The Musculoskeletal System
Bones And Cartilage
The skeleton consists of cartilage and bone. Cartilage has a role in the growth and repair of bone, and in the adults forms the articular skeleton responsible for the movement of joints. Bone is a specialised form of connective tissue which performs the function of providing mechanical support and is also a mineral reservoir for calcium homeostasis. There are 206 bones in the human body, and depending upon their size and shape may be long, flat, tubular etc.
Table of Contents
Read And Learn More: Systemic Pathology Notes
Normal Structure Of Bone
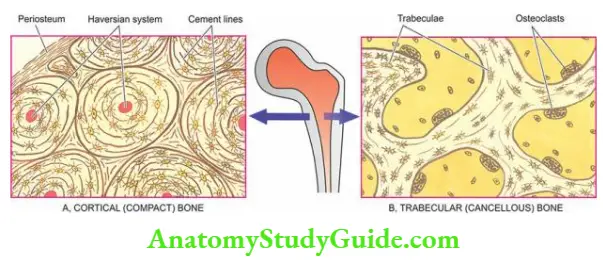
Bone is divided into 2 components:
- Cortical or compact bone comprises 80% of the skeleton and is the dense outer shell responsible for structural rigidity. It consists of haversian canals with blood vessels surrounded by concentric layers of mineralised collagen-forming osteons which are joined together by cement lines.
- Trabecular or cancellous bone composes 20% of the skeleton and has trabeculae traversing the marrow space. Its main role is in mineral homeostasis.
Musculoskeletal System Diseases
Bones And Cartilage Histology: Bone consists of large quantities of extracellular osteoid matrix which is loaded with calcium hydroxyapatite and a relatively small number of bone cells which are of 3 main types: osteoblasts, osteocytes and osteoclasts.
1. Osteoblasts: Osteoblasts are uninucleate cells found abundantly along the new bone-forming surfaces. They synthesise bone matrix. The serum levels of bone-related alkaline phosphatase (other being hepatic alkaline phosphatase) are a marker for osteoblastic activity.
Its levels are raised at puberty during period of active bone growth and in pathologic conditions associated with high osteoblastic activity such as in fracture repair and Paget’s disease of the bone.
2. Osteocytes: Osteocytes are those osteoblasts which get incorporated into the bone matrix during its synthesis. Osteocytes are found within small spaces called lacunae lying in the bone matrix. The distribution of the osteocytic lacunae is a reliable parameter for distinguishing between woven and lamellar bone.
- Woven bone is immature and rapidly deposited. It contains a large number of closely-packed osteocytes and consists of an irregular interlacing pattern of collagen fibre bundles in the bone matrix. Woven bone is seen in foetal life and in children under 4 years of age.
- Lamellar bone differs from the woven bone in having smaller and less numerous osteocytes and fine and parallel lamellar sheets of collagen fibres. Lamellar bone usually replaces woven bone or pre-existing cartilage.
3. Osteoclasts: Osteoclasts are large multinucleate cells of mononuclear-macrophage origin and are responsible for bone resorption. The osteoclastic activity is determined by bone-related serum acid phosphatase levels (other being prostatic acid phosphatase).
Osteoclasts are found along the endosteal surface of the cortical (compact) bone and the trabeculae of the trabecular (cancellous) bone.
4. Osteoid matrix: of bone consists of 90-95% of collagen type I and comprises nearly half of the total body’s collagen. Virtually the whole of the body’s hydroxyproline and hydroxylysine reside in the bone.
The architecture of bone collagen reflects the rate of its synthesis and may be woven or lamellar, as described above.
Musculoskeletal System Diseases
Bone Formation And Resorption: Bone deposition is the result of osteoblasts while bone resorption is the function of osteoclasts. The bone formation may take place directly from collagen called membranous ossification seen in certain flat bones, or may occur through an intermediate stage of cartilage termed endochondral ossification found in the metaphysis of long bones.
- In either case, firstly an uncalcified osteoid matrix is formed by osteoblasts which are then mineralised in 12-15 days. This delay in mineralisation results in the formation of about 15 µm thick osteoid seams at calcification fronts (About >1 µm of matrix osteoid is formed daily).
- Uncalcified osteoid appears eosinophilic in H & E stains and does not stain with the von Kossa reaction, while mineralised osteoid is basophilic in appearance and stains black with the von Kossa reaction (a stain for calcium).
- Areas of active bone resorption have scalloped edges of bone surface called Howship’s lacunae and contain multinucleated osteoclasts.
- In this way, osteoblastic formation and osteoclastic resorption continue to take place into adult life in a balanced way termed bone modelling. The important role of vitamin D1, parathyroid hormone and calcitonin in calcium metabolism has already been discussed.
Normal Structure Of Cartilage:
Cartilage consists of 2 components: cartilage matrix and chondrocytes.
Cartilage matrix: Like bone, cartilage consists of organic and inorganic materials. The inorganic material of cartilage is calcium hydroxyapatite similar to that in bone matrix but the organic material of the cartilage is distinct from the bone.
- It consists of a very high content of water (80%) and the remaining 20% consists of type II collagen and proteoglycans. The high water content of the cartilage matrix is responsible for the function of articular cartilage and lubrication.
- Proteoglycans are macromolecules having proteins complexed with polysaccharides termed glycosaminoglycans.
- Cartilage glycosaminoglycans consist of chondroitin sulfate and keratan sulfate, the former being most abundant comprising 55-90% of cartilage matrix varying on the age of the cartilage.
Chondrocytes: Primitive mesenchymal cells which form bone cells from chondroblasts which give rise to chondrocytes. However, calcified cartilage is removed by the osteoclasts. Depending upon the location and structural composition, cartilage is of 3 types:
- Hyaline cartilage is the basic cartilaginous tissue comprising articular cartilage of joints, cartilage in the growth plates of developing bones, costochondral cartilage, cartilage in the trachea, bronchi and larynx and the nasal cartilage. Hyaline cartilage is the type found in most cartilage-forming tumours and in the fracture callus.
- Fibrocartilage is a hyaline cartilage that contains more abundant type II collagen fibres. It is found in the annulus fibrosus of the intervertebral disc, menisci, insertions of joint capsules, ligament and tendons. Fibrocartilage may also be found in some cartilage-forming tumours and in the fracture callus.
- Elastic cartilage is hyaline cartilage that contains abundant elastin. Elastic cartilage is found in the pinna of ears, epiglottis and arytenoid cartilage of the larynx
Normal Structure of Bone and Cartilage:
- Bone is composed of cortical or compact bone (80%) which is the dense outer shell, and trabecular or cancellous bone (20%) which has trabeculae traversing the marrow space.
- Bone consists of extracellular osteoid matrix and bone cells (osteoblasts, osteocytes and osteoclasts).
- Cartilage consists of cartilage matrix and chondrocytes and lacks blood vessels, lymphatics and nerves. It may have focal areas of calcification.
Musculoskeletal System Diseases
Infection, Necrosis, Fracture Healing
Osteomyelitis:
- An infection of the bone is termed osteomyelitis (myelo = marrow). A number of systemic infectious diseases may spread to the bone such as enteric fever, actinomycosis, mycetoma (Madura’s foot), syphilis, tuberculosis and brucellosis.
- However, two of the conditions which produce significant pathologic lesions in the bone, namely pyogenic osteomyelitis and tuberculous osteomyelitis, are described below.
Pyogenic Osteomyelitis:
Pyogenic or suppurative osteomyelitis is usually caused by bacterial infection and rarely by fungi. The profile of patients in developing and developed countries is different: In the developing countries of the world, it may occur by haematogenous route, most commonly in the long bones of infants and young children (5-15 years of age) (called haematogenous osteomyelitis)
- On the other hand, in the developed world, where the institution of antibiotics is early and prompt, the haematogenous spread of infection to the bone is uncommon; instead, a direct extension of infection from the adjacent area, frequently involving the jaws and skull, is the more common mode of spread.
- Bacterial osteomyelitis may be a complication at all ages in patients with compound fractures, surgical procedures involving prosthesis or implants, gangrene of a limb in diabetics, debilitation and immunosuppression.
- Though any etiologic agent may cause osteomyelitis, Staphylococcus aureus is implicated in a vast majority of cases. Less frequently, other organisms such as streptococci, Escherichia coli, Pseudomonas, Klebsiella and anaerobes are involved.
- Mixed infections are common in posttraumatic cases of osteomyelitis. There may be transient bacteraemia preceding the development of osteomyelitis so blood cultures may be positive.
- Clinically, the child with acute haematogenous osteomyelitis has a painful and tender limb. Fever, malaise and leucocytosis generally accompany the bony lesion. A radiologic examination confirms the bony destruction.
- Occasionally, osteomyelitis remains undiscovered until it becomes chronic. Draining sinus tracts may form which may occasionally be the site for the development of squamous carcinoma. Persistence, neglect and chronicity of osteomyelitis over a longer period of time may lead to the development of amyloidosis.
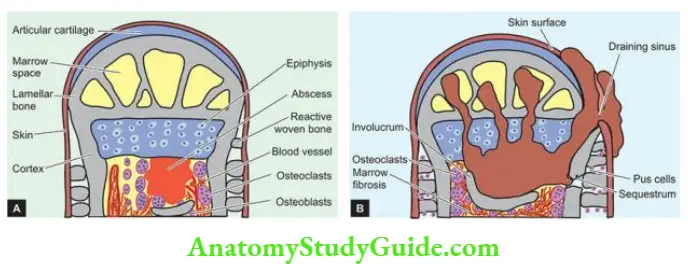
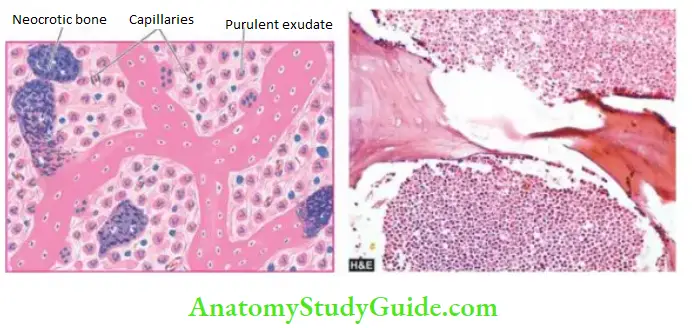
Morphologic Features: Depending upon the duration, osteomyelitis may be acute, subacute or chronic. The basic pathologic changes in any stage of osteomyelitis are suppuration, ischaemic necrosis, healing by fibrosis and bony repair. The sequence of pathologic changes is as under:
- The infection begins in the metaphyseal end of the marrow cavity which is largely occupied by pus. At this stage, microscopy reveals congestion, oedema and exudate of neutrophils.
- The tension in the marrow cavity is increased due to pus and results in the spread of infection along the marrow cavity, into the endosteum, and into the Haversian and Volkmann’s canal, causing periostitis.
- The infection may reach the subperiosteal space forming subperiosteal abscesses. It may penetrate through the cortex creating draining skin sinus tracts.
- A combination of suppuration and impaired blood supply to the cortical bone results in erosion, thinning and infarction necrosis of the cortex called a sequestrum.
- With the passage of time, there is the formation of new bone beneath the periosteum present over the infected bone. This forms an encasing sheath around the necrosed bone and is known as an involucrum. Involucrum has an irregular surface and has perforations through which discharging sinus tracts pass.
- Long-continued neo-osteogenesis gives rise to a dense sclerotic pattern of osteomyelitis called chronic sclerosing nonsuppurative osteomyelitis of Garré.
- Occasionally, acute osteomyelitis may be contained to a localised area and walled off by fibrous tissue and granulation tissue. This is termed Brodie’s abscess.
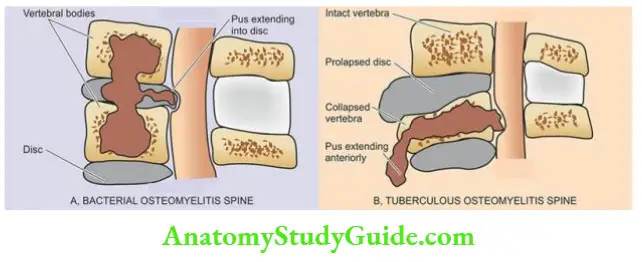
- In vertebral pyogenic osteomyelitis, the infection begins from the disc (discitis) and spreads to\ involve the vertebral bodies.
Complications: Osteomyelitis may result in the following complications:
- Septicaemia
- Acute bacterial arthritis
- Pathologic fractures
- Development of squamous cell carcinoma in long-standing cases
- Secondary amyloidosis in long-standing cases
- Vertebral osteomyelitis may cause vertebral collapse with paravertebral abscess, epidural abscess, cord compression and neurologic deficits.
Tuberculous Osteomyelitis:
- Tuberculous osteomyelitis, though rare in developed countries, continues to be a common condition in underdeveloped and developing countries of the world. The tubercle bacilli, M. tuberculosis, reach the bone marrow and synovium most commonly by haematogenous dissemination from an infection elsewhere, usually from the lungs, and infrequently by direct extension from the pulmonary or gastrointestinal tuberculosis.
- The disease affects adolescents and young adults more often. Most frequently involved are the spine and bones of extremities.
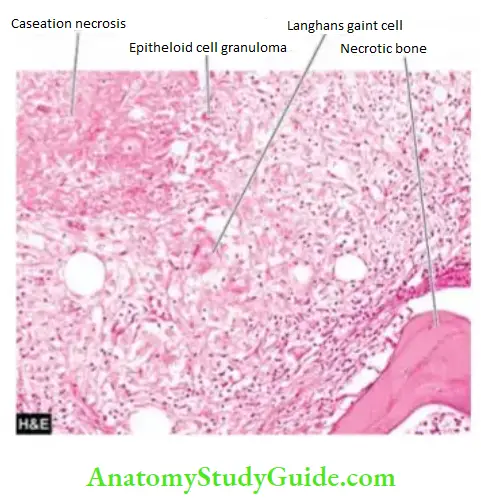
Morphologic Features: The bone lesions in tuberculosis have the same general histological appearance as in tuberculosis elsewhere and consist of central caseation necrosis surrounded by tuberculous granulation tissue and fragments of necrotic bone.
- The tuberculous lesions appear as a focus of bone destruction and replacement of the affected tissue by caseous material and formation of multiple discharging sinuses through the soft tissues and skin. Involvement of joint spaces and intervertebral discs is frequent.
- Tuberculosis of the spine, Pott’s disease, often commences in the vertebral body and may be associated with compression fractures and destruction of intervertebral discs, producing permanent damage and paraplegia.
- Extension of caseous material along with pus from the lumbar vertebrae to the sheaths of the psoas muscle produces a psoas abscess or lumbar cold abscess. The cold abscess may burst through the skin and form a sinus. Long-standing cases may develop systemic amyloidosis.
Avascular Necrosis (Osteonecrosis):
Avascular necrosis of the bones or osteonecrosis results from ischaemia. It is a relatively common condition.
Etiology And Pathogenesis: Some of the common causes are as follows:
- Fracture or dislocation
- Sickle cell disease
- Corticosteroid administration
- Radiation therapy
- Chronic alcoholism
- Idiopathic
The mechanism of osteonecrosis in many cases remains obscure, while in others it is by an interruption in the blood supply to the bones induced by direct trauma, compression, or thromboembolic obstruction.
Morphologic Features: There are pathological fractures of the involved bone due to infarcts. The most common sites are the ones where the disruption in blood supply is at arterial circulation. The infarcts mainly involve the medulla of the long bone in the diaphysis. This is because the nutrient arteries supply blood to sinusoids of the medulla and the inner cortex after penetrating the cortex, while the cortex is relatively unaffected due to collateral circulation.
Grossly, the lesional area shows a wedge-shaped area of infarction in the subchondral bone under the convex surface of the joint.
Microscopically, the infracted medulla shows saponified marrow fat. The overlying cartilage and the cortex of the long bones are relatively unaffected.
Long-term sequelae of osteonecrosis include the occurrence of malignant tumours in this location such as osteosarcoma, malignant fibrous histiocytoma and fibrosarcoma etc.
Fracture Healing:
Fracture of the bone initiates a series of tissue changes which eventually lead to the restoration of normal structure and function of the affected bone. Fracture of a bone is commonly associated with injury to the soft tissues. The various types of fractures and their mechanism of healing are discussed along with the healing of specialised tissues.
Infection, Necrosis, Fracture Healing:
- Suppurative or pyogenic osteomyelitis is usually caused by bacterial infection and rarely by fungi. It may be by the haematogenous route or by the direct spread of infection.
- Tuberculous osteomyelitis continues to be a common condition in developing countries of the world, either by haematogenous spread or by direct spread from adjacent focus.
- Avascular necrosis or osteonecrosis results from ischaemia and may occur be idiopathic or result from steroid administration, alcoholism etc.
- Healing of fractures occurs by callus formation, either as a primary or secondary union.
Disorders Of Bone Growth And Development:
A number of abnormalities of the skeleton are due to disordered bone growth and development and are collectively termed skeletal dysplasias. These include both local and systemic disorders.
- Local defects involve a single bone or a group of bones such as absence or presence in diminished form, fused with neighbouring bones (example syndactyly), and formation of extra bones (example supernumerary ribs).
- However, more importantly, skeletal dysplasias include systemic disorders involving particular epiphyseal growth plates. These include achondroplasia (a disorder of chondroblasts), osteogenesis imperfecta (a disorder of osteoblasts), osteopetrosis (a disorder of osteoclasts) and foetal rickets (a disorder of mineralisation).
- Though multiple exostoses (osteochondromas) is a hereditary lesion, it is described later along with solitary sporadic exostosis.
Achondroplasia:
Achondroplasia is an autosomal dominant genetic abnormality. There is selective interference with normal endochondral ossification at the level of epiphyseal cartilaginous growth plates of long bones. Thus, the long bones are abnormally short but the skull grows normally leading to a relatively large skull. Achondroplasia is the commonest cause of dwarfism.
Osteogenesis Imperfecta:
- Osteogenesis imperfecta is an autosomal dominant or recessive disorder of synthesis of type I collagen that constitutes 90-95% of bone matrix. The disorder, thus, involves not only the skeleton but other extra-skeletal tissues as well-containing type I collagen such as sclera, eyes, joints, ligaments, teeth and skin.
- The skeletal manifestations of osteogenesis imperfecta are due to defective osteoblasts which normally synthesise type I collagen. This results in thin or nonexistent cortices and irregular trabeculae (too little bone) so the bones are very fragile and liable to multiple fractures. The growth plate cartilage is, however, normal.
- The condition may be evident at birth (osteogenesis imperfecta congenital) when it is more severe or may appear during adolescence (osteogenesis imperfecta tarda) which is a less incapacitating form.
- Extraskeletal lesions of osteogenesis imperfecta include blue and translucent sclerae, hearing loss due to bony abnormalities of the middle and inner ear, and imperfect teeth.
Osteopetrosis:
- Osteopetrosis, also called marble bone disease, is an autosomal dominant or recessive disorder of increased skeletal mass or osteosclerosis caused by a hereditary defect in osteoclast function. The condition may appear in 2 forms: autosomal recessive (malignant infantile form) and autosomal dominant (benign adult form).
- Failure of normal osteoclast function of bone resorption coupled with continued bone formation and endochondral ossification results in net overgrowth of calcified dense bone (too much bone) which occupies most of the available marrow space.
- Despite the increased density of the bone compared with normal bone, there is poor structural support so the skeleton is susceptible to fractures.
- Besides the skeletal abnormalities, the infantile malignant form is characterised by effects of marrow obliteration such as anaemia, neutropenia, thrombocytopenia, hepatosplenomegaly with extramedullary haematopoiesis, hydrocephalus and neurologic involvement with consequent deafness, optic atrophy and blindness. Metabolically, hypocalcaemia occurs due to defective osteoclast function.
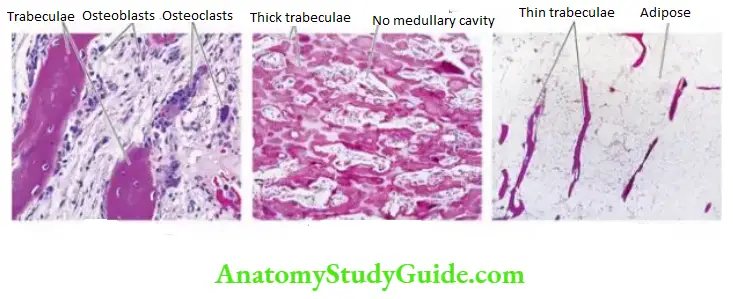
Histologically, the number of osteoclasts is increased which have dysplastic, bizarre and irregular nuclei and are dysfunctional.
Disorders of Bone Growth and Development:
- Disordered bone growth and development cause skeletal dysplasias which may be as local or systemic defects.
- Achondroplasia is a form of dwarfism in which there is selective interference with normal endochondral ossification at the level of epiphyseal cartilaginous growth plates of long bones.
- Osteogenesis imperfecta is an autosomal dominant or recessive disorder of synthesis of type I collagen involving the skeleton (too little bone) and extra-skeletal tissues.
- Osteopetrosis or marble bone disease, is an autosomal disorder of increased skeletal mass (too much bone) caused by a hereditary defect in osteoclast function.
Metabolic And Endocrine Bone Diseases:
A large number of metabolic and endocrine disorders produce generalised skeletal disorders. These include the following:
- Osteoporosis Results from the quantitative reduction in otherwise normal bone.
- Osteomalacia and rickets are Characterised by a qualitative abnormality in the form of impaired bone mineralisation due to deficiency of vitamin D in adults and children respectively.
- Scurvy is Caused by a deficiency of vitamin C resulting in subperiosteal haemorrhages.
- Hyperparathyroidism Resulting in the formation of giant cell-rich brown tumours.
- Pituitary dysfunctions Hyperpituitarism causes gigantism and acromegaly and hypopituitarism results in dwarfism.
- Thyroid dysfunctions Hyperthyroidism causes osteoporosis and hypothyroidism leading to cretinism.
- Renal osteodystrophy Occurs in chronic renal failure and results in features of osteitis fibrosa cystica, osteomalacia and areas of osteosclerosis.
- Skeletal fluorosis Occurs due to excess sodium fluoride content in the soil and water in an area.
Many of the conditions listed above have been discussed in respective chapters already; others are considered below.
Osteoporosis:
- Osteoporosis or osteopenia is a common clinical syndrome involving multiple bones in which there is quantitative reduction of bone tissue mass. This reduction in bone mass results in a fragile skeleton associated with an increased risk of fractures and consequent pain and deformity.
- The condition is particularly common in elderly people and more frequent in postmenopausal women. The condition may remain asymptomatic or may cause only backache. However, more extensive involvement is associated with fractures, particularly of the distal radius, femoral neck and vertebral bodies.
- Osteoporosis may be difficult to distinguish radiologically from other osteopenias such as osteomalacia, osteogenesis imperfecta, osteitis fibrosa of hyperparathyroidism, renal osteodystrophy and multiple myeloma.
- Radiologic evidence becomes apparent only after more than 30% of bone mass has been lost. Levels of serum calcium, inorganic phosphorus and alkaline phosphatase are usually within normal limits.
- Many non-invasive techniques are now available for the measurement of bone mass example DEXA and SEXA scans (dual-energy and single-energy X-ray absorptiometry), quantitative CT and ultrasound.
Pathogenesis: Osteoporosis is conventionally classified into 2 major groups: primary and secondary.
- Primary osteoporosis: results primarily from osteopenia without an underlying disease or medication. Primary osteoporosis is further subdivided into 2 types: idiopathic type found in the young and juveniles and is less frequent, and involutional type seen in postmenopausal women and ageing individuals and is more common.
- The exact mechanism of primary osteoporosis is not known but there is a suggestion that it is the result of excessive osteoclastic resorption and slow bone formation. A number of risk factors have been attributed to causing this imbalance between bone resorption and bone formation. These include the following:
- Genetic factors are more marked in whites and Asians than blacks.
- Sex is more frequent in females than in males.
- Reduced physical activity as in old age.
- Deficiency of sex hormones oestrogen deficiency in women as in postmenopausal osteoporosis and androgen deficiency in men.
- Combined deficiency of calcitonin and oestrogen.
- Hyperparathyroidism primary and secondary.
- Deficiency of vitamin D.
- Local factors which may stimulate osteoclastic resorption or slow osteoblastic bone formation.
- The exact mechanism of primary osteoporosis is not known but there is a suggestion that it is the result of excessive osteoclastic resorption and slow bone formation. A number of risk factors have been attributed to causing this imbalance between bone resorption and bone formation. These include the following:
- Secondary osteoporosis: is attributed to a number of factors and conditions (for example immobilisation, chronic anaemia, acromegaly, hepatic disease, hyperparathyroidism, hypogonadism, thyrotoxicosis and starvation), or as an effect of medication (for example administration of glucocorticoids, anticonvulsant drugs and a large dose of heparin).
Morphologic Features: Except for disuse or immobilisation of osteoporosis which is localised to the affected limb, other forms of osteoporosis have systemic skeletal distribution. The most commonly encountered osteoporotic fractures are vertebral crush fracture, femoral neck fracture and wrist fracture. There is an enlargement of the medullary cavity and thinning of the cortex.
Histologically, osteoporosis may be active or inactive type.
- Active osteoporosis is characterised by increased bone resorption and the formation of accelerated turnover. There is an increase in the number of osteoclasts with increased resorptive surfaces as well as an increased quantity of osteoid with increased osteoblastic surfaces. The width of osteoid seams is normal.
- Inactive osteoporosis has the features of minimal bone formation and reduced resorptive activity i.e. reduced turnover.
- Histological changes of inactive osteoporosis include decreased number of osteoclasts with decreased resorptive surfaces, and normal or reduced amount of osteoid with decreased osteoblastic surface. The width of osteoid seams is usually reduced.
Brown Tumour Of Hyperparathyroidism:
- Hyperparathyroidism of primary or secondary type results in oversecretion of parathyroid hormone which causes increased osteoclastic resorption of the bone. General aspects of hyperparathyroidism are discussed. Here, skeletal manifestations of hyperparathyroidism are considered.
- Severe and prolonged hyperparathyroidism results in osteitis fibrosa cystic. The lesion is generally induced as a manifestation of primary hyperparathyroidism, and less frequently, as a result of secondary hyperparathyroidism such as in chronic renal failure (renal osteodystrophy).
- The clinical manifestations of bone disease in hyperparathyroidism are its susceptibility to fracture, skeletal deformities, joint pains and dysfunctions as a result of deranged weight bearing.
- The bony changes may disappear after the cure of primary hyperparathyroidism such as the removal of functioning adenoma. The chief biochemical abnormality of excessive parathyroid hormone is hypercalcaemia, hypophosphataemia and hypercalciuria.
Morphologic Features: The bone lesions of primary hyperparathyroidism affect the long bones more severely and may range from the minor degree of generalised bone rarefaction to prominent areas of bone destruction with cyst formation or brown tumours.
Grossly, there are focal areas of erosion of cortical bone and loss of lamina dura at the roots of teeth.
Histologically, the following sequential changes appear over a period of time:
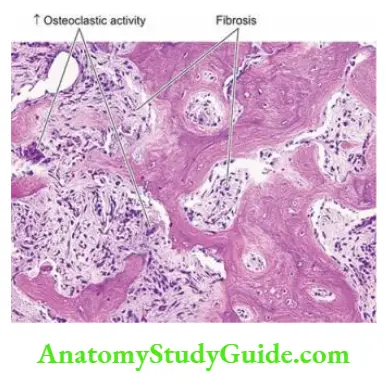
- The earliest change is demineralisation and increased bone resorption beginning at the subperiosteal and endosteal surface of the cortex and then spreading to the trabecular bone.
- There is the replacement of bone and bone marrow by fibrosis along with an increased number of bizarre osteoclasts at the surfaces of moth-eaten trabeculae and cortex (osteitis fibrosa).
- As a result of increased resorption, microfractures and microhemorrhages occur in the marrow cavity leading to the development of cysts (osteitis fibrosa cystica).
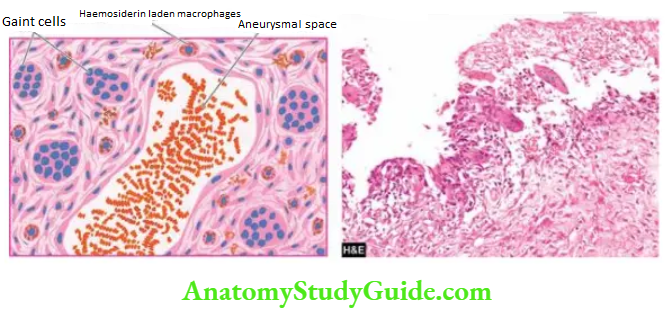
- Haemosiderin-laden macrophages and multinucleate giant cells appear at the areas of haemorrhages producing an appearance of a giant cell-rich brown tumour of hyperparathyroidism requiring differentiation from giant cell tumour or osteoclastoma.
- However, the so-called brown tumours, unlike osteoclastoma, are not true tumours but instead, regress or disappear on surgical removal of hyperplastic or adenomatous parathyroid tissue.
Renal Osteodystrophy (Metabolic Bone Disease):
Renal osteodystrophy is a loosely used term that encompasses a number of skeletal abnormalities appearing in cases of chronic kidney disease and in patients treated with dialysis for several years. Renal osteodystrophy is more common in children than in adults.
Clinical symptoms of bone disease in advanced renal failure appear in less than 10% of patients but radiologic and histologic changes are observed in a fairly large proportion of cases.
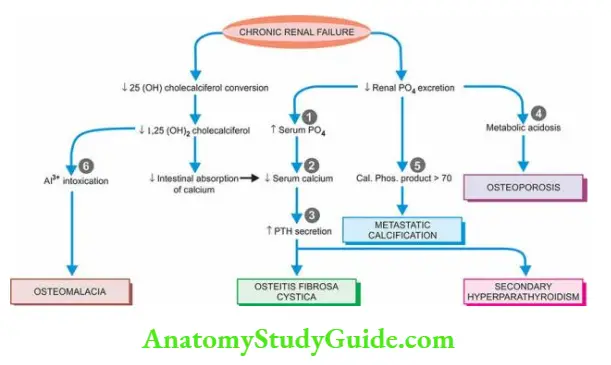
Pathogenesis Renal osteodystrophy involves two main events hyperphosphataemia and hypocalcaemia which, in turn, leads to parathormone elaboration and resultant osteoclastic activity and major lesions of renal osteodystrophy osteomalacia (rickets in children), secondary hyperparathyroidism, osteitis fibrosa cystica, osteosclerosis and metastatic calcification. The mechanisms underlying renal osteodystrophy are schematically illustrated and briefly outlined below
- Hyperphosphataemia In chronic kidney disease, there is impaired renal excretion of phosphate, causing phosphate retention and hyperphosphataemia. Hyperphosphataemia, in turn, causes hypocalcaemia which is responsible for secondary hyperparathyroidism.
- Hypocalcaemia may also result from the following
- Due to renal dysfunction, there is decreased conversion of vitamin D metabolite 25(OH) cholecalciferol to its active form 1,25 (OH)2 cholecalciferol. Reduced intestinal absorption of calcium.
- Parathormone secretion Hypocalcaemia stimulates the secretion of parathormone, eventually leading to secondary hyperparathyroidism which, in turn, causes increased osteoclastic activity.
- Metabolic acidosis As a result of decreased renal function, acidosis sets in which may cause osteoporosis and bone decalcification.
- Calcium phosphorus product >70 When the product of the biochemical value of calcium and phosphate is higher than 70, metastatic calcification may occur at extraosseous sites.
- Dialysis-related metabolic bone disease Long-term dialysis employing the use of aluminium-containing dialysate is currently considered to be a major cause of metabolic bone lesions.
-
- Aluminium interferes with the deposition of calcium hydroxyapatite in bone and results in osteomalacia, secondary hyperparathyroidism and osteitis fibrosa cystic. In addition, accumulation of β2-microglobulin amyloid in such cases causes dialysis-related amyloidosis.
Morphologic Features: The following skeletal lesions can be identified in renal osteodystrophy:
- Mixed osteomalacia-osteitis fibrosis is the most common manifestation of renal osteodystrophy resulting from disordered vitamin D metabolism and secondary hyperparathyroidism.
- Pure osteitis fibrosa results from metabolic complications of secondary hyperparathyroidism.
- Pure osteomalacia of renal osteodystrophy is attributed to aluminium toxicity.
- Renal rickets resembling the changes seen in children with nutritional rickets with widened osteoid seams may occur.
- Osteosclerosis is characterised by enhanced bone density in the upper and lower margins of vertebrae.
- Metastatic calcification is seen at extraosseous sites such as in medium-sized blood vessels, periarticular tissues, myocardium, eyes, lungs and gastric mucosa.
Skeletal Fluorosis:
- Fluorosis of bones occurs due to high sodium fluoride content in soil and water consumed by people in some geographic areas and is termed endemic fluorosis. Such endemic regions exist in some tropical and subtropical areas; in India, it exists in some parts of Punjab and Andhra Pradesh.
- The condition affects farmers who consume drinking water from wells. Non-endemic fluorosis results from occupational exposure in manufacturing industries of aluminium, magnesium, and superphosphate.
Pathogenesis: In fluorosis, fluoride replaces calcium as the mineral in the bone and gets deposited without any regulatory control.
- This results in heavily mineralised bones which are thicker and denser but are otherwise weak and deformed (just as in osteopetrosis).
- In addition, there are also deposits of fluoride in soft tissues, particularly as nodules in the interosseous membrane. The patient develops skeletal deformities and mottling of teeth.
Morphologic Features Grossly, the long bones and vertebrae develop nodular swellings which are present both inside the bones and on the surface.
Microscopically, these nodules are composed of heavily mineralised irregular osteoid admixed with fluoride which requires confirmation chemically.
Paget’S Disease Of Bone (Osteitis Deformans):
Paget’s disease of bone or osteitis deformans was first described by Sir James. Paget’s disease of bone is an osteolytic and osteosclerotic bone disease of uncertain aetiology involving one (monostotic) or more bones (polyostotic). The condition affects predominantly males over the age of 50 years. Though the aetiology remains obscure, the following factors have been implicated
- There has been some evidence that osteitis deformans are a form of slow-virus infection by paramyxovirus (for example respiratory syncytial virus, measles) in osteoclasts. However, the virus has not been cultured from the osteoclasts of Paget’s disease.
- Autosomal dominant inheritance and genetic susceptibility have been proposed on the basis of observation of a 7-10 fold higher prevalence of disease in first-degree relatives. The susceptibility gene located on chromosome 18q encodes for a member of the tumour necrosis factor called RANK (receptor activator of nuclear factor κB).
Clinically, the monostotic form of the disease may remain asymptomatic and the lesion is discovered incidentally or on radiologic examination.
Polyostotic form, however, is more widespread and may produce pain, fractures, skeletal deformities, and occasionally, sarcomatous transformation. Typically, there is a marked elevation of serum alkaline phosphatase and normal to high serum calcium levels.
Morphologic Features: Monostotic Paget’s disease involves most frequently the tibia, pelvis, femur, skull and vertebra, while the order of involvement in polyostotic Paget’s disease is: vertebrae, pelvis, femur, skull, sacrum and tibia. Three sequential stages are identified in Paget’s disease:
- Initial osteolytic stage This stage is characterised by areas of osteoclastic resorption produced by an increased number of large osteoclasts.
- Mixed osteolytic-osteoblastic stage In this stage, there is an imbalance between osteoblastic laying down of new bone and osteoclastic resorption so that mineralisation of the newly-laid matrix lags behind, resulting in the development of characteristic mosaic pattern or jigsaw puzzle appearance of osteoid seams or cement lines.
- The narrow space between the trabeculae and cortex is filled with collagen which gradually becomes less vascular.
- Quiescent osteosclerotic stage After many years, excessive bone formation results and thus the bone becomes more compact and dense producing osteosclerosis. However, newly formed bone is poorly mineralised, soft and susceptible to fractures. Radiologically, this stage produces a characteristic cotton-wool appearance of the affected bone.
Metabolic and Endocrine Bone Diseases and Paget’s Disease:
- Osteoporosis or osteopenia is a common clinical condition involving multiple bones in which there is a quantitative reduction of bone tissue mass resulting in a fragile skeleton associated with an increased risk of fractures.
- Severe and prolonged hyperparathyroidism results in the formation of giant cell-rich brown tumours (osteitis fibrosa cystica).
- Renal osteodystrophy is the appearance of skeletal abnormalities appearing in cases of chronic kidney disease. It involves hyperphosphataemia and hypocalcaemia.
- Skeletal fluorosis is due to the consumption of high sodium fluoride content present in soil and water. In this, fluoride replaces calcium as the mineral in the bone and gets deposited without any regulatory control.
- Paget’s disease of bone or osteitis deformans is an osteolytic and osteosclerotic bone disease of uncertain aetiology involving one (monostotic) or more bones (polyostotic).
Tumour-Like Lesions Of Bone:
In the context of bones, several non-neoplastic conditions resemble true neoplasms clinically,
radiologically and morphologically and need to be distinguished from them. gives a list of such tumour-like lesions. A few common conditions are described below.
Fibrous Dysplasia:
Fibrous dysplasia is not an uncommon tumour-like lesion of the bone. It is a benign condition, possibly of developmental origin, characterised by the presence of a localised area of replacement of bone by fibrous connective tissue with a characteristic whorled pattern and containing trabeculae of woven bone. Radiologically, the typical focus of fibrous dysplasia has a well-demarcated ground-glass appearance.
Three types of fibrous dysplasia are distinguished monostotic, polyostotic, and Albright syndrome. The spectrum of the phenotype of the disease is due to activating mutation in the GNAS1 gene, which encodes for α-subunits of the stimulatory G-protein, GSα.

- Monostotic fibrous dysplasia affects a solitary bone and is the most common type, comprising about 70% of all cases. The condition affects either sex and most patients are between 20 and 30 years of age.
- The bones most often affected, in descending order of frequency, are ribs, craniofacial bones (especially maxilla), femur, tibia and humerus. The condition generally remains asymptomatic and is discovered incidentally, but infrequently may produce tumour-like enlargement of the affected bone.
- Polyostotic fibrous dysplasia Polyostotic form of fibrous dysplasia affecting several bones constitutes about 25% of all cases. Both sexes are affected equally but the lesions appear at a relatively earlier age than the monostotic form. The most frequently affected bones are craniofacial, ribs, vertebrae and long bones of the limbs.
- Approximately a quarter of cases with polyostotic forms have more than half of the skeleton involved by disease. The lesions may affect one side of the body or may be distributed segmentally in a limb. Spontaneous fractures and skeletal deformities occur in the childhood polyostotic form of the disease.
- Albright syndrome Also called McCune-Albright syndrome, is a form of polyostotic fibrous dysplasia associated with endocrine dysfunctions and accounts for less than 5% of all cases.
- Unlike monostotic and polyostotic varieties, Albright syndrome is more common in females. The syndrome is characterised by polyostotic bone lesions, skin pigmentation (café-audit macular spots) and sexual precocity, and infrequently other endocrinopathies.
Morphologic Features: All forms of fibrous dysplasia have an identical pathologic appearance.
Grossly, the lesions appear as sharply demarcated, localised defects measuring 2-5 cm in diameter, present within the cancellous bone, having a thin and smooth overlying cortex. The epiphyseal cartilages are generally spared in the monostotic form but involved in the polyostotic form of the disease.
The cut section of the lesion shows the replacement of normal cancellous bone of the marrow cavity by gritty, grey-pink, rubbery soft tissue which may have areas of haemorrhages, myxoid change and cyst formation.
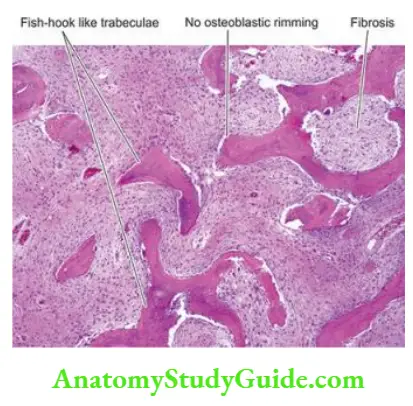
Histologically, the lesions of fibrous dysplasia have characteristic benign-looking fibroblastic tissue arranged in a loose, whorled pattern in which there are irregular and curved trabeculae of woven (non-lamellar) bone in the form of fish-hook appearance or Chinese letter shapes.
Characteristically, there are no osteoblasts rimming the trabeculae of the bone, suggesting a maturation defect in the bone. Rarely, malignant change may occur in fibrous dysplasia, most often an osteogenic sarcoma.
Fibrous Cortical Defect (Metaphyseal Fibrous Defect, Non-Ossifying Fibroma):
Fibrous cortical defect or metaphyseal fibrous defect is a rather common benign tumour-like lesion occurring in the metaphyseal cortex of long bones in children. The most commonly involved bones are the upper or lower end of the tibia or the lower end of the femur. The lesion is generally solitary but rarely there may be multiple and bilaterally symmetrical defects.
Radiologically, the lesion is eccentrically located in the metaphysis and has a sharply-delimited border. The pathogenesis of fibrous cortical defects is unknown. Possibly, it arises as a result of some developmental defect at the epiphyseal plate or could be a tumour of histiocytic origin because of its close resemblance to fibrohistiocytic tumours.
Clinically, the fibrous cortical defect causes no symptoms and is usually discovered accidentally when an X-ray of the region is done for some other reason.
Morphologic Features Grossly, the lesion is generally small, less than 4 cm in diameter, granular and brown. A larger lesion (5-10 cm) occurring usually in response to trauma is referred to as non-ossifying fibroma.
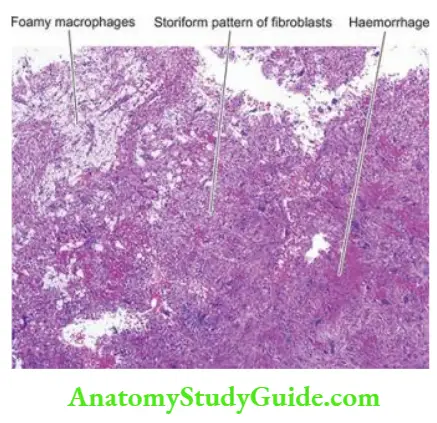
Microscopically, fibrous cortical defect consists of cellular masses of fibrous tissue showing the storiform pattern. There are numerous multinucleate osteoclast-like giant cells, haemosiderin-laden macrophages and foamy cells; hence the lesion is also termed histiocytic xanthogranuloma or fibrous xanthoma of bone.
Solitary (Simple, Unicameral) Bone Cyst:
A solitary, simple or unicameral bone cyst is a benign condition occurring in children and adolescents, most frequently located in the metaphyses at the upper end of the humerus and femur. The cyst expands the bone causing thinning of the overlying cortex.
Possibly, the lesion arises due to local disorders of bone growth and development. Clinically, solitary bone cysts may remain asymptomatic or may cause pain and fracture.
Morphologic Features Grossly, a simple cyst of the bone is generally unilocular with a smooth inner surface. The cavity is filled with clear fluid.
Histologically, the cyst wall consists of thin collagenous tissue having scattered osteoclast giant cells and newly formed reactive bony trabeculae. Fracture alters the appearance and produces sanguineous fluid in the cavity, and haemorrhages, haemosiderin deposits and macrophages in the cyst wall.
Aneurysmal Bone Cyst:
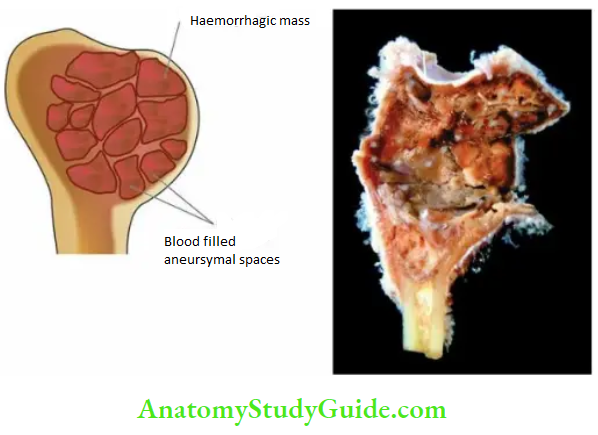
- An aneurysmal bone cyst, true to its name, is an expanding osteolytic lesion filled with blood (aneurysm = dilatation, distension). The condition is seen more commonly in young patients under 30 years of age. The most frequently involved bones are shafts or metaphyses of long bones or the vertebral column.
- The radiographic appearance shows a characteristic ballooned-out expansile lesion underneath the periosteum. Clinically, the aneurysmal bone cyst may enlarge over a period of years and produce pain, tenderness and pathologic fracture.
- The pathogenesis is not clear but it has been suggested by some authors that the condition probably arises from persistent local alteration in haemodynamics. 17p13 translocation has been identified in these cases.
Morphologic Features Grossly, the lesion consists of a large haemorrhagic mass covered over by thinned-out reactive bone.
Histologically, the cyst consists of blood-filled aneurysmal spaces of variable size, some of which are endothelium-lined. The spaces are separated by connective tissue septa-containing osteoid tissue, numerous osteoclast-like multinucleate giant cells and trabeculae of bone. The condition has to be distinguished histologically from giant cell tumour or osteoclastoma and telangiectatic osteosarcoma.
Tumour-like Lesions of Bone:
- Fibrous dysplasia is a benign condition having the presence of a localised area of replacement of bone by fibrous connective tissue with a characteristic whorled pattern. It is of three types monostotic, polyostotic, and Albright syndrome.
- Fibrous cortical defect or metaphyseal fibrous defect occurs in the metaphyseal cortex of long bones in children, most commonly on the upper or lower end of the tibia or lower end of the femur and resembles fibrohistiocytic tumours.
- A solitary, simple or unicameral bone cyst is a benign condition occurring in children and adolescents, frequently located in the metaphyses at the upper end of the humerus and femur.
- An aneurysmal bone cyst is an expanding osteolytic lesion filled with blood and requires distinction from a giant cell tumour of bone.
Tumours Of Bone And Cartilage:
- Bone and cartilage tumours are comparatively infrequent in adults (<1% of all cancers) but some of the highly malignant bone tumours are more common in children. Bone tumours may be primary or metastatic. Since the histogenesis of some bone tumours is obscure, the WHO has recommended a widely accepted classification of bone and cartilage tumours based on both histogenesis and histologic criteria into primary tumours (benign and malignant) arising from different tissue components (osseous and non-osseous, indigenous to the bone) and metastatic tumours.
- However, in the following discussion, only osseous bone tumours are considered, while non-osseous bone tumours are described elsewhere in the book. The anatomic origin of common primary bone tumours is illustrated.
- The aetiology of most bone tumours is unknown. However, some bone tumours occur in the setting of inherited syndromes but they are morphologically not different from corresponding sporadic tumours.
- The molecular and genetic basis of some bone tumours has become available for devising targeted therapies in future.
- It may be mentioned here that the diagnosis of any bone lesion is established by a combination of clinical, radiological and pathological examination, supplemented by relevant biochemical and haematological investigations.
- These include serum levels of calcium, phosphorus, alkaline phosphatase and acid phosphatase. Specific investigations like plasma and urinary proteins and the bone marrow examination in case of myeloma, urinary catecholamines in metastatic neuroblastoma and haematologic profile in lymphoma and leukaemic involvement of the bone, are of considerable help.
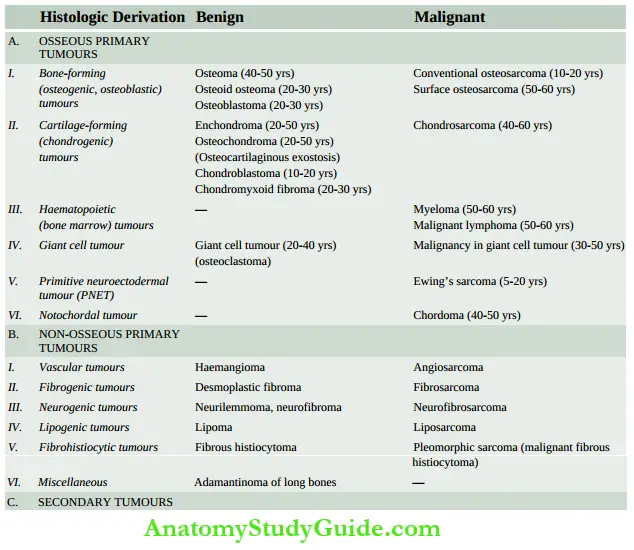
Bone-Forming Tumours:
- Bone-forming or osteoblastic groups of bone tumours are characterised by the common property of synthesis of osteoid or bone, or both, directly by the tumour cells (osteogenesis).
- The formation of reactive bone and endochondral ossification should not be construed as osteogenesis.
- Benign bone-forming tumours include osteoma, osteoid osteoma and osteoblastoma, while the malignant counterpart is osteosarcoma (osteogenic sarcoma).
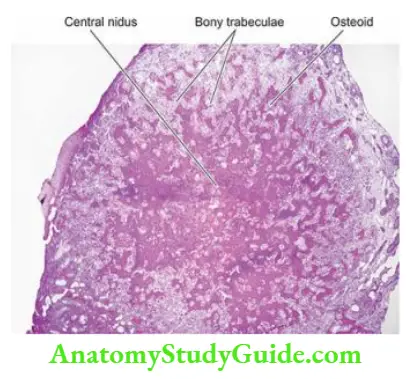
Osteoma:
- An osteoma is a rare benign, slow-growing lesion, regarded by some as a hamartoma rather than a true neoplasm. Similar lesions may occur following trauma, subperiosteal haematoma or local inflammation. Osteoma is almost exclusively restricted to flat bones of the skull and face.
- It may grow into paranasal sinuses or protrude into the orbit. An osteoma may form a component of Gardner’s syndrome. Radiologic appearance is of a dense ivory-like bony mass.
Microscopically, the lesion is composed of well-differentiated mature lamellar bony trabeculae separated by fibrovascular tissue.
Osteoid Osteoma and Osteoblastoma:
Osteoid osteoma and osteoblastoma (or giant osteoid osteoma) are closely related to benign tumours occurring in children and young adults. Osteoid osteoma is more common than osteoblastoma. There are no clear-cut histologic criteria to distinguish the two. The distinction between them is based on clinical features, size and radiographic appearance.
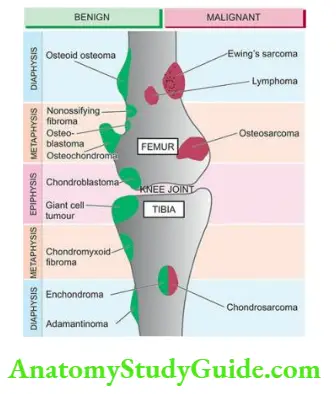
- Osteoid osteoma is a small (usually less than 1 cm) tumour located in the cortex of a long bone, associated characteristically with nocturnal pain. The tumour is clearly demarcated having a surrounding zone of reactive bone formation which radiographically appears as a small radiolucent central focus or nidus surrounded by dense sclerotic bone. The pain is possibly due to increased elaboration of prostaglandin E2 by proliferating osteoblasts.
- Osteoblastoma, on the other hand, is larger in size (usually more than 1 cm), painless, located in the medulla, commonly in the vertebrae, ribs, ilium and long bones, and there is the absence of reactive bone formation.
Histologically, the distinction between osteoid osteoma and osteoblastoma is not obvious. In either case, the lesion consists of trabeculae of osteoid, rimmed by osteoblasts and separated by highly vascularised connective tissue stroma. Later, some of the trabeculae are mineralised and calcified.
Osteosarcoma:
- Osteosarcoma or osteogenic sarcoma is the most common primary malignant tumour of the bone. The tumour is characterised by the formation of osteoid or bone, or both, directly by sarcoma cells. The tumour arises from primitive osteoblast-forming mesenchyme.
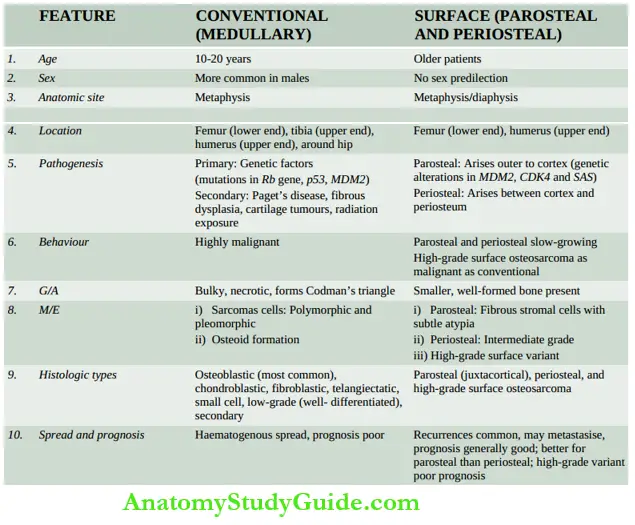
- Depending upon their locations within the bone, osteosarcomas are classified into 2 main categories: conventional (central, medullary or classic) and surface (parosteal and periosteal).
Conventional Osteosarcoma:
This is the more common, and classic type and is generally referred to as ‘osteosarcoma’ if not specified. The tumour occurs in young patients between the age of 10 and 20 years. Males are affected more frequently than females.
The tumour arises in the metaphysis of long bones. The most common sites, in descending order of frequency, are the lower end of the femur and upper end of the tibia ( around the knee joint about 60%); the upper end of the humerus (10%); pelvis and the upper end of the femur (around hip joint about 15%); and less often in jaw bones, vertebrae and skull. Rarely, osteosarcoma may occur in extraskeletal soft tissues.
Pathogenesis: Based on the pathogenesis, osteosarcoma is divided into 2 types: primary and secondary.
- Primary osteosarcoma is more common and occurs in the absence of any known underlying disease. Its aetiology is unknown but there is evidence linking this form of osteosarcoma with genetic factors (for example hereditary mutation of chromosome 13 in common with retinoblastoma locus), periods of active bone growth (occurrence of the tumour in younger age), and with certain environmental influences (example radiation, oncogenic virus).
- Cases of hereditary retinoblastoma have a very high prevalence risk of development of osteosarcoma implicating the RB gene in their pathogenesis. About 20% of sporadic osteosarcomas show a mutation in the p53 tumour suppressor gene; some have overexpression of the MDM2 gene and mutation in cyclin D1, p16 and CDK4.
- Secondary osteosarcoma, on the other hand, develops following pre-existing bone disease example in cartilage tumours (dedifferentiated chondrosarcoma), Paget’s disease of bone, fibrous dysplasia, post-radiation, chronic osteomyelitis and bone infarcts. The tumour has a more aggressive behaviour than the primary osteosarcoma.
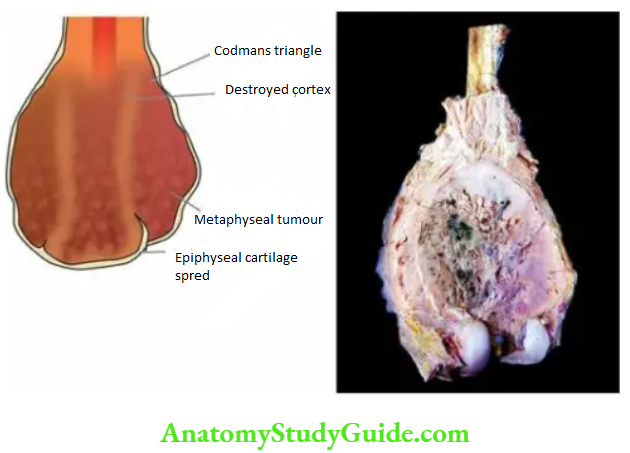
Clinical Features: Conventional osteosarcoma is a highly malignant tumour. The tumour arises centrally in the metaphysis, extends longitudinally for a variable distance into the medullary cavity, and expands laterally on either side breaking through the cortex and lifting the periosteum.
- If the periosteum is breached, the tumour grows relentlessly into the surrounding soft tissues. The only tissue which is able to stop its spread, albeit temporarily, is the cartilage of the epiphyseal plate.
- The radiographic appearance is quite a distinctive characteristic sunburst pattern due to osteogenesis within the tumour and the presence of Codman’s triangle formed at the angle between the elevated periosteum and the underlying surface of the cortex.
- Clinically, conventional osteosarcoma presents with pain, tenderness and an obvious swelling of the affected extremity. Serum alkaline phosphatase level is generally raised but calcium and phosphorus levels are normal.
- The tumour metastasises rapidly and widely to distant sites by the haematogenous route and disseminates commonly to the lungs, other bones, the brain and various other sites.
Morphologic Features Grossly, the tumour appears as a grey-white, bulky mass at the metaphyseal end of a long bone of the extremity. The articular end of the bone is generally uninvolved in the initial stage. Codman’s triangle, though identified radiologically, may be obvious on macroscopic examination.
The cut surface of the tumour is grey-white with areas of haemorrhages and necrotic bone. Tumours which form an abundance of osteoid, bone and cartilage may have hard, gritty and mucoid areas.
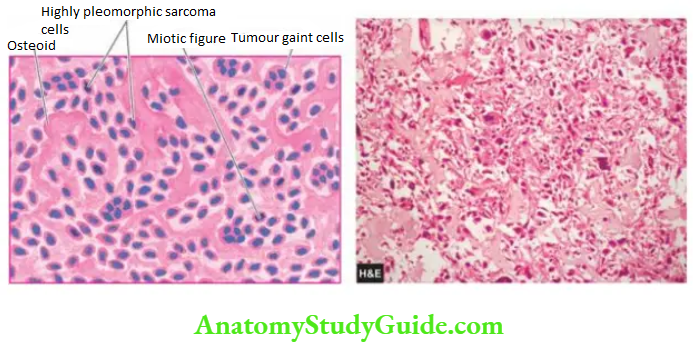
Histologically, conventional osteosarcoma shows considerable variation in pattern from case to case and even within a tumour from one area to the other. However, the following two features characterise all classic forms of conventional osteosarcomas:
- Sarcoma cells Tumour cells of osteosarcomas are undifferentiated mesenchymal stromal cells which show marked pleomorphism and polymorphism variation in size as well as shape. The tumour cells may have various shapes such as spindled, round, oval and polygonal and bizarre tumour giant cells. The tumour cells have variable sizes and show hyperchromatism and atypical mitoses.
- Histochemically, these tumour cells are positive for alkaline phosphatase. Immunohistochemically, sarcoma cells of osteosarcoma express vimentin, osteocalcin, osteonectin and type I collagen.
- Osteogenesis Most often, anaplastic sarcoma cells form osteoid matrix and bone directly that is found interspersed in the areas of tumour cells.
These are called osteoblastic osteosarcomas. At times, the tumour cells may produce cartilage or fibrous tissue and are correspondingly called chondroblastic and fibroblastic osteosarcomas.
Histologic Variants: A few histologic variants of osteosarcoma have been described in the WHO classification:
- Telangiectatic osteosarcoma The tumour in this variant presents with pathological fractures. The tumour has large, cavernous, dilated vascular channels. This variant has a more aggressive course.
- Small cell osteosarcoma This variant has small, uniform tumour cells similar to the tumour cells of Ewing’s sarcoma or lymphoma but osteogenesis by these tumour cells is the distinguishing feature.
- Low-grade (or well-differentiated) osteosarcoma Although the generally classic form of osteosarcoma is a highly malignant tumour, rarely a well-differentiated variant having minimal cytologic atypia resembling parosteal osteosarcoma may be seen.
- Secondary osteosarcoma Secondary osteosarcoma developing in one of the conditions listed above is more often chondroblastic osteosarcoma and is more aggressive than primary osteosarcoma.
Surface Osteosarcoma:
About 5% of osteosarcomas occur on the surface of the bone and include 3 variants: parosteal and periosteal (slow-growing tumours), and high-grade surface osteosarcoma. Genetic alterations in MDM2, CDK4 and SAS genes have been described in these variants.
1. Parosteal or juxtacortical osteosarcoma: It is an uncommon form of slow-growing osteosarcoma having its origin from the metaphysis on the external surface of the bone (parosteal or juxtacortical means outer to cortex). The tumour should be distinguished from the more common medullary osteosarcoma because of its better prognosis and different presentation.
The tumour occurs in the older age group (3rd to 4th decade), has no sex predilection and is slowly growing. Its common locations are the metaphysis of long bones, more frequently the lower end of the femur and the upper end of the humerus. X-ray examination usually reveals a dense bony mass attached to the outer cortex of the affected long bone.
Grossly, the tumour is lobulated and circumscribed, with calcified mass in the subperiosteal location.
Microscopically, the features which characterise the usual osteosarcoma (sarcomatous stroma and production of neoplastic osteoid and bone) are present, but the tumour shows a high degree of structural differentiation, and there are generally well-formed bony trabeculae. These features account for distinctly better prognosis in these cases.
2. Periosteal osteosarcoma: It is a rarer form of osteosarcoma than the parosteal type and arises between the cortex and the overlying periosteum. Its common location is the diaphysis of the tibia or the femur. It occurs in young adults (average age 25 years).
Microscopically, periosteal osteosarcoma has cartilaginous differentiation and a higher degree of anaplasia than that seen in parosteal osteosarcoma but has a lower grade than conventional osteosarcoma it is an intermediate-grade sarcoma.
3. High-grade surface osteosarcoma: It represents less than 1% of osteogenic sarcomas.
Grossly, these tumours often have soft areas that resemble fish flesh.
Microscopically, it shows features similar to classical intraosseous high-grade osteosarcoma. sums up the contrasting features of conventional (central or medullary) and surface osteosarcomas.
Cartilage-Forming Tumours:
Cartilage-forming or chondroblasts tumours are composed of frank cartilage or derived from cartilage-forming cells. This group comprises several benign lesions (osteochondromas, enchondroma, chondroblastoma and chondromyxoid fibroma), and a malignant counterpart, chondrosarcoma.
Osteochondromas:
Osteochondromas also called osteocartilaginous exostoses, are the commonest of benign cartilage-forming lesions. Though designated and discussed with neoplasms, exostosis or osteochondroma is not a true tumour but is regarded as a disorder of growth and development.
It may occur as a solitary sporadic exostosis or there may be multiple hereditary exostoses. Hereditary forms are due to germline mutation in EXT1 or EXT2 gene while sporadic type is due to mutated EXT1 only
Exostoses arise from metaphyses of long bones as exophytic lesions, most commonly lower femur and upper tibia (around the knee) and upper humerus but may also be found in other bones such as the scapula or ilium. They are discovered most commonly in late childhood or adolescence and are more frequent in males.
They may remain asymptomatic and be discovered as an incidental radiographic finding or may produce obvious deformity. Both solitary and multiple exostoses may undergo transformation into chondrosarcoma but the risk is much greater with multiple hereditary exostoses.
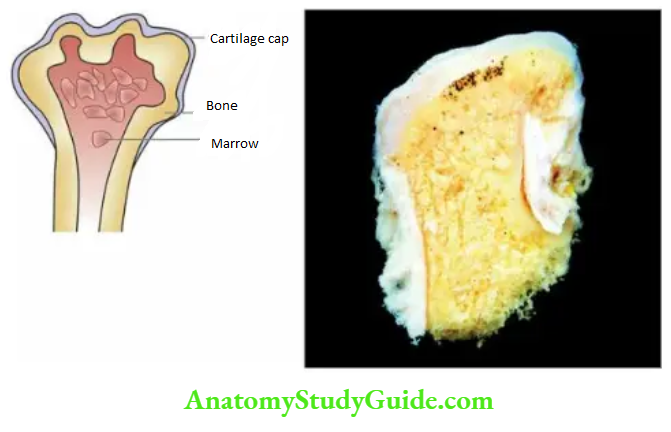
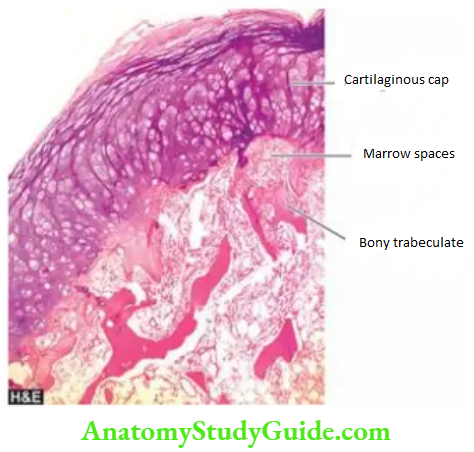
Morphologic Features Grossly, osteochondromas have a broad or narrow base (may be either sessile or pedunculated) which is continuous with the cortical bone. They protrude exophytic ally as mushroom-shaped, cartilage-capped lesions enclosing well-formed cortical bone and marrow.
Microscopically, they are composed of an outer cap composed of mature cartilage resembling epiphyseal cartilage and the inner mature lamellar bone and bone marrow.
Enchondroma:
- Enchondroma is the term used for the benign cartilage-forming tumour that develops centrally within the interior of the affected bone, while chondroma refers to the peripheral development of lesion similar to osteochondromas. Enchondromas may occur singly or they may be multiple, forming a non-hereditary disorder called enchondromatosis or Ollier’s disease.
- The coexistence of multiple enchondromas with multiple soft tissue haemangiomas constitutes a familial syndrome called Maffucci’s syndrome.
- The most common locations for enchondromas are short tubular bones of the hands and feet, and less commonly, they involve the ribs or the long bones. They may appear at any age and in either sex. Enchondromas, like osteochondromas, may remain asymptomatic or may cause pain and pathologic fractures.
- X-ray reveals a radiolucent, lobulated tumour mass with spotty calcification. Malignant transformation of solitary enchondroma is rare but multiple enchondromas may develop into chondrosarcoma.
Morphologic FeatureS Grossly, the enchondroma is a lobulated, bluish-grey, translucent, cartilaginous mass lying within the medullary cavity.
Histologically, the tumour has a characteristic lobulated appearance. The lobules are composed of normal adult hyaline cartilage separated by vascularised fibrous stroma. Foci of calcification may be evident within the tumour. Enchondroma is distinguished from chondrosarcoma by the absence of invasion into surrounding tissues and lack of cellular features of malignancy.
Chondroblastoma:
- Chondroblastoma is a relatively rare benign tumour arising from the epiphysis of long bones adjacent to the epiphyseal cartilage plate. The most commonly affected bones are the upper tibia and lower femur (i.e. about the knee) and upper humerus.
- The tumour usually occurs in patients under 20 years of age with male preponderance (male-female ratio 2:1). The radiographic appearance is of a sharply-circumscribed, lytic lesion with multiple small foci of calcification.
- Chondroblastoma may be asymptomatic or may produce local pain, tenderness and discomfort. The behaviour of the tumour is benign though it may recur locally after curettage.
Morphologic Features Grossly, chondroblastoma is a well-defined mass, up to 5 cm in diameter, lying in the epiphysis. The tumour is surrounded by a thin capsule of dense sclerotic bone. The Cut surface reveals a soft chondroid tumour with foci of haemorrhages, necrosis and calcification.
Histologically, the tumour is highly cellular and is composed of small, round to polygonal mononuclear cells resembling chondroblasts with characteristic coffee-bean nuclei and multinucleate osteoclast-like giant cells.
There are small areas of cartilaginous intercellular matrix and focal calcification described as chicken-wire calcification.
Chondromyxoid Fibroma:
Chondromyxoid fibroma is an uncommon benign tumour of cartilaginous origin arising in the metaphysis of long bones. The most common locations are the upper end of the tibia and the lower end of the femur around the knee joint. The majority of tumours appear in 2nd to 3rd decades of life with male preponderance.
Radiographically, the tumour appears as a sharply-outlined radiolucent area with foci of calcification and expansion of the affected end of the bone.
The lesion may be asymptomatic or may cause pain, swelling and discomfort in the affected joint. The lesions may recur after curettage. Thus, there are many similarities with chondroblastoma.
Morphologic Features Grossly, chondromyxoid fibroma is a sharply-demarcated, grey-white lobulated mass, not exceeding 5 cm in diameter, lying in the metaphysis. The tumour is often surrounded by a layer of dense sclerotic bone.
The cut surface of the tumour is soft to firm and lobulated but calcification within the tumour is not as common as with other cartilage-forming tumours.
Histologically, the tumour has an essentially lobulated pattern. The lobules are separated by fibrous tissue and a variable number of osteoclast-like giant cells.
The lobules themselves are composed of immature cartilage consisting of spindle-shaped or stellate cells with abundant myxoid or chondroid intercellular matrix.
In view of the close histogenetic relationship between chondromyxoid fibroma and chondroblastoma, occasional tumours show a combination of histological features of both.
Chondrosarcoma:
Chondrosarcoma is a malignant tumour of chondroblasts. In frequency, it is next in frequency to
osteosarcoma but is relatively slow-growing and thus has a much better prognosis than that of
osteosarcoma. Two types of chondrosarcoma are distinguished: central and peripheral.
- Central chondrosarcoma is more common and arises within the medullary cavity of diaphysis or metaphysis. This type of chondrosarcoma is generally primary i.e. occurs de novo.
- Peripheral chondrosarcoma arises in the cortex or periosteum of the metaphysis. It may be primary or secondary occurring on a pre-existing benign cartilaginous tumour such as osteocartilaginous exostoses (osteochondromas) or multiple enchondromatosis.
Both forms of chondrosarcoma usually occur in patients between 3rd and 6th decades of life with slight male preponderance. In contrast to benign cartilaginous tumours, the majority of chondrosarcomas are found more often in the central skeleton (in the pelvis, ribs and shoulders); sometimes around the knee joint.
Radiologic appearance is of hugely expansile and osteolytic growth with foci of calcification. Clinically, the tumour is slow-growing and comes to attention because of pain and gradual enlargement over the years.
Lower grades of the tumour recur following surgical removal but higher grades cause metastatic dissemination, commonly to the lungs, liver, kidney and brain.
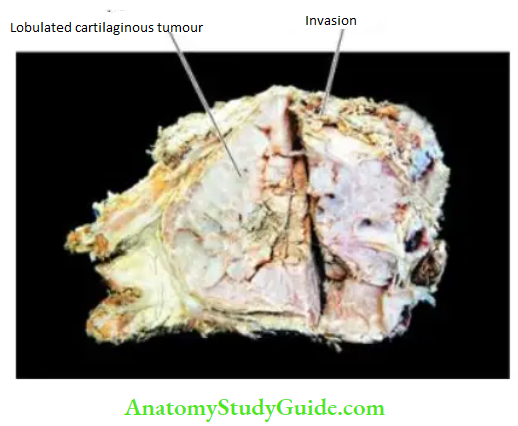
Morphologic Features Grossly, chondrosarcoma may vary in size from a few centimetres to extremely large and lobulated masses of firm consistency. The cut section of the tumour shows a translucent, bluish-white, gelatinous or myxoid appearance with foci of ossification.
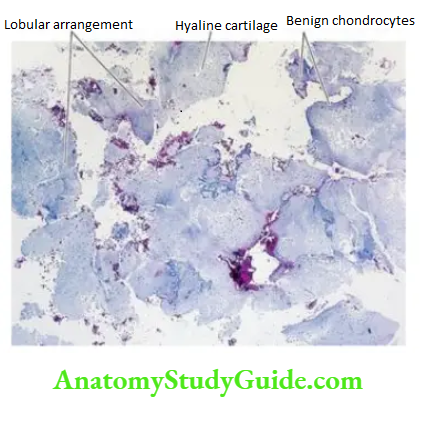
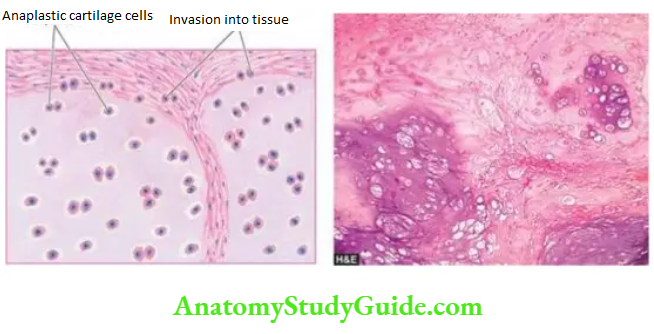
Histologically, the two hallmarks of chondrosarcoma are invasive character and the formation of lobules of anaplastic cartilage cells. These tumour cells show cellular features of malignancy such as hyperchromatism, pleomorphism, two or more cells in the lacunae and tumour giant cells.
However, sometimes the distinction between a well-differentiated chondrosarcoma and a benign chondroma may be difficult and in such cases, location, clinical features and radiological appearance are often helpful.
A few uncommon variants of chondrosarcoma are mesenchymal chondrosarcoma, dedifferentiated chondrosarcoma and clear cell chondrosarcoma.
Giant Cell Tumour (Osteoclastoma):
- Giant cell tumour or osteoclastoma is a distinctive neoplasm with uncertain histogenesis and hence is classified separately. The tumour arises in the epiphysis of long bones close to the articular cartilage.
- The most common sites of involvement are the lower end of the femur and the upper end of the tibia (i.e. about the knee), the lower end of the radius and the upper end of the fibula.
- Giant cell tumour occurs in patients between 20 and 40 years of age with no sex predilection. Clinical features at presentation include pain, especially on weight-bearing and movement, noticeable swelling and pathological fracture.
- Radiologically, a giant cell tumour appears as a large, lobulated and osteolytic lesion at the end of an expanded long bone with a characteristic ‘soap bubble’ appearance.
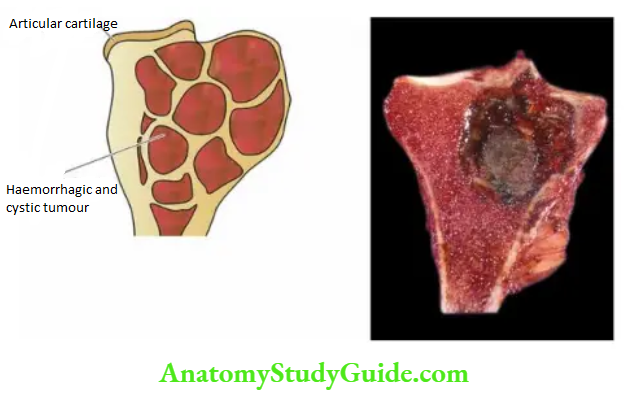
Morphologic Features Grossly, a giant cell tumour is eccentrically located in the epiphyseal end of a long bone which is expanded. The tumour is well-circumscribed, dark tan and covered by a thin shell of subperiosteal bone. The cut surface of the tumour is characteristically haemorrhagic, necrotic, and honey-combed due to focal areas of cystic degeneration.
Histologically, the hallmark features of giant cell tumours are the presence of a large number of multinucleate osteoclast-like giant cells regularly scattered throughout the stromal mononuclear cells:
- Giant cells often contain as many as 100 benign nuclei and have many similarities to normal osteoclasts. These cells have very high acid phosphatase activity.
- Stromal cells are mononuclear cells and are the real tumour cells and their histologic appearance determines the biologic behaviour of the tumour. Typically, they are uniform, plump, spindle-shaped or round to oval cells with numerous mitotic figures.
- Other features of the stroma include its scanty collagen content, rich vascularity, areas of haemorrhages and the presence of macrophages.
The giant cell tumour of the bone has certain peculiarities which deserve further elaboration. These are its cell of origin, its differentiation from other giant cell lesions and its biological behaviour.
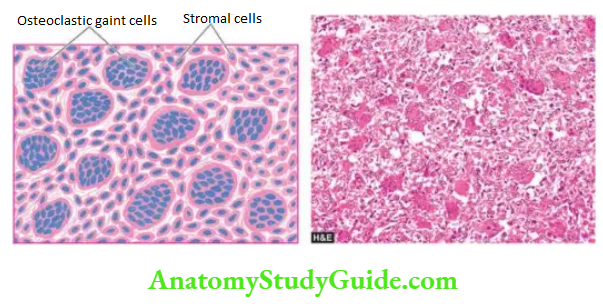
Cell Of Origin: Though designated as giant cell tumour or osteoclastoma, the actual tumour cells are round to spindled mononuclear cells while proliferated osteoclastic giant cells are seen as background cells. Histogenesis of stromal cells is uncertain but possibly they are of mesenchymal origin.
Molecular profiling of giant cell tumours suggests that RANK (receptor activator of nuclear factor κB), a physiologic growth factor for osteoclastic proliferation, is expressed in stromal cells. Giant osteoclastic cells are believed to be formed by the RANK/RANKL signalling pathway.
Other Giant Cell Lesions: This peculiar tumour with the above description is named ‘giant cell tumour’ but giant cells are present in several other benign tumours and tumour-like lesions from which the giant cell tumour is to be distinguished.
These benign giant cell lesions are chondroblastoma, brown tumour of hyperparathyroidism, reparative giant cell granuloma, aneurysmal bone cyst, simple bone cyst and metaphyseal fibrous defect (non-ossifying fibroma).
Biologic Behaviour: Giant cell tumours are best described as aggressive and recurrent tumours. About 40 to 60% of them recur after curettage, sometimes after a few decades of initial resection. Approximately 4% of cases result in distant metastases, mainly to the lungs.
- Metastases are histologically benign and there is usually a history of repeated curettages and recurrences. Thus, attempts at histologic grading of giant cell tumours do not always yield satisfactory results.
- One of the factors considered significant in the malignant transformation of this tumour is the role of radiotherapy resulting in the development of post-radiation bone sarcoma.
Ewing’S Sarcoma And Primitive Neuroectodermal Tumour (Es/Pnet):
Ewing’s sarcoma (ES) is a highly malignant small round cell tumour occurring in patients between the age of 5 and 20 years with a predilection for occurrence in females. Since its first description by James Ewing in 1921, the histogenesis of this tumour has been a debatable issue.
At different times, the possibilities suggested for the cell of origin have been endothelial, pericytic, bone marrow, osteoblastic, and mesenchymal; currently, it is settled for its origin from primitive neuroectodermal cells. Now, Ewing’s sarcoma includes 3 variants:
- classic (skeletal) Ewing’s sarcoma;
- soft tissue Ewing’s sarcoma; and
- primitive neuroectodermal tumour (PNET).
- The three are linked together by a common neuroectodermal origin and by a common cytogenetic translocation abnormality t(11; 22) (q24; q12).
- This suggests a phenotypic spectrum in these conditions varying from undifferentiated Ewing’s sarcoma to PNET positive for rosettes and neural markers (neuron-specific enolase, S-100). However, PNET ultimately has a worse prognosis.
- The skeletal Ewing’s sarcoma arises in the medullary canal of diaphysis or metaphysis. The common sites are shafts and metaphysis of long bones, particularly the femur, tibia, humerus and fibula, although some flat bones such as the pelvis and scapula may also be involved.
- Clinical features include pain, tenderness and swelling of the affected area accompanied by fever, leucocytosis and elevated ESR. These signs and symptoms may lead to an erroneous clinical diagnosis of osteomyelitis.
- However, X-ray examination reveals a predominantly osteolytic lesion with patchy subperiosteal reactive bone formation producing a characteristic ‘onion-skin’ radiographic appearance.
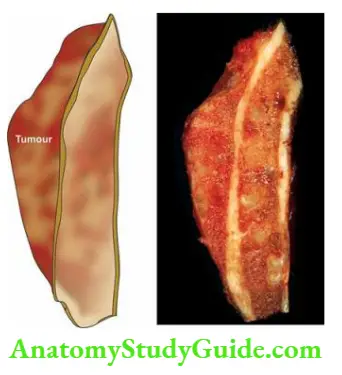
Morphologic Features Grossly, Ewing’s sarcoma is typically located in the medullary cavity and produces expansion of the affected diaphysis (shaft) or metaphysis, often extending into the adjacent soft tissues. The tumour tissue is characteristically grey-white, soft and friable.
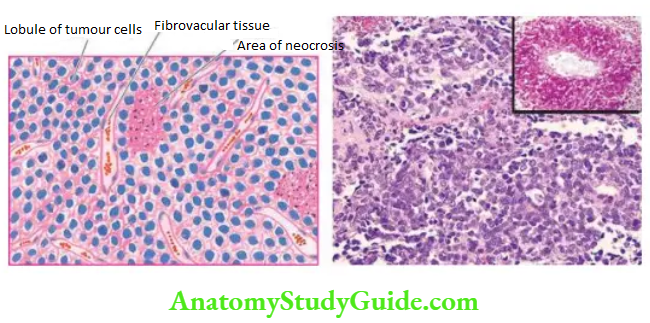
Histologically, Ewing’s tumour is a member of small round cell tumours which includes other tumours such as PNET, neuroblastoma, embryonal rhabdomyosarcoma, lymphoma leukaemias, and metastatic small cell carcinoma. Ewing’s tumour shows the following histologic characteristics:
- Pattern: The tumour is divided by fibrous septa into irregular lobules of closely-packed tumour cells. These tumour cells are characteristically arranged around capillaries forming pseudorosettes.
- Tumour cells: The individual tumour cells comprising the lobules are small and uniform resembling lymphocytes and have ill-defined cytoplasmic outlines, scanty cytoplasm and round nuclei having ‘salt and pepper’ chromatin and frequent mitoses.
- Based on these cytological features the tumour is also called round cell tumour or small blue cell tumour. The cytoplasm contains glycogen that stains with periodic acid-Schiff (PAS) reactions.
- A consistently expressed cell surface marker by tumour cells of the ES/PNET group is CD99 which is a product of the MIC-2 gene located on the X and Y chromosomes.
- Other features: Tumour is richly vascularised and lacks the intercellular network of reticulin fibres. There may be areas of necrosis and acute inflammatory cell infiltration. Focal areas of reactive bone formation may be present.
Ewing’s sarcoma metastasises early by a haematogenous route to the lungs, liver, other bones and brain. The involvement of other bones has prompted a suggestion of the multicentric origin of Ewing’s sarcoma.
The prognosis of Ewing’s sarcoma used to be dismal (5-year survival rate less than 10%). But currently, the use of a combined regimen consisting of radiotherapy and systemic chemotherapy has improved the outcome greatly (5-year survival rate 40-80%).
Chordoma:
- Chordoma is a slow-growing malignant tumour arising from remnants of notochord. Notochord is the primitive axial skeleton which subsequently develops into the spine. Normally, remnants of notochord are represented by notochordal or physaliphorous (physalis = bubble, photos = bearing) cells present in the nucleus pulposus and a few clumps within the vertebral bodies.
- Chordomas thus occur in the axial skeleton, particularly the sacrum and coccyx (50%), sphenooccipital region (35%), and less often in the spine (15%). Chordoma is usually found in patients over the age of 40 years with no sex predilection. Radiographically, the tumour usually appears as an osteolytic lesion.
- Symptoms of spinal cord compression may be present. The tumour grows slowly and infiltrates adjacent structures but metastases develop rarely. Recurrences after local excision are frequent and the tumour almost invariably proves fatal.
Morphologic Features Grossly, the tumour is soft, lobulated, translucent and gelatinous with areas of haemorrhages.
Microscopically, chordoma is composed of highly vacuolated physaliphorous cells surrounded by a sea of intercellular mucoid material. Histologic differentiation between chordoma and chondrosarcoma or mucin-secreting carcinoma may sometimes be difficult and is facilitated by positive cytokeratin and S-100 immunostaining in the former.
Metastatic Bone Tumours:
- Metastases to the skeleton are more frequent than the primary bone tumours. Metastatic bone tumours are exceeded in frequency by only 2 other organs lungs and liver. Most skeletal metastases are derived from haematogenous spread.
- Bony metastases of carcinomas predominate over sarcomas. Some of the common carcinomas metastasising to the bones are from the breast, ovary, prostate, lung, kidney, liver, stomach, thyroid, cervix, body of the uterus, urinary bladder, testis, melanoma and neuroblastoma of adrenal gland.
- Examples of sarcomas which may metastasise to the bone are embryonal and alveolar rhabdomyosarcoma, Ewing’s sarcoma and osteosarcoma.
- Skeletal metastases may be single or multiple. The most commonly involved bones are the spine, pelvis, femur, skull, ribs and humerus. The usual radiographic appearance is of an osteolytic lesion. Osteoblastic bone metastases occur in cancer of the prostate, carcinoid tumour and small cell carcinoma of the lung.
- Metastatic bone tumours generally reproduce the microscopic picture of the primary tumour. Many times, evidence of skeletal metastases is the first clinical manifestation of an occult primary cancer in the body.


Tumours of Bone and Cartilage:
- Primary bone tumours are less common and are classified on the basis of histogenesis and histologic criteria into osseous and non-osseous tumours. Both groups may have benign and malignant tumours.
- Common benign bone-forming osseous tumours are osteoma, osteoid osteoma and osteoblastoma.
- Osteosarcoma is a more common malignant bone-forming tumour. Conventional (or classic or medullary) osteosarcoma occurs in young patients between the age of 10 and 20 years, arising in the metaphysis. The most common site is around the knee joint and is a highly malignant tumour.
- Surface osteosarcomas include 2 slow-growing types (parosteal and periosteal) and high-grade surface osteosarcoma; these tumours occur in older age group than conventional type. Parosteal and periosteal surface osteosarcomas have a better prognosis while the prognosis of the high-grade type is like that of conventional osteosarcoma.
- Osteochondromas are the commonest of benign cartilage-forming lesions. Others are enchondroma, chondroblastoma and chondromyxoid fibroma.
- Chondrosarcoma occurs in the 4th to 6th decade, is seen more often in the central skeleton, and may be a slow-growing or high-grade malignant tumour.
- A giant cell tumour is an aggressive and recurrent tumour arising in the epiphysis, often occurring in young adults.
- Ewing’s sarcoma arises from primitive neuroectodermal cells, seen in the age group of 5- 20 years, and occurs in the diaphysis. It is a malignant small round cell tumour that responds well to the combination of radiotherapy and chemotherapy.
- Metastatic bone tumours are more common and may arise from various carcinomas and some sarcomas.
Joints
Normal Structure:
The joints are of 2 types diarthrodial or synovial joints with a joint cavity, and synarthrodial or nonsynovial joints without a joint cavity. Most of diseases of joints affect diarthrodial or synovial joints.
- In diarthrodial joints, the ends of two bones are held together by a joint capsule with ligaments and tendons inserted at the outer surface of the capsule. The articular surfaces of bones are covered by hyaline cartilage which is thicker in weight-bearing areas than in non-weight-bearing areas.
- The joint space is lined by a synovial membrane or synovium which forms synovial fluid that lubricates the joint during movements. The synovium may be smooth or thrown into numerous folds and villi.
- The synovial membrane is composed of an inner layer of 1-4 cell thick synoviocytes and an outer layer of loose vascular connective tissue. On electron microscopy, two types of synoviocytes are distinguished: type A and type B.
- Type A synoviocytes are more numerous and are related to macrophages and produce degradative
enzymes, while type B synthesise hyaluronic acid. Diseases of joints are numerous and joints are also involved in several systemic disorders. - In the following discussion, only those joint diseases which are morphologically significant are described. Synovial tumours are discussed in the next chapter together with other soft tissue tumours.
Degenerative Joint Disease (Osteoarthritis):
Osteoarthritis (OA), also called osteoarthrosis or degenerative joint disease (DJD), is the most common form of chronic disorder of synovial joints. It is characterised by progressive degenerative changes in the articular cartilages over the years, particularly in weight-bearing joints.
Types And Pathogenesis: OA occurs in 2 clinical forms—primary and secondary.
- Primary OA occurs in the elderly, more commonly in women than in men. The process begins by the end of 4th decade and then progressively and steadily increases producing clinical symptoms. Little is known about the aetiology and pathogenesis of primary OA.
- The condition may be regarded as a reward for longevity. Probably, wear and tear with repeated minor trauma, heredity, obesity, and ageing per se, all contribute to focal degenerative changes in the articular cartilage of the joints.
- Genetic factors favouring susceptibility to develop OA have been observed; genetic mutations in proteins which regulate cartilage growth have been identified example FRZB gene.
- Secondary OA may appear at any age and is the result of any previous wear and tear phenomena involving the joint such as previous injury, fracture, inflammation, loose bodies and congenital dislocation of the hip.
The molecular mechanism of damage to the cartilage in OA appears to be the breakdown of collagen type II, probably by IL-1, TNF and nitric oxide.
Morphologic Features: As mentioned above, the weight-bearing joints such as hips, knees and vertebrae are most commonly involved but interphalangeal joints of fingers may also be affected. The pathologic changes occur in the articular cartilages, adjacent bones and synovium:
- Articular cartilages The regressive changes are most marked in the weight-bearing regions of articular cartilages. Initially, there is a loss of cartilaginous matrix (proteoglycans) resulting in progressive loss of normal metachromasia.
- This is followed by focal loss of chondrocytes, and at other places, proliferation of chondrocytes forming clusters. Further progression of the process causes loosening, flaking and fissuring of the articular cartilage resulting in the breaking off of pieces of cartilage exposing subchondral bone.
- Protrusion and dislodgement of cartilage into joint space may result in loose bodies or joint mice formation. Radiologically, this progressive loss of cartilage is apparent as narrowed joint space.
- Bone The denuded subchondral bone appears like polished ivory. There is the death of superficial osteocytes and increased osteoclastic activity causing rarefaction, microcyst formation and occasionally microfractures of the subjacent bone.
- These changes result in remodelling of bone and changes in the shape of joint surface leading to flattening and mushroom-like appearance of the articular end of the bone. The margins of the joints respond to cartilage damage by osteophyte or spur formation.
- These are cartilaginous outgrowths at the joint margins which later get ossified. Osteophytes give the appearance of lipping of the affected joint. Loosened and fragmented osteophytes may form free ‘joint mice’ or loose bodies.
- Synovium Initially, there are no pathologic changes in the synovium but in advanced cases there is low-grade chronic synovitis and villous hypertrophy. There may be some amount of synovial effusion associated with chronic synovitis.
- The manifestations of OA are most conspicuous in large joints such as hips, knees and back. However, the pattern of joint involvement may be related to the type of physical activity such as ballet dancers’ toes, karate fingers etc.
- A minor degree of OA may remain asymptomatic. In symptomatic cases, clinical manifestations are pain that gets aggravated by joint use and is relieved by rest and diminished mobility. Patients may get joint stiffness after a period of inactivity but that usually lasts for brief spells of time.
- Degenerative changes in the interphalangeal joints lead to hard bony and painless enlargements in the form of nodules at the base of the terminal phalanx called Heberden’s nodes.
- These nodes are more common in females and heredity seems to play a role. In the spine, osteophytes of OA may cause compression of cervical and lumbar nerve root with pain, muscle spasms and neurologic abnormalities.
Inflammatory Joint Diseases:
Inflammatory disorders of joints may be immune-mediated (rheumatoid arthritis, SLE, reactive arthritis in rheumatic fever etc), infectious (various microbes example gonococci, M. tuberculosis) crystal-induced (example gout) arthritis, or may be idiopathic. Besides, pigmented villonodular tenosynovitis, bursal cysts, and ganglion are other inflammatory synovial conditions. Common examples among these are discussed here.
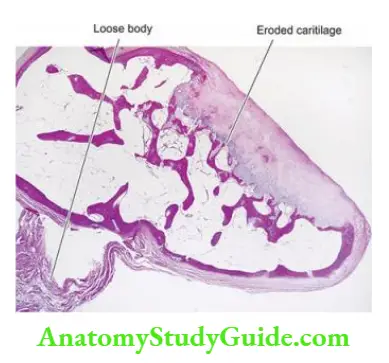
Rheumatoid Arthritis:
- Rheumatoid arthritis (RA) is a chronic multisystem disease of unknown cause. Though the most prominent manifestation of RA is an inflammatory arthritis of the peripheral joints, usually with a symmetrical distribution, its systemic manifestations include haematologic, pulmonary, neurological and cardiovascular abnormalities.
- RA is a common disease having peak incidence in 3rd to 4th decades of life, with 3-5 times higher preponderance in females. The condition has a high association with HLA-DR4 and HLADR1 and has familial aggregation.
- This association is particularly marked in those RA cases that have cyclic citrullinated polypeptide (CCP) antibodies. The onset of the disease is insidious, beginning with a prodrome of fatigue, weakness, joint stiffness, vague arthralgias and myalgias.
- This is followed by pain and swelling of joints, usually in a symmetrical fashion, especially involving joints of hands, wrists and feet. Unlike migratory polyarthritis of rheumatic fever, RA usually persists in the involved joint.
- Approximately 20% of patients develop rheumatoid nodules located over the extensor surfaces of the elbows and fingers. Extra-articular manifestations infrequently produce symptoms, but when present complicate the diagnosis.
Etiopathogenesis: Present concept of aetiology and pathogenesis proposes that RA occurs in an immunogenetically predisposed individual to the effect of microbial agents acting as trigger antigens.
The role of superantigens which are produced by several microorganisms with a capacity to bind to HLA-DR molecules (MHC-II region) has also emerged.
Immunologic derangements: A number of observations in patients and experimental animals indicate the role of immune processes, particularly autoimmune phenomena, in the development of RA. These include the following:
- Detection of circulating autoantibody called rheumatoid factor (RF) against Fc portion of autologous IgG in about 80% of cases of RA. RF antibodies are heterogeneous and consist of IgM and IgG classes.
- The presence of antigen-antibody complexes (IgG-RF complexes) in the circulation as well as in the synovial fluid.
- The presence of other autoantibodies such as antinuclear factor (ANF), antibodies to collagen type 2, and antibodies to cytoskeleton.
- Antigenicity of proteoglycans of human articular cartilage.
- The presence of γ-globulin, particularly IgG and IgM, in the synovial fluid.
- Association of RA with amyloidosis.
- Activation of cell-mediated immunity as observed by the presence of numerous inflammatory cells in the synovium, chiefly CD4+ T lymphocytes and some macrophages.
Trigger events: Though the above hypothesis of a possible role of autoimmunity in the aetiology and pathogenesis of RA is generally accepted, controversy continues as regards the trigger events which initiate the destruction of articular cartilage. Various possibilities which have been suggested are as follows:
- The existence of an infectious agent such as mycoplasma, Epstein-Barr virus (EBV), cytomegalovirus (CMV) or rubella virus, either locally in the synovial fluid or systemic infection sometime prior to the attack of RA.
- The possible role of HLA-DR4 and HLA-DR1 molecules in the initiation of immunologic damage.
Thus, the proposed events in the immunopathogenesis of RA can be summed up as under:
- In response to antigenic exposure (e.g. infectious agent) in a genetically predisposed individual (HLA-DR), CD4+ T-cells are activated.
- These cells elaborate cytokines, the important ones being tumour necrosis factor (TNF)-α, interferon (IF)-γ, interleukin (IL)-1 and IL-6.
- These cytokines activate endothelial cells, B lymphocytes and macrophages.
- Activation of B-cells releases IgM antibody against IgG (i.e. anti-IgG); this molecule is termed rheumatoid factor (RF).
- IgG and IgM immune complexes trigger inflammatory damage to the synovium, small blood vessels and collagen.
- Activated endothelial cells express adhesion molecules which stimulate the collection of inflammatory cells.
- Activation of macrophages releases more cytokines which cause damage to joint tissues and vascularisation of cartilage termed pannus formation.
- Eventually, damage and destruction of bone and cartilage are followed by fibrosis and ankylosis producing joint deformities.
Laboratory Findings: These include the following investigations:
- About 80% of cases are seropositive for RA factor. However, RA factor titres are elevated in certain unrelated diseases too such as viral hepatitis, cirrhosis, sarcoidosis and leprosy.
- HLA-DR4 and HLA-DR1 association in familial cases.
- Anti-CCP antibodies for evaluation of the association of RA factor with HLA.
- Other laboratory findings include mild normocytic and normochromic anaemia, elevated ESR, mild leucocytosis and hypergammaglobulinaemia.
- Advanced cases show characteristic radiologic abnormalities such as narrowing of joint space and ulnar deviation of the fingers and radial deviation of the wrist.
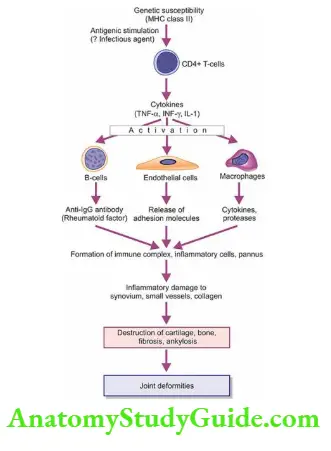
Morphologic Features: The predominant pathologic lesions are found in the joints and tendons, and in advanced cases, extra-articular lesions are encountered.
Articular Lesions: Ra involves first the small joints of hands and feet and then symmetrically affects the joints of wrists, elbows, ankles and knees. The proximal interphalangeal and metacarpophalangeal joints are affected most severely. Frequently cervical spine is involved but the lumbar spine is spared.
Histologically, the characteristic feature is diffuse proliferative synovitis with the formation of pannus. The microscopic changes are as under:
- Numerous folds of large villi of the synovium.
- Marked thickening of the synovial membrane due to oedema, congestion and multilayering
of synoviocytes. - Intense inflammatory cell infiltrate in the synovial membrane with predominance of lymphocytes, plasma cells and some macrophages, at places forming lymphoid follicles.
- Foci of fibrinoid necrosis and fibrin deposition.
The pannus progressively destroys the underlying cartilage and subchondral bone. This invasion of the pannus results in demineralisation and cystic resorption of the underlying bone. Later, fibrous adhesions or even bony ankylosis may unite the two opposing joint surfaces. In addition, persistent inflammation causes weakening and even rupture of the tendons.
Extra-Articular Lesions: Nonspecific inflammatory changes are seen in the blood vessels (acute vasculitis), lungs, pleura, pericardium, myocardium, lymph nodes, peripheral nerves and eyes. But one of the characteristic extra-articular manifestations of RA is the occurrence of rheumatoid nodules in the skin.
- Rheumatoid nodules are particularly found in the subcutaneous tissue over pressure points such as the elbows, occiput and sacrum but can also be seen in the cartilage of the joint.
- The centre of these nodules consists of an area of fibrinoid necrosis and cellular debris, surrounded by several layers of palisading large epithelioid cells, and peripherally there are numerous lymphocytes, plasma cells and macrophages.
- Similar nodules may be found in the lung parenchyma, pleura, heart valves, myocardium and other internal organs.
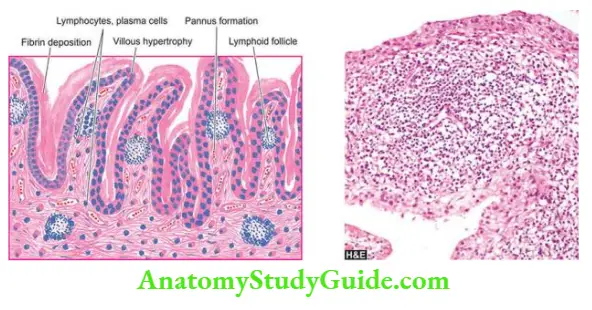
1. Juvenile RA: found in adolescent patients under 16 years of age is characterised by acute onset of fever and predominant involvement of knees and ankles. Pathologic changes are similar but RF is rarely present.
2. Felty’s syndrome: consists of polyarticular RA associated with splenomegaly and hypersplenism and consequent haematologic derangements.
3. Ankylosing spondylitis or rheumatoid spondylitis: is rheumatoid involvement of the spine, particularly sacroiliac joints, in young male patients.
The condition has a strong HLA-B27 association and may have associated inflammatory diseases such as inflammatory bowel disease, anterior uveitis and Reiter’s syndrome.
Suppurative Arthritis:
- Infectious or suppurative arthritis is invariably an acute inflammatory involvement of the joint. Bacteria usually reach the joint space from the bloodstream but other routes of infection by direct contamination of an open wound or lymphatic spread may also occur.
- Immunocompromised and debilitated patients are increasingly susceptible to suppurative arthritis. The common causative organisms are gonococci, meningococci, pneumococci, staphylococci, streptococci, H. influenzae and gram-negative bacilli.
- Clinically, the patients present with manifestations of any local infection such as redness, swelling, pain and joint effusion. Constitutional symptoms such as fever, neutrophilic leucocytosis and raised ESR are generally associated.
Morphologic Features: The haematogenous infectious joint involvement is more often monoarticular rather than polyarticular. The large joints of lower extremities such as the knee, hip and ankle, and shoulder and sternoclavicular joints are particularly favoured sites.
The process begins with hyperaemia, synovial swelling and infiltration by polymorphonuclear and mononuclear leucocytes along with the development of effusion in the joint space.
There may deformation of inflammatory granulation tissue and onset of fibrous adhesions between the opposing articular surfaces resulting in permanent ankylosis.
Tuberculous Arthritis:
Tuberculous infection of the joints results most commonly from haematogenous dissemination of the organisms from pulmonary or other focus of infection. Another route of infection is direct spread from tuberculous osteomyelitis close to the joint. Uncommon in the West, the disease is seen not infrequently in developing countries more commonly in children and also in adults.
Morphologic Features: Tuberculous involvement of the joints is usually
monoarticular type but tends to be more destructive than suppurative arthritis. The most commonly involved sites are the spine, hip joint and knees, and less often other joints are affected. Tuberculosis of the spine is termed Pott’s disease or tuberculous spondylitis.
Grossly, the affected articular surface shows deposition of grey-yellow exudate and occasionally tubercles are present. The joint space may contain tiny grey-white loose bodies and an excessive amount of fluid.
Histologically, the synovium is studded with solitary or confluent caseating tubercles. The
underlying articular cartilage and bone may be involved by extension of tuberculous granulation tissue and cause necrosis (caries).
Gout And Gouty Arthritis:
Gout is a disorder of purine metabolism manifested by the following features, occurring singly or
in combination:
- Increased serum uric acid concentration (hyperuricaemia).
- Recurrent attacks of a characteristic type of acute arthritis in which crystals of monosodium
urate monohydrate may be demonstrable in the leucocytes present in the synovial fluid. - Aggregated deposits of monosodium urate monohydrate (tophi) in and around the joints of the extremities.
- Renal disease involving interstitial tissue and blood vessels.
- Uric acid nephrolithiasis.
The disease usually begins in 3rd decade of life and affects men more often than women. A family history of gout is present in a fairly large proportion of cases indicating the role of inheritance in hyperuricaemia. Clinically, the natural history of gout comprises 4 stages: asymptomatic hyperuricaemia, acute gouty arthritis, asymptomatic intervals of inter critical periods, and chronic tophaceous stage. In addition, gout nephropathy and urate nephrolithiasis may occur.
Types And Pathogenesis: Fundamental biochemical hallmark of gout is hyperuricaemia. A serum uric acid level in excess of 7 mg/dl, which represents the upper limit of solubility of monosodium urate in serum at 37°C at blood pH, is associated with an increased risk of the development of gout. Thus, the pathogenesis of goutispathogenesis of hyperuricaemia.
Hyperuricaemia and gout may be classified into 2 types: metabolic and renal, each of which may be primary or secondary. Primary refers to cases in which the underlying biochemical defect causing hyperuricaemia is not known, while secondary denotes cases with known causes of hyperuricaemia.
- Hyperuricaemia of metabolic origin This group comprises about 10% of cases of gout which are characterised by the overproduction of uric acid. There is either an accelerated rate of purine biosynthesis de novo or an increased turnover of nucleic acids.
- The causes of primary metabolic gout include a number of specific enzyme defects in purine metabolism which may be either of unknown cause or are inborn errors of metabolism. The secondary metabolic gout is due to either increased purine biosynthesis or a deficiency of glucose-6-phosphatase.
- Hyperuricaemia of renal origin About 90% of cases of gout are the result of reduced renal excretion of uric acid. Altered renal excretion could be due to reduced glomerular filtration of uric acid, enhanced tubular reabsorption or decreased secretion.
The causes of gout of renal origin include diuretic therapy, drug-induced (for example aspirin, pyrazinamide, nicotinic acid, ethambutol and ethanol), adrenal insufficiency, starvation, diabetic ketosis, and disorders of parathyroid and thyroid.
Renal disease per se rarely causes secondary hyperuricaemia such as in polycystic kidney disease and leads to urate nephropathy.
Morphologic Features: The pathologic manifestations of gout include: acute gouty arthritis, chronic tophaceous arthritis, tophi in soft tissues, and renal lesions as under:
1. Acute gouty arthritis: This stage is characterised by acute synovitis triggered by the precipitation of a sufficient amount of needle-shaped crystals of monosodium urate from serum or synovial fluid. There is joint effusion containing numerous polymorphs, macrophages and microcrystals of urates.
- The mechanism of acute inflammation appears to include phagocytosis of crystals by leucocytes, activation of the kallikrein system, activation of the complement system and urate-mediated disruption of lysosomes within the leucocytes leading to the release of lysosomal products in the joint effusion.
- Initially, there is monoarticular involvement accompanied by intense pain, but later it becomes polyarticular along with constitutional symptoms like fever.
- Acute gouty arthritis is predominantly a disease of the lower extremities, affecting most commonly the great toe. Other joints affected, in order of decreasing frequency, are the instep, ankles, heels, knees, wrists, fingers and elbows.
2. Chronic tophaceous arthritis: Recurrent attacks of acute gouty arthritis lead to progressive evolution into chronic arthritis. The deposits of urate encrust the articular cartilage.
There is synovial proliferation, pannus formation and progressive destruction of articular cartilage and subchondral bone. Deposits of urates in the form of tophi may be found in the periarticular tissues.
3. Tophi in soft tissue: A tophus (meaning ‘a porous stone’) is a mass of urates measuring a few millimetres to a few centimetres in diameter. Tophi may be located in the periarticular tissues as well as subcutaneously such as on the hands and feet. Tophi are surrounded by inflammatory reactions consisting of macrophages, lymphocytes, fibroblasts and foreign body giant cells.
4. Renal lesions: Chronic gouty arthritis frequently involves the kidneys. Three types of renal
lesions are described in the kidneys: acute urate nephropathy, chronic urate nephropathy and uric acid nephrolithiasis.
- Acute urate nephropathy is attributed to the intratubular deposition of monosodium urate crystals resulting in acute obstructive uropathy.
- Chronic urate nephropathy refers to the deposition of urate crystals in the renal interstitial tissue.
- Uric acid nephrolithiasis is related to hyperuricaemia resulting in hyperuricaciduria.
Pseudogout (Pyrophosphate Arthropathy):
Pseudogout refers to an inflammatory joint involvement due to the deposition of calcium pyrophosphate in the joint space. The condition is seen in middle-aged and elderly individuals of either sex. The pain is usually less severe and involvement of the big toe is rare.
The pathogenesis is unclear but several factors have been implicated. These include associated metabolic diseases (for example hyperparathyroidism, hypothyroidism, gout, ochronosis, Wilson’s disease and haemochromatosis), hereditary, familial occurrence, rheumatoid arthritis and osteoarthritis.
Morphologic Features: The involvement may be monoarticular or polyarticular but large joints such as knees, hips and shoulders are more often affected.
The joint effusion contains crystals of calcium pyrophosphate. There is an acute inflammatory response and deposits of rhomboid crystals on the articular cartilage, ligaments, tendons and joint capsule, termed chondrocalcinosis.
Pigmented Villonodular Synovitis And Tenosynovial Giant Cell Tumour (Nodular Tenosynovitis):
The terms ‘pigmented villonodular synovitis’ and ‘nodular tenosynovitis’ represent diffuse and localised forms respectively of the same underlying process.
The localised form of the lesion is also termed angiofibroma or benign synovioma. When the giant cells are numerous in localised tenosynovitis, the condition is called giant cell tumour of a tendon sheath.
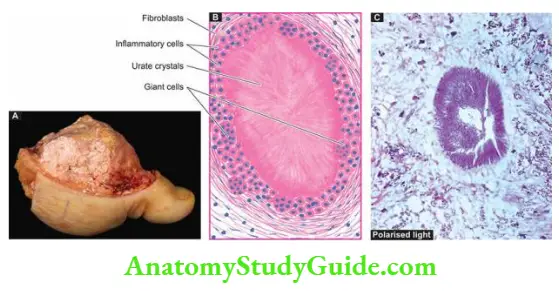
The origin and histogenesis of these conditions are unknown. They were initially regarded as inflammatory in origin and hence the name synovitis. But currently, cytogenetic studies have shown clonal proliferation of cells indicating that these lesions are neoplastic.
Clinically, they present with pain, swelling and limitation of movement of the affected joint and may be easily mistaken for rheumatoid or infective arthritis. The lesions are adequately treated by excision but recurrences are common.
Morphologic Features: Though the two conditions have many morphologic similarities, they are best described separately.
1. Giant cell tumour of tendon sheath (Nodular tenosynovitis): Localised nodular tenosynovitis is seen most commonly in the tendons of fingers.
Grossly, it takes the form of a solitary, circumscribed, pedunculated, small and lobulated nodule, measuring less than 2 cm in diameter. It is closely attached to and sometimes grooved by the underlying tendon. In the section, the lesion is yellowish-brown.
Histologically, it is well encapsulated and is composed of sheets of small oval to spindle shaped cells, foamy xanthoma cells, scattered multinucleate giant cells and irregular bundles of collagen. Many of the spindle-shaped cells are haemosiderin-laden.
2. Pigmented villonodular tenosynovitis: This is a diffuse form of synovial overgrowth seen most commonly in the knee and hip.
Grossly, the synovium has a characteristic sponge-like reddish-brown or tan appearance with
intermingled elongated villous projections and solid nodules.
Histologically, the changes are modified by recurrent injury. The enlarged villi are covered by
hyperplastic synovium and abundant sub-synovial infiltrate of lymphocytes, plasma cells and macrophages, many of which are lipid-laden and haemosiderin-laden. Multinucleate giant cells are scattered in these areas.
Cyst Of Ganglion:
- A ganglion is a small, round or ovoid, movable, subcutaneous cystic swelling of the synovium. The most common location is the dorsum of the wrist but may be found on the dorsal surface of the foot near the ankle.
- The Histogenesis of the ganglion is disputed. It may be the result of herniated synovium, embryologically displaced synovial tissue, or post-traumatic degeneration of connective tissue.
Morphologic Features Grossly, a ganglion is a small cyst filled with clear mucinous fluid. It may or may not communicate with the joint cavity or tendon where it is located. Microscopically, the cyst has a wall composed of dense or oedematous connective tissue which is sometimes lined by synovial cells but more often has indistinct lining.

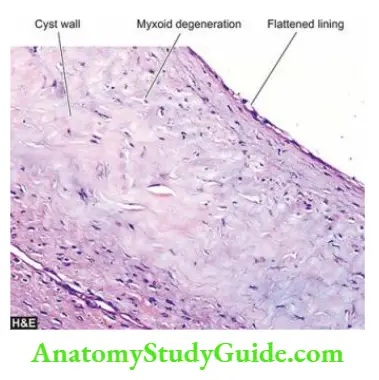
Bursitis:
Inflammation of bursa is termed bursitis or bursal cyst. Bursae are synovial-lined sacs found over bony prominences. Bursitis occurs following mechanical trauma or inflammation. It may result following a single injury such as olecranon bursitis and prepatellar bursitis, but is more often due to repeated injuries from excessive pressure such as in a housemaid’s knee or tennis elbow.
Morphologic Features Grossly, the bursal sac is thick-walled and may contain watery, mucoid or granular brown material.
Histologically, the bursal wall is composed of dense fibrous tissue lined by inflammatory granulation tissue. The wall is infiltrated by lymphocytes, plasma cells and macrophages and may show focal calcium deposits.
Diseases of Joints:
- Osteoarthritis or degenerative joint disease is a chronic disorder of synovial joints characterised by progressive degenerative changes in the articular cartilages over the years.
- Rheumatoid arthritis is a chronic multisystem disease of unknown cause occurring in the 3rd to 4th decade of life, and is more common in women. It occurs in immunogenetically predisposed individuals to the effect of some microbial infection as the trigger event.
- Suppurative arthritis is an acute inflammatory involvement of the joint by bacteria, usually from the bloodstream. Tuberculous arthritis occurs from haematogenous spread of infection from another focus.
- Gout is a disorder of purine metabolism manifested by hyperuricaemia, recurrent attacks of characteristic acute arthritis. There is a formation of tophi in and around the joints. It may be associated with uric acid nephrolithiasis.
- Pigmented villonodular synovitis and nodular tenosynovitis are diffuse and localised forms respectively of the same underlying process. When the giant cells are numerous in localised tenosynovitis, the condition is called the giant cell tumour of the tendon sheath.
- A ganglion is a small, round or ovoid, movable, subcutaneous cystic swelling due to degeneration of synovium.
- Bursal cyst or bursitis is inflammation of the sac-like synovial tissue.
Skeletal Muscles
Normal Structure:
Striated skeletal muscles consist of bundles of fibres called fascicles, each of which is surrounded by a connective tissue sheath termed perimysium. Perimysium contains blood vessels and nerve supply of the muscle fascicles. Each muscle fibre is enveloped by a delicate fibrous stroma called endomysium.
- Individual muscle fibre is an elongated multinucleated syncytium-like cell about 100 µm in diameter and several centimetres in length. The muscle nuclei are spindle-shaped and lie at the periphery of the fibre under the sarcolemma, the plasma membrane of muscle fibre. The cytoplasm of the muscle fibre contains myofilaments which are contractile elements.
- Myofilaments are of 2 types myosin comprising thick filaments and actin constituting thin filaments. These together produce cross-striations in muscle fibres seen in longitudinal sections on light microscopy. Sarcomeres are the partitions of myofilaments into equal zones.
- Each sarcomere represents the distance between consecutive Z bands and contains the central A (anisotropic) band, and the lateral I (isotropic) bands.
- The major functions of striated skeletal muscle are to convert chemical energy into mechanical energy, to act as a store of energy and proteins, and to play a role in the metabolism of the body. The muscle, however, cannot function as a contractile organ without a nerve supply. For this purpose, there are motor units, each of which consists of the following:
- Motor neuron cell body located in the spinal cord anterior horn, or a cranial nerve nucleus.
- The axon of the motor neuron in the peripheral or cranial nerve.
- The neuromuscular junction.
- The muscle fibres are innervated by the motor neuron.
- The muscle fibre contraction occurs by action potential generated by chemical transmission of the impulse across the synaptic gap by acetylcholine.
- Skeletal muscles are affected by a number of systemic diseases and pathologic processes such as ischaemia and toxic (Zenker’s) necrosis; atrophy and hypertrophy; degeneration and regeneration; and polymyositis, dermatomyositis, and various forms of infective myositis (example viral myositis, pyogenic myositis, gas gangrene and parasitic involvements such as cysticercosis).
- Some of these conditions have been considered already in different chapters. Here, three important groups of specific diseases myopathies (mainly muscular dystrophies), neuromuscular diseases (mainly myasthenia gravis and denervation atrophy), and inflammatory myopathies (polymyositis, dermatomyositis, inclusion body myositis) are discussed here.
Myopathic Diseases (Myopathies):
Myopathies are primary skeletal muscle diseases having structural changes or functional impairment. These conditions may have the following clinical manifestations:
- Intermittent or persistent chronic muscle weakness
- Muscle pains (myalgias), cramps and stiffness
- Enlargement or atrophy of muscle
Based on aetiology and pathogenesis, myopathies are divided into the following broad groups:
- Hereditary myopathies or muscular dystrophies
- Congenital myopathies (for example nemaline myopathy)
- Disorders of muscle energy metabolism (for example glycogen storage and glycolytic defects)
- Mitochondrial myopathies (for example ragged red fibre diseases)
- Disorders of muscle membrane excitability or channelopathies (for example calcium channel disorders)
- Endocrine and metabolic myopathies (for example hypo- and hyperthyroidism)
- Myopathies of systemic diseases (for example respiratory, cardiac etc)
- Drug-induced myopathies (for example cholesterol-lowering drugs, glucocorticoids)
Only hereditary myopathies also termed muscular dystrophies, are briefly considered below.
Muscular Dystrophies:
- Muscular dystrophies are a group of genetically-inherited primary muscle diseases, having in common, progressive and unremitting muscular weakness. Eight forms of muscular dystrophies are described: Duchenne’s, Becker’s, myotonic, facioscapulohumeral, limb-girdle, EmeryDreifuss, congenital and oculopharyngeal type.
- Each type of muscular dystrophy is a distinct entity having differences in inheritance pattern, age at onset, defective gene or protein, clinical features, other organ system involvements and clinical course.
- These differences are summarised. However, in general, muscular dystrophies manifest in childhood or in early adulthood. A family history of neuromuscular disease is elicited in many cases.
Investigations: In general, limited investigations are done in a suspected case of myopathy which include the following:
- Serum enzyme determinations Most commonly determined is CK-MM isoenzyme which is elevated (CK-MB is myocardial bound and is raised in cardiac muscle diseases). Other enzymes which may be raised in myopathies are AST, ALT and LDH.
- Electrodiagnostic studies These include EMG, nerve stimulation and nerve conduction studies.
- Molecular studies and DNA analysis can be carried out to establish a definitive diagnosis.
- Muscle biopsy In All cases without an identifiable genetic defect, biopsy from a skeletal muscle (most often quadriceps, biceps or deltoid) remains the most important diagnostic test. The biopsy is subjected to various studies light microscopy, histochemistry, immunohistochemistry, and electron microscopy.
On light microscopy, common to all forms of muscular dystrophies are: muscle fibre necrosis, regenerative activity, replacement by, interstitial fibrosis and adipose tissue
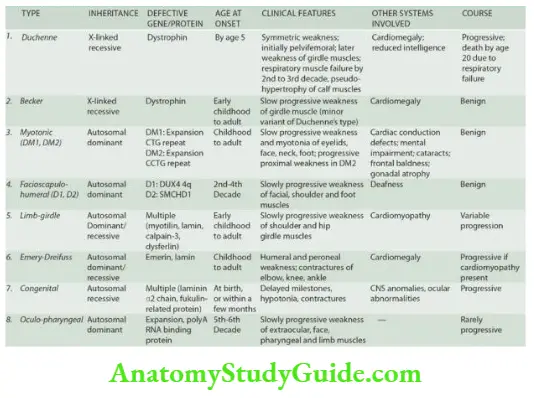
The group of neurogenic diseases affecting skeletal muscles is characterised by a combination of muscular weakness and fatiguability. The most common of these is myasthenia gravis; others are congenital myasthenia, an acquired Eaton-Lambert syndrome associated with carcinoma of the lung, and denervation atrophy.
A classification of neuromuscular disorders based on the part of the motor unit involved is given Myasthenia gravis and denervation atrophy are described below.
Myasthenia Gravis:
- Myasthenia gravis (MG) is a neuromuscular disorder of autoimmune origin in which the acetylcholine receptors (AChRs) in the motor end-plates of the muscles are damaged due to antibody-mediated immune attack and thus the number of AChRs is reduced.
- The term ‘myasthenia’ means ‘muscular weakness’ and ‘gravis’ implies ‘serious’; thus both together denote the clinical characteristics of the disease. MG may be found at any age but adult women are affected more often than adult men in the ratio of 3:2.
- The condition presents clinically with muscular weakness and fatiguability, initially in the ocular musculature but later spreads to involve the trunk and limbs. There is about 10% mortality in MG which is due to severe generalised disease and involvement of respiratory muscles.
- Several other autoimmune diseases have been found associated with MG such as autoimmune thyroiditis, rheumatoid arthritis, SLE, pernicious anaemia and collagen-vascular diseases.

Pathogenesis: The pathogenesis of MG is best understood in the context of normal muscle metabolism
- Normally, acetylcholine is synthesised in the motor nerve terminal and stored in vesicles that are released spontaneously when an action potential reaches the nerve terminal. Acetylcholine from released vesicles combines with AChRs, initiating an action potential which is propagated along the muscle fibre triggering muscle contraction.
- In MG, the basic defect is a reduction in the number of available AChRs at the postsynaptic muscle membrane. In addition, the postsynaptic folds are flattened. These changes result in decreased neuromuscular transmission leading to failure to trigger muscle action potentials and consequent weakened muscle contraction.
- The neuromuscular abnormalities in MG are mediated by an autoimmune response. About 85-90% of patients of MG have anti-AChR-antibodies in their sera. These antibodies reduce the number of available AChRs either by blocking the active sites of the receptors or by damaging the post-synaptic muscle membrane in collaboration with complement.
- The exact mechanism of how the autoimmune response is initiated is not completely understood but the thymus appears to play a role in this process. The majority of patients of MG may have either thymoma or thymic hyperplasia; thymectomy is helpful in ameliorating the condition. The thymus possibly sensitises B cells to produce anti-AChR antibodies.
Investigations: Based on clinical features, diagnosis of MG is suspected but it is confirmed by the following investigations:
- Anti-AChR antibodies Serum of approximately 85-90% cases of MG shows antibodies against AChRs.
- Electrodiagnostic testing Nerve stimulation test may help in establishing the diagnosis of MG.
- Muscle biopsy Biopsy plays a role in evaluating patients with neuromuscular disease.
Grossly, the muscles appear normal until late in the course of the disease when they become wasted.
By light microscopy, a few clumps of lymphocytes may be found around small blood vessels. Degenerating muscle fibres are present in half the cases.
Electron microscopy reveals a reduction in the synaptic area of the motor axons due to flattening or simplification of postsynaptic folds. The number of AChRs is greatly reduced. By immunohistochemistry combined with electron microscopy, it is possible to demonstrate the complexity of IgG and complement at the neuromuscular junctions.
Denervation Atrophy:
- If the muscle or a part of the muscle is deprived of its motor nerve supply, the affected muscle undergoes atrophy. In demyelination, on the other hand, there is a conduction block in the nerve impulse but no denervation and hence muscle atrophy does not occur.
- Denervating diseases are characterised by axonal degeneration and consequent muscle atrophy. These include amyotrophic lateral sclerosis (ALS) as an example of anterior horn cell disease, and peripheral neuropathy causing injury to myelinated axon.
- (It may be interesting to recall here that Stephen Hawking, a British-born famous international cosmologist, who died in March 2018 at 74 suffered from early-onset slowly-progressive ALS that gradually paralysed him).
- The clinical manifestations of denervation atrophy are the combination of muscular weakness and reduced muscle bulk. In amyotrophic lateral sclerosis, there are characteristic fasciculations of muscles of the shoulder and tongue.
Morphologic Features: Denervation atrophy is pathologically characterised by groups of small angulated muscle fibres alternating with groups of plump, normal or even hypertrophic fibres with intact innervation. Further progression of the process may produce superimposed changes in muscular dystrophy.
Inflammatory Myopathies:
This group includes three conditions having overlapping clinical features of progressive skeletal
muscle weakness: polymyositis, dermatomyositis and inclusion body myositis, but this condition have distinctive age at presentation and some other features.
Etiology And Pathogenesis: All three forms of inflammatory myositis appear to have an autoimmune aetiology. This is supported by the following:
- Association with other autoimmune (collagen) diseases.
- Presence of various autoantibodies against nuclear and cytoplasmic antigens in 20% of cases.
- Presence of humoral immune mechanism in dermatomyositis as observed by B cell infiltration in the lesions.
- In polymyositis and inclusion body myositis, T-cell mediated cytotoxicity is implicated as seen by CD8+ T cells along with macrophages in the lesions.
- Some non-immune factors such as viral infection with coxsackie B, influenza, Epstein-Barr, CMV etc in triggering autoimmune mechanism has been suggested.
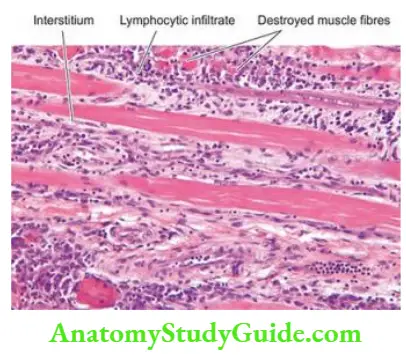
Morphologic Features: There is symmetric involvement of skeletal muscles such as those of the pelvis, shoulders, neck, chest and diaphragm.
Histologically, vacuolisation and fragmentation of muscle fibres and numerous inflammatory cells are present. In the late stage, muscle fibres are replaced by fat and fibrous tissue.
Clinical Features: It is a multi-system disease characterised by:
- progressive muscle weakness, mainly proximal,
- skin rash, typically with heliotropic erythema and periorbital oedema,
- dysphagia due to involvement of pharyngeal muscles,
- respiratory dysfunction, and
- association with deep-seated malignancies.
Diseases of Skeletal Muscles and Neuromuscular Junction:
- Myopathies are primary skeletal muscle diseases resulting in chronic muscle weakness. These include hereditary or muscular dystrophies, congenital, mitochondrial, inflammatory, endocrine, channelopathies, metabolic and toxic myopathies.
- Muscular dystrophies have various forms: Duchenne’s, Becker’s, myotonic, facioscapulohumeral, limb-girdle, Emery-Dreifuss, congenital and oculopharyngeal type. Each type has a distinct inheritance pattern, age at onset, defective gene or protein, clinical features, other organ system involvements and clinical course.
- Neurogenic diseases affecting skeletal muscles have a combination of muscular weakness and fatiguability. Examples include myasthenia gravis (due to reduced AChRs from autoimmune damage) and denervation atrophy.
- Inflammatory myopathies are a group of 3 diseases having common clinical features of progressive skeletal muscle weakness manifesting at different ages polymyositis (>18 years), dermatomyositis (children and young adults) and inclusion body myositis (>50 years).
Leave a Reply