
Investigations and Diagnosis:
Conventional plain radiograph: It is useful along with CT or MRI, for detecting complications of osteopenia (reduced bone mass; pre-osteoporosis), such as fractures. However, radiography is relatively insensitive for detection of early disease as it requires a substantial amount of bone loss (about 30%) to be apparent on X-ray images. The radiographic features of osteoporosis are cortical thinning and increased radiolucency.
Bone biopsy: Tetracycline labeling of the skeleton on bone biopsy determines the rate of skeletal growth. However, the current use of BMD tests, in combination with hormonal evaluation and biochemical markers of bone remodeling, has replaced the bone biopsy.
Biochemical markers: They provide an index of the overall rate of bone remodeling. Markers for bone formation include serum bone-specific alkaline phosphatase, serum osteocalcin, serum propeptide of type I procollagen. Markers for bone resorption urine and serum cross-linked N-telopeptide, urine and serum cross-linked C-telopeptide and urine total free deoxypyridinoline.
Bone mineral density (BMD): It is a measure of the mineral content of bone. It is mostly calcium, but also comprises potassium, manganese, magnesium, strontium, selenium, and other minerals. The diagnosis of osteoporosis can be made by measuring the bone mineral density (BMD).
- Dual energy X-ray absorptiometry (DXA): It is the most commonly used technique to measure BMD. Two low dose X-rays are directed to the area of interest. It measures areal bone density (mineral/surface area rather than a true volumetric density), usually spine (lumbar) and hip (proximal femur). It uses low doses of radiation and is the ‘gold standard’ for the diagnosis of osteoporosis. Results from DXA are expressed as either the T-score or the Z-score.
- T-score that is 2.5 SD or more below the young adult mean BMD is defined as osteoporosis, provided that other causes of low BMD have been ruled out (such as osteomalacia)
- T-score that is 1 to 2.5 SD below the young adult mean is termed low bone mass (osteopenia)
- Normal bone density is defined as a value within 1 SD of the mean value in the young adult reference population
- Z-score is a comparison of the patient’s BMD to an age-matched population.
- A Z-score of -2 or lower is considered below the expected range for age
Quantitative ultrasound: This has many advantages in assessing osteoporosis being quick, easy and cheaper than other methods and does not need ionizing radiation. It is used as a screening procedure prior to DXA assessment and is not used for diagnostic purposes. The calcaneus is the most common skeletal site for quantitative ultrasound assessment.
Quantitative CT scanning: It assesses true volumetric mineral density in mg/cmIt gives separate estimates of BMD for trabecular and cortical bone. It is more expensive and requires higher radiation than other techniques.
Other investigations: These may be necessary to exclude other diseases or identify contributory factors associated with osteoporosis. These include complete blood count, thyroid function test, vitamin D levels and myeloma screen.
Additionally renal function test, serum calcium, phosphorus, serum PTH, cortisol may be indicated.
Treatment of Osteoporosis:
The aim of treatment is to reduce the risk of fracture. Treatment plan for osteoporosis includes:
- Diagnosis Of Individuals At Highest Risk
- Exclusion Of Secondary Causes Of Osteoporosis
- Selecting appropriate treatment.
Prevention:
Lifestyle changes: Methods to prevent osteoporosis include:
- Nutrition: Sufficient daily calcium intake (1200–1500 mg/day) and vitamin D.
- Cessation of smoking.
- Reduction in alcohol intake.
- Increase weight bearing exercise: They increase bone mass density and also helps to prevent falls. At least 30 minutes weight bearing exercise 3x per week is needed to increase bone density. Osteoporosis orthosis helps to prevent spine fractures and support the building up of muscles.
- Fall prevention can help prevent osteoporosis complications.
Pharmacological Management:
Question 68. Write short note on drugs used in osteoporosis.
Answer:
Drug treatments can reduce the risk of fracture by up to 50%.
The two main classes of drugs used namely:
- Antiresorptive Agents
- Anabolic agents.
1. Antiresorptive agents: They block bone resorption by inhibiting the activity of osteoclasts. These include bisphosphonates, denosumab, raloxifene, and calcitonin.
- Bisphosphonates: These drugs are essentially analogues of pyrophosphate and include alendronic acid, risedronate, clodronate, pamidronate, etidronate. Usually taken weekly, and with plenty of water. Poorly absorbed from the gut.
- Should be taken on an empty stomach, as they can bind to calcium in food, after which, the drug cannot be absorbed.
- Only about 10% of the oral dose is absorbed under normal circumstances.
- Adverse drug reactions: GI upset, esophagitis, bone pain, headache and fever rarely osteonecrosis of jaw. Oral and IV bisphosphonates should not be used in CKD and eGFR <30 to 35 mL/min.
- Hormone-replacement therapy (HRT)
- No longer used as mainstream treatment. Used if other treatments are not tolerated or effective.
- It may be useful in women in post-menopausal osteoporosis. Most effective when started early in menopause, and to be continued for >5 years.
- Disadvantages:
- Bone loss continues, and is possibly increased upon stopping HRT
- Increases The Risks Of Cardiovascular Disease and stroke.
- Estrogen analogs: Estrogen replacement therapy is good for prevention of osteoporosis. However, it is not recommended unless there are other indications for its use. In hypogonadal men testosterone has been shown to give improvement in bone quantity and quality.
- Raloxifene, Bazedoxifene: It is a selective estrogen receptor modulators (SERMs) that act on the estrogen receptors throughout the body in a selective manner. Raloxifene has an advantage of reducing the risk of invasive breast cancer. SERMs have been proven effective in clinical trials.
- Calcitonin: It directly inhibits osteoclastic bone resorption activity via the calcitonin receptor on the surface of osteoclasts. It acts by inhibiting actin cytoskeleton which is needed for the osteoclastic activity.
2. Bone anabolic agents: They stimulate bone formation by acting primarily on osteoblasts. These include parathyroid hormone and teriparatide.
Teriparatide and abaloparatide: It acts like parathyroid hormone and stimulates osteoblasts and increases their activity. Mostly used for patients with established osteoporosis, those with low BMD or several risk factors for fracture or cannot tolerate the oral bisphosphonates. It is given as a daily injection with the use of a pen-type injection device.
Sodium fluoride: It produces skeletal changes such as pronounced bone density with increased number and thickness of trabeculae, cortical thickening, and partial obliteration of the medullary space.
3. Other agents:
- RANKL inhibitors: Denosumab is a fully human monoclonal antibody that mimics the activity of osteoprotegerin. It binds to RANKL, thereby preventing RANKL from interacting with RANK and reducing its bone resorption.
- Romosozumab, a monoclonal anti-sclerostin antibody has been approved
- Strontium ranelate: It is an alternative oral treatment, belonging to a class of drugs called ‘dual action bone agents’ (DABAs). It has proven efficacy, especially in the prevention of vertebral fracture. Strontium ranelate is taken as a 2 g oral suspension daily.
Supplements: Dietary supplements are given as adjuncts to pharmacological therapy.
These include:
- Calcium and
- Vitamin D.
Antiphospholipid Antibody Syndrome (Hughes Syndrome):
Question 69. Write short note on antiphospholipid antibody (APLA) syndrome and antiphospholipid antibodies.
Answer:
- Antiphospholipid antibody (APLA) syndrome or antiphospholipid syndrome (APS) is characterized by the presence of a family of antibodies in the patient’s plasma known as antiphospholipid antibodies (autoantibodies directed against phospholipids) and a syndrome of hypercoagulability. This prothrombotic disorder is also associated with thrombocytopenia.
- The term antiphospholipid antibody is misleading because these antibodies react with plasma proteins which themselves bind and form complexes with negatively charged phospholipids. So, in contrast to the terminology antiphospholipid, there is no direct binding of antibodies to phospholipids.
Types of Antiphospholipid Antibody Syndrome:
- Primary antiphospholipid syndrome: They do not have any predisposing cause and their only manifestation is hypercoagulable state.
- Secondary antiphospholipid syndrome: These are found in association with autoimmune diseases, like systemic lupus erythematosus. Because of this association, they were formerly known as lupus anticoagulant syndrome.
Clinical Features:
It is the most common acquired hematologic cause of recurrent thromboembolic events. Clinical manifestations chiefly are due to a direct or indirect effect of venous or arterial thrombosis and/or pregnancy morbidity
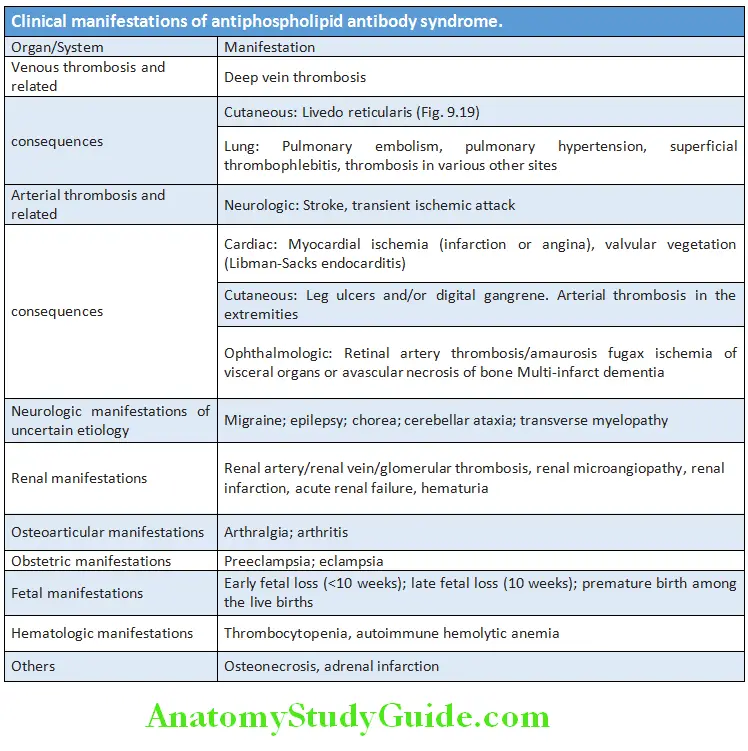
Diagnosis:
Types of antiphospholipid antibodies: Antiphospholipid antibodies are a heterogeneous group of autoantibodies directed against phospholipids-binding proteins. Many plasma proteins (e.g. prothrombin, b2-glycoprotein) bind to anionic phospholipids resulting in different antiphospholipid antibodies. Antiphospholipid antibodies (APAs) may include
- Lupus Anticoagulant (La) Antibody
- Anticardiolipin Antibody (Acl),
- Anti-β2-glycoprotein-I (anti-β2-GPI) antibody.
- Lupus anticoagulant (LA) antibody:
- It is named lupus as it was first detected in patients with systemic lupus erythematosus (SLE).
- It was called anticoagulant because it paradoxically prolongs the clotting times by coagulation assays in vitro (e.g. prolongation of APTT).
- However, the term lupus anticoagulant is a misnomer, because it is more frequently found in patients without lupus and is not anticoagulant and is associated with a hypercoagulable state rather than bleeding.
- For LA screening, two or more phospholipid-dependent coagulation tests are necessary.
- These include:
- Activated Partial Thromboplastin Time
- Dilute Russell Viper Venom Time (Drvvt) And
- Kaolin clotting time.
- Anticardiolipin antibody (aCL): They target cardiolipin (a bovine cardiac protein) and are detected by immunoassays.
- Anti-β 2-glycoprotein-I (anti-b 2-GPI) antibody: They are directed against b 2-glycoprotien I and may be causal in APLA syndrome.
Signifiance:
LA antibodies are more specific, whereas aCL antibodies are more sensitive for antiphospholipid syndrome. IgG aCL antibodies are more specific than IgM type.
Antiphospholipid antibodies may be detected in 10% of normal population and 30–50% of SLE patients. They can also be found in patients with infections such as human immunodeficiency virus and during therapy with medications (e.g. chlorpromazine).
Criteria for Diagnosis:
APLA Syndrome:
Management:
Treatment of Venous Thrombosis:
- Patients with APS have a high risk of recurrent thromboembolism. Hence, they should receive antithrombotic therapy.
- Anticoagulation with unfractionated heparin or low-molecular-weight heparin (LMWH), and oral warfarin, for acute venous thromboembolism (VTE). This is followed by long-term oral warfarin to a target international normalized ratio (INR) of 2 to 3, alone or in combination with 80 mg of aspirin daily. Duration of treatment is probably lifelong.
- Catastrophic APSis treated with anticoagulation, glucocorticoids, and, in severe cases, plasma exchange and/or intravenous immune globulin (IVIG).
Prevention of Recurrence of Arterial Thrombosis:
- Aspirin 325 mg/day or warfarin (INR between 1.4 to 2.8).
Prophylaxis of any Thrombotic Episode:
- Aspirin for preventing thrombosis in females with previous pregnancy loss.
- Modification of other risk factors (e.g. hypercholesterolemia, smoking).
Management of Pregnancy in patients with Antiphospholipid Syndrome:
- Attempted conception: Start aspirin 80 mg daily.
- Confirmed intrauterine pregnancy by ultrasound, start heparin (5,000–10,000 units every 12 hours) or low-molecular-weight heparin in prophylactic doses (enoxaparin 1 mg/kg, dalteparin 5000 units once a day) and continue till late in the third trimester.
- Females with a history of pregnancy losses: The goal is to prevent recurrent pregnancy loss. Two or more early pregnancy losses, or one or more late pregnancy losses without any previous history of thrombosis should receive combination of aspirin and heparin (unfractionated or low molecular weight) during pregnancy.
Treatment:
- Intravenous heparin along with high-dose corticosteroids.
- Early addition of plasma exchange and/or intravenous immunoglobulin (400 mg/kg qd) or CD20 monoclonal antibody rituximab, ofatumumab, veltuzumab, and ocrelizumab (375 mg/m 2 per week for 4 weeks) in patients who do not respond promptly to heparin and corticosteroids.
Antiphospholipid antibody syndrome classification criteria (revised Sapporo classification criteria):
Vascular thrombosis:
- Arterial, venous, or small vessel
Pregnancy morbidity:
- One or more fetal deaths
- One or more premature births due to severe
- preeclampsia or placental insufficiency
- Three or more first trimester losses
Plus:
- Lupus anticoagulant
- Anticardiolipin IgG or IgM (medium to high titer)
- Anti-beta 2 glycoprotein-1 IgG or IgM on two occasions 12 weeks (or more) apart
- A definite diagnosis of antiphospholipid syndrome is made if the patient has one clinical and one laboratory criteria.
- Prognosis: Despite treatment, mortality is about 50%.
Inflammatory Muscle Diseases:
Question 70. Write short note on inflammatory muscle diseases.
Answer:
Common Inflmmatory Muscle Disease:
Idiopathic inflammatory myopathies consist of three major types:
- Polymyositis
- Dermatomyositis
- Inclusion body myositis.
Polymyositis and Dermatomyositis:
Question 71. Write short note on polymyositis and dermatomyositis.
Differentiate polymyositis from dermatomyositis.
Idiopathic Polymyositis:
Onset: Gradual. Commonly between 40 and 60 years of age.
Clinical Manifestations:
- Symmetrical proximal muscle weakness:
- Sites affected: Usually affects the lower limbs (hips and thighs) than the upper limbs (shoulder girdle muscles). The distal muscles, facial muscles are not usually affected.
- Others: Involvement of pharyngeal, laryngeal and respiratory muscles can lead to dysphonia and respiratory failure.
- Systemic features: Fever, weight loss and fatigue are common.
- Investigation: Muscle biopsy shows fiber necrosis, regeneration and inflammatory cell infiltrate.
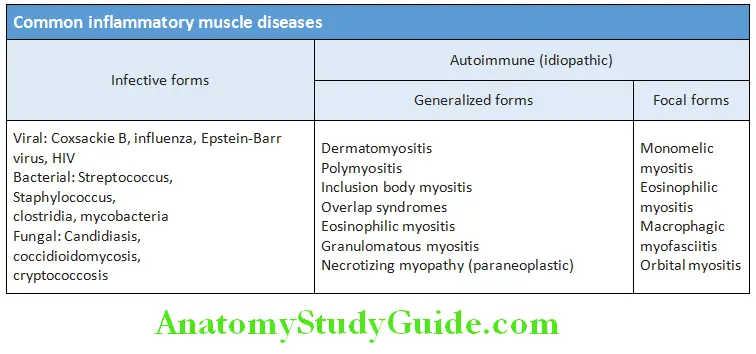
Idiopathic Dermatomyositis:
Question 72. Write short note on dermatomyositis.
Answer:
- Dermatomyositis characterized by polymyositis along with characteristic skin changes. The skin changes may occur before or after the muscle syndrome.
- Skin changes: These include
- Gottron’s papules: Scaly, erythematous or violaceous, psoriasiform plaques occurring over the extensor surfaces of PIP and DIP joints.
- Heliotrope rash: Violaceous discoloration of the eyelid may be seen along with periorbital edema.
- Shawl sign or the V sign: Occurrence of erythematous macules distributed in a ‘shawl’ pattern over the upper back, chest and shoulders.
- Holster sign: Poikiloderma on the lateral aspects of the thighs.
- Periungual telangiectasias: Periungual nail-fold capillaries are enlarged and tortuous.
- Mechanic’s hand: Fissured, scaly and hyperkeratotic hand (in cases of antisynthetase syndrome).
- Others: Ulcerative vasculitis and calcinosis (calcium deposition) of the subcutaneous tissue seen in about 25% of cases.
- In the long-term, muscle fibrosis and contractures of joints occur.
- Increased risk of malignancy: Few patients with dermatomyositis above 60 years of age have an underlying malignancy.
- The most common being ovarian and gastric carcinoma, and lymphoma.
- Amyopathic dermatomyositis or dermatomyositis sine myositis: Presence of pathognomonic skin lesion without muscle involvement.
- Investigation: Muscle biopsy shows necrosis of muscle (single fibers or in groups), perifascicular atrophy, and infiltration with lymphocytes.
Lists the differences between dermatomyositis, polymyositis and inclusion-body myositis:
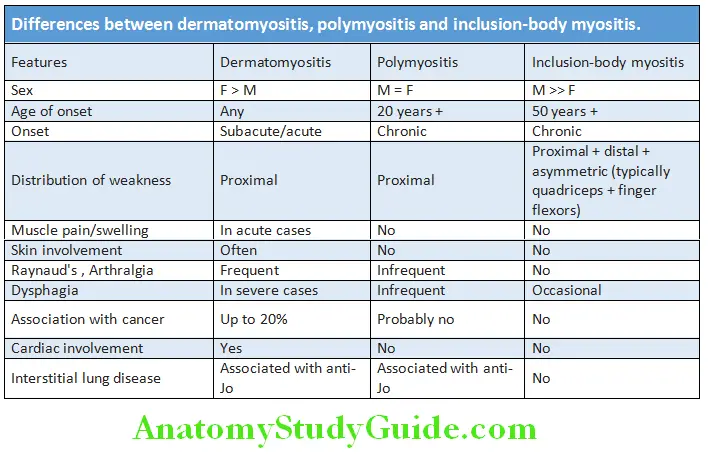
Diagnosis:
- ESR and CRP: Usually raised.
- Serum autoantibody studies: Antinuclear antibodies frequently positive.
- Muscles enzymes: Raised levels of serum creatine kinase (CK), aldolase, serum glutamic oxaloacetic transaminase (SGOT), lactate dehydrogenase (LDH) and serum glutamic pyruvate transaminase (SGPT).
- Autoantibodies directed against cytoplasmic RNA synthetases, other cytoplasmic proteins and RNP found in about 30% of patients. Only antihistidyl-tRNA synthetase (anti-Jo-1 antibody) is a diagnostic marker and is found in 20% of patients with polymyositis/dermatomyositis.
- Electromyography (EMG): It can confirm the presence of myopathy and exclude neuropathy.
- MRI: It can be used to detect abnormally inflamed muscle.
- Needle muscle biopsy: Characteristic changes: fiber necrosis, regeneration and lymphocytic inflammatory cell infiltrate.
- Open biopsy helps in more thorough assessment.
- Screening for malignancy: These include CXR (chest X-ray), mammography, pelvic/abdominal ultrasound, PET scan, urine microscopy and circulating tumor markers.
Management:
- Corticosteroids: The treatment of choice is prednisolone (dose of 1 mg/kg daily) till patient shows significant improvement. High-dose intravenous methylprednisolone (1 g/day for 3 days) may be necessary in patients with respiratory or pharyngeal weakness.
- Additional immunosuppressive therapy: If no improvement with prednisolone, azathioprine (2–3 mg/kg/day) may be added. It also has some steroid-sparing effect. Other steroid-sparing agents include methotrexate, azathioprine, ciclosporin, cyclophosphamide mycophenolate mofetil, tacrolimus and MMF.
- Intravenous immunoglobulin: It may be effective in refractory or rapidly progressive cases.
Leave a Reply