Neurology
Introduction And Symptomatology
Weakness and Paralysis Categories
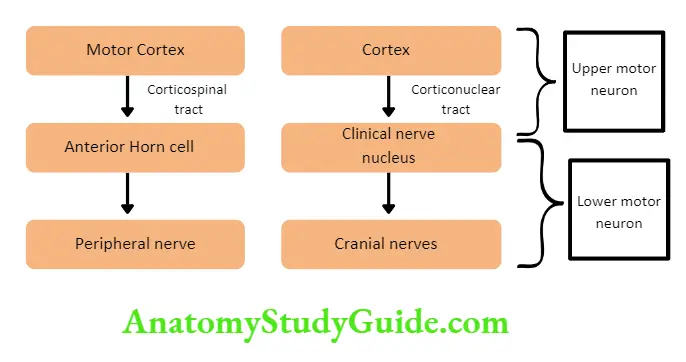
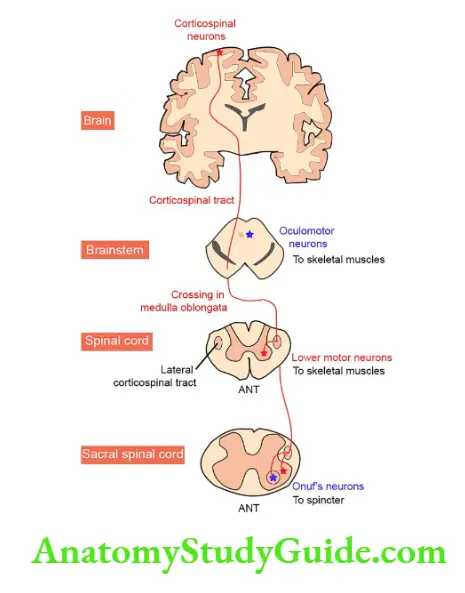
- Upper motor neurons: They consist of corticospinal interneurons which arise from the motor cortex and descend to the spinal cord where they activate the lower motor neurons (anterior horn cells) through synapses.
- Lower motor neurons: The term “motor neuron” is usually used only to the efferent neurons that actually innervate muscles (the lower motor neurons).
- A motor neuron consists of a nerve cell (neuron) that is located in the anterior horn cell of the spinal cord and its fibers (axon) project outside the spinal cord to directly or indirectly control effector organs, mainly muscles and glands.
- Motor neuron axons are efferent nerve fibers and carry signals from the spinal cord to the effectors to produce effects.
Read And Learn More: General Medicine Question And Answers
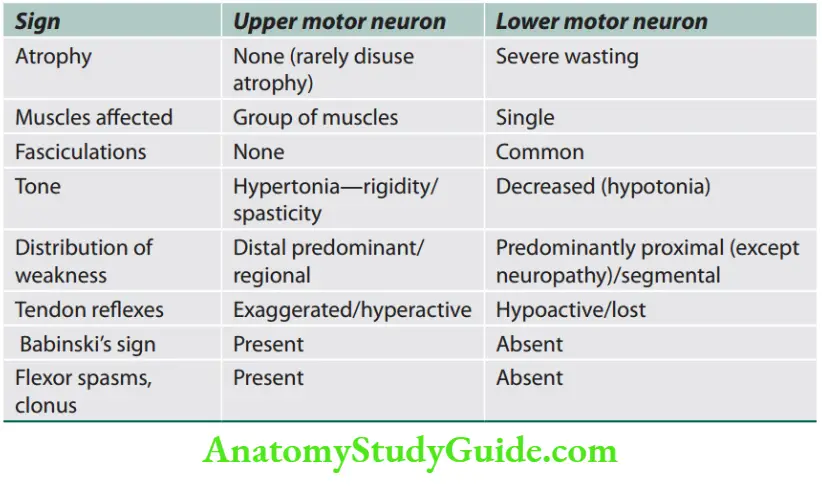
Signs of Upper and Lower Motor Neuron Disease
Question 1. List the differences between upper motor neuron disease and lower motor neuron disease.
Answer:
The differences between upper motor neuron disease and lower motor neuron disease
Tone
Muscle tone is a partial state of contraction of a skeletal muscle to maintain its optimal length during resting conditions, even.
Increased tone: Associated with the disease of upper motor neurons due to loss of inhibition of γ-motor neurons above the site of the lesion.
Hypotonia: Causes of hypotonia.

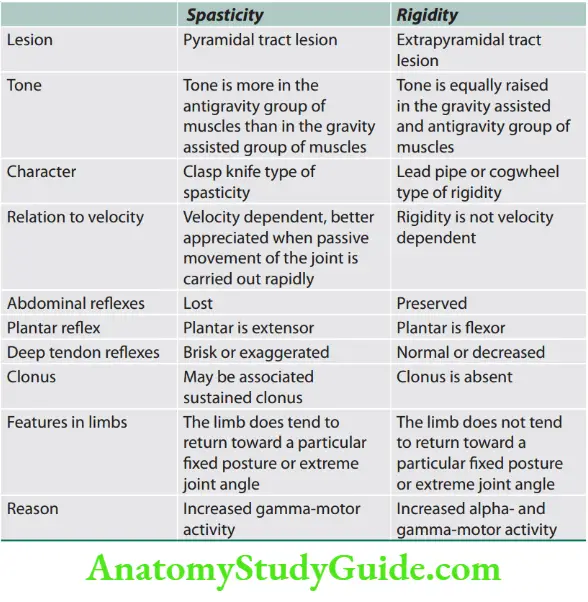
Question 2. List the causes of hypotonia. (or) Enumerate the differences between spasticity and rigidity.
Answer:
The differences between spasticity and rigidity are listed.
Other causes of hypertonia: Tetanus, seizure (tonic phase), tetany, catatonia, paratonia (gegenhalten, mitgehen).
Fasciculation and Fibrillation
Question 3. What is fasciculation and fibrillation?
Answer:
When a motor unit (group of muscle fibers) becomes diseased, especially in anterior horn cell diseases, it may discharge spontaneously, producing fasciculations that may be seen or felt clinically or recorded by electromyography (EMG).
Fasciculations are visible, fine, and fast, sometimes vermicular contractions of fine muscle fibers that occur spontaneously and intermittently.
When motor neurons or their axons degenerate, the denervated muscle fibers also may discharge spontaneously.
These single muscle fiber discharges, or fibrillation potentials, cannot be seen or felt but can be recorded with EMG.
Causes of fasciculation: Amyotrophic lateral sclerosis, progressive spinal muscular atrophy, post-polio syndrome, hyperthyroidism, organophosphorus poisoning, drugs (e.g., atropine, lithium), mercury, benign fasciculation.
Myotonia
Question 4. What is myotonia?
Answer:
- Myotonia is characterized by continued, involuntary muscle contraction even after cessation of voluntary effort (i.e., muscle contraction continues beyond the period of time required for a particular movement to be made and there is failure of normal muscle relaxation).
- It is best seen in the face and hand muscles. When the patient is asked to smile and then relax his facial muscle, a delay in relaxation of the muscle is noted and the smile remains fixed for a longer duration (transverse smile). Similarly, when the patient is asked to grip the examiner’s fingers and then let go immediately, a delay in the relaxation of the grip is noted.
- Myokymia is a vermicular or continuous rippling movement of a group of muscle fibers that can be seen in neuropathies [Guillain–Barré syndrome (GBS)], plexopathies, and Isaac’s syndrome.
Causes of myotonia
- Myotonic dystrophy type 1
- Myotonic dystrophy type 2/proximal myotonic myopathy
- Myotonia congenita
- Paramyotonia congenita
- Hyperkalemic periodic paralysis
Ataxia
Question 5. Write a short note on the causes of ataxia.
Answer:
Ataxia is a disorder characterized by unsteadiness and impaired coordination of regulating body posture and the rate, range, force, and direction of movement. Types of ataxia
Causes of hypotonia.
- Lesions of the motor side of the reflex arc: Poliomyelitis, polyneuritis, peripheral nerve injuries
- Lesions of the sensory side of the reflex arc: Tabes dorsalis, herpes zoster, carcinomatous neuropathy
- Combined motor and sensory lesion: Syringomyelia, cord or root compression, gross cord destruction
- Lesions of the muscle (myopathies), neuromuscular junction (NMJ) (myasthenia)
- State of neuronal shock in upper motor neuron lesion
- Cerebellar lesions
- Chorea
- Periodic paralysis
- Rapid eye movement (REM) sleep
- Benzodiazepine overdose, neuromuscular blockers
Reflexes
Plantar Response/Reflx
Question 6. Write a short note on plantar reflex and extensor plantar reflex/Babinski’s sign.
Answer:
Plantar response/reflex is a nociceptive, superficial reflex. Its segmental innervation is the S1 segment of the spinal cord.It was first described by Babinski.
Plantar response/reflex Technique
- Position the patient in a supine with hip and knee extended.
- Fix the ankle joint by holding it and stroke (gentle but firm pressure) the outer aspect of the sole with a blunt point (tip of a key). The stroke is directed forward and then curves inward along the metatarsophalangeal joints from the little to the big toe and stopped short of the base of the great toe (root value S1).
Plantar response/reflex Interpretation
The normal response is great toe will flex at the metatarsophalangeal joint accompanied by flexion of other toes. The normal response should not be termed “negative Babinski’s sign”.
Plantar response/reflex Abnormal responses
Absent: No response is seen. Plantar response/reflex may be absent when there is a loss of sensation of the sole (L5–S1), thick sole, paralysis of the extensor hallucis, and lesions of a reflex arc.
Extensor: Extension (dorsiflexion) of the great toe with or without fanning of others’ toes (abduction) is known as Babinski’s sign (mediated by L5).
Fanning of toes without great toe extension has no significance. When fully developed it is accompanied by dorsiflexion of the ankle, flexion of the hip and knee joint, and slight abduction of the thigh with contraction of the tensor fascia lata.
Plantar response/reflex causes are:
Plantar response/reflex Physiological: It may be normally extensor in infants below 6 months, during deep sleep, under general anesthesia.
Plantar response/reflex Pathological: Lesion of corticospinal (pyramidal) tract above S1 segment, deep coma, transiently after seizure, alcohol intoxication, hypoglycemia, and metabolic encephalopathy.
Alternative ways to elicit Babinski’s sign are Chaddock’s (lateral malleolus), Gordon’s (calf), Oppenheim’s (anterior tibia), Schaeffer’s (Achilles tendon), Gonda’s (press down 4th toe), Stransky’s (adduct little toe), Bing’s (pinprick on dorsolateral foot).
Deep Tendon Reflex
Vertigo
Question 7. Write a short note on the causes of vertigo.
Answer:
vertigo Definition: Vertigo is defined as an abnormal perception (hallucination/illusion) of movement (a sensation of rotation or tipping) of either the environment or self (body or part of it).
The individual feels that the surroundings are spinning or moving. BPPV is the most common cause.
The perceived movement may be falling down, or rotating or there is a sensation of spinning of the outside world. It is often accompanied by nausea or vomiting.
vertigo Mechanism: It develops because of conflicting visual, proprioceptive, and vestibular information about a person’s position in space.
Lesions causing vertigo
Gait
- Observation to be noted while the patient walks:
- The posture of the body while walking,
- The regularity of the movement,
- The position and movement of the arms,
- The relative ease and smoothness of the movement of the legs,
- The distance between the feet both in forward and lateral directions,
- The ability to maintain a straight course,
- The ease of turning,
- Stopping, and
- Position of feet and posture just before initiation of gait.
Types of ataxia.
- Cerebellar: vasculitis, multiple sclerosis, infection bleeding, infarction, tumors, direct injury, toxins (e.g., alcohol), genetic disorders
- Sensory: Posterior column diseases, large fiber neuropathy
- Optic
- Vestibular
- Frontal lobe ataxia (Bruns ataxia)
- Mixed
- Psychogenic
- Pseudoataxia
Gait Cycle

Abnormalities of Gait
Question 8. Write a short note on gait abnormalities with examples.
Answer:
Neurogenic gait disorders should be differentiated from those due to skeletal abnormalities (characterized by pain-producing an antalgic gait, or limp).
Gait abnormalities incompatible with any anatomical or physiological deficit may be due to functional disorders.
Pyramidal (circumduction/hemiplegic) gait Lesions of the upper motor neuron produce a characteristic extension of the affected leg.
There is a tendency for the toes to strike the ground on walking and outward throwing/swing of lower limbs.
This movement occurring at the hip joint is called circumduction. There is a leaning toward the opposite normal side.
The arm of the affected side is adducted at the shoulder and flexed at the elbow, wrist, and fingers.
In hemiplegia/hemiparesis, there is a clear asymmetry between affected and normal sides on walking, but in paraparesis, both lower legs swing slowly from the hips in extension and are stiffly dragged over the ground (walking in mud).
Foot drop (high stepping/slapping gait)
In normal walking, the heel is the first part of the foot to hit the ground.
A lower motor neuron lesion affecting the leg will cause weakness of ankle dorsiflexion, resulting in a less controlled descent of the foot, which makes a slapping noise as it hits the ground.
In severe cases, the foot will have to be lifted higher at the knee to allow room for the inadequately dorsiflexed foot to swing through, resulting in a high-stepping gait.
Cause: for example, common peroneal nerve palsy.
Myopathic gait/waddling gait (primary muscle disease)
- During walking, alternating transfer of the body’s weight through each leg needs adequate hip abduction.
Causes: Weakness of proximal lower limb muscles (e.g., polymyositis, muscular dystrophy) causes difficulty rising from sitting.
The hips are not properly fixed by these muscles and trunk movements are exaggerated, and walking becomes a waddle or rolling. The pelvis is poorly supported by each leg.
This may be seen with bilateral congenital dislocation of the hip (Trendelenburg gait). The patient walks on a broad base with exaggerated lumbar lordosis.
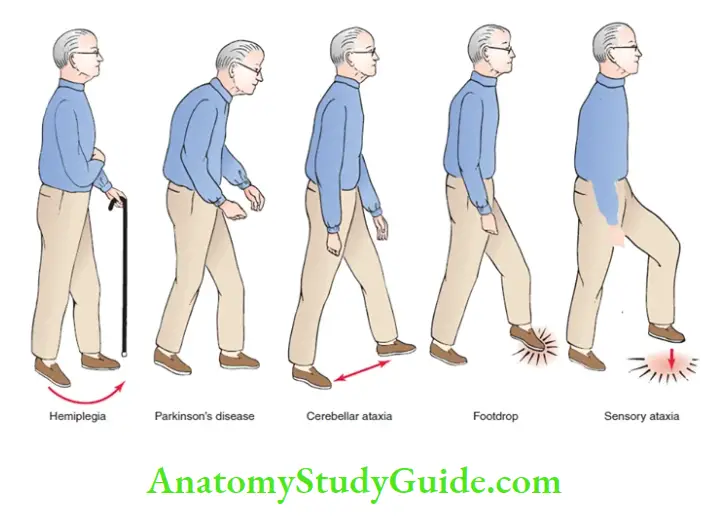
Ataxic gait (cerebellar ataxia: broad-based gait) In this type of gait, the patient, is unstable, tremulous and reels in any direction (including backward) and walks on a broad base.
Ataxia describes this incoordination. The patient finds difficulty in executing tandem walking.
Ataxic Gait Causes: Lesions of the cerebellum, vestibular apparatus, or peripheral nerves.
When walking, the patient tends to veer to the side of the affected cerebellar lobe.
When the disease involves the cerebellar vermis, the trunk becomes unsteady without limb ataxia, with a tendency to fall backward or sideways, and is termed truncal ataxia.
Apraxic gait
- In an apraxic gait, the acquired walking skills become disorganized. On examination of the legs, the power, cerebellar function, and proprioception are normal. Leg movement is normal when sitting or lying and the patient can carry out complex motor tasks (e.g., bicycling motion). But the patient cannot initiate and organize the motor act of walking. The feet appear stuck to the floor and the patient cannot walk.
- Apraxic gait Causes: Diffuse bilateral hemisphere disease or diffuse frontal lobe disease (e.g., tumor, hydrocephalus, and infarction).
Marche a petits pas
- It is characterized by small, slow steps and marked instability. In contrast to the festination found in Parkinson’s disease, it lacks increasing pace and freezing.
- Cause: Small vessel cerebrovascular disease, and accompanying bilateral upper motor neuron signs.
Extrapyramidal gait (shuffling gait)/Festinant gait
- It is characterized by stooped posture and gait difficulties with problems initiating walking and controlling the pace of the gait.
- Patients make a series of small, flat-footed shuffles and become stuck while trying to start walking or when walking through doorways (freezing).
- The center of gravity will be moved forward to aid propulsion and difficulty stopping.
- It is characterized by muscular rigidity throughout extensors and flexors.
- Power is preserved, the pace is shortened and slows to a shuffle, and its base remains narrow.
- There is a stoop and diminished arm swinging and gait becomes festinant (hurried) with short rapid steps.
- A patient will be having difficulty in turning quickly and initiating movement. Retropulsion, i.e., small backward steps are taken involuntarily when a patient halts.
- Cause: Parkinsonism.
Scissoring gait
It may be seen classically with cerebral palsy due to bilateral spasticity.
Sensory ataxia gait (stamping gait)
- It is characterized by broad-based, high stepping, stamping gait and ataxia due to loss of proprioception (position sense).
- This type of ataxia becomes more prominent by the removal of sensory input (e.g., walking with eyes closed) and becomes worse
in the dark. Romberg’s test is positive. - Causes: Peripheral sensory (large fiber) lesions (e.g., polyneuropathy), posterior column lesion (vitamin B12 deficiency
or tabes dorsalis).
Choreiform gait (hyperkinetic gait)
- The patient will display irregular, jerky, involuntary movements in all extremities. Walking may accentuate their baseline movement disorder.
- Causes: Sydenham’s chorea, Huntington’s disease, and other forms of chorea, athetosis, or dystonia.
Abnormal Speech And Language
Abnormal Speech And Language Definitions:
Phonation: It is the production of vocal sounds without word formation.
Speech: It consists of words that are articulate vocal sounds that symbolize and communicate ideas. Speech is the articulation and phonation of language sounds.
Language: It refers to the selection and serial ordering of words according to learned rules by which a person can use spoken or written modalities to communicate with others and to express cerebral activities involved with thinking and learning.
It can be by speech (auditory symbols), writing (graphic symbols), or gestures and pantomime (motor symbols).
Aphasias
Question 9. Describe the clinical features and distinguish, based on clinical examination, the various disorders of speech.
Answer:
Aphasia is loss or defective language content of speech resulting from damage to the speech centers within the dominant (usually left in 97%) hemisphere.
A language disturbance occurring after a right hemisphere lesion in a right-hander is known as crossed aphasia.
It includes a defect in or loss of the power of expression by speech, writing, or gestures or a defect in or loss of the ability to comprehend spoken or written language or to interpret gestures.
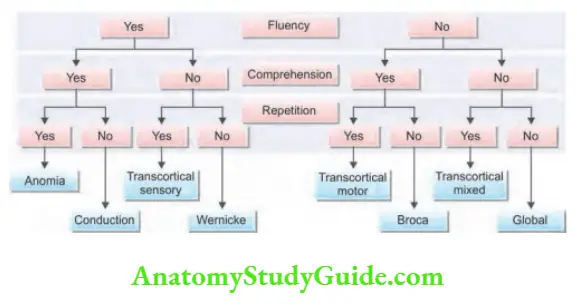
Aphasia may be categorized according to whether the speech output is fluent or nonfluent.
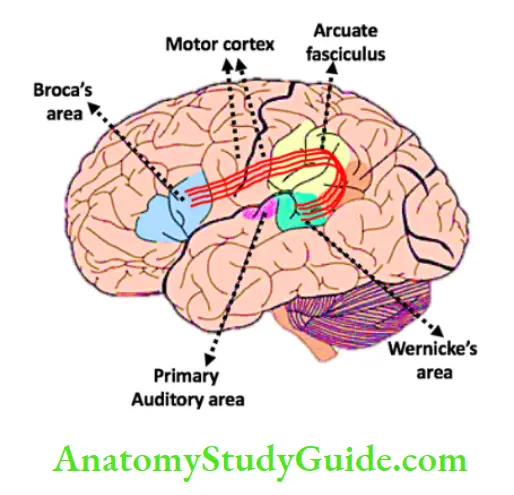
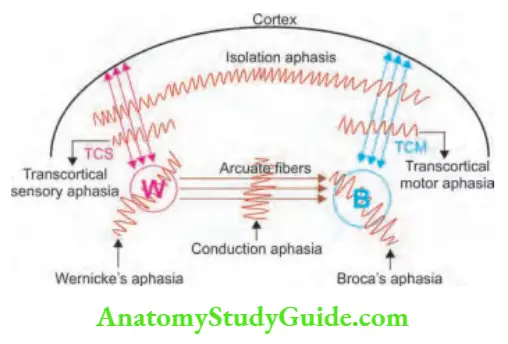
- Fluent aphasias (receptive aphasias) are impairments mostly due to the input or reception of language, with difficulties either in auditory verbal comprehension or in the repetition of words, phrases, or sentences spoken by others. For example, Wernicke’s aphasia.
- Nonfluent aphasias (expressive aphasias) are difficulties in articulating, with relatively good auditory, verbal comprehension. For example, Broca’s aphasia.
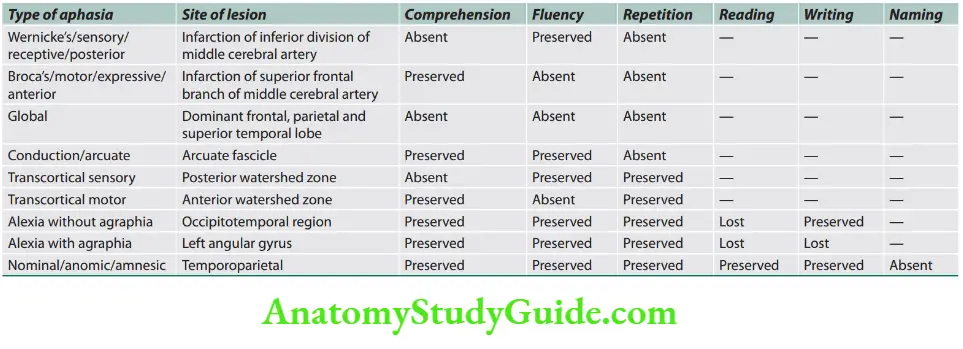
Categories/Varieties of Aphasia
Alexia: It is the impairment of visual word recognition, in the context of intact auditory word recognition and writing ability.
Agraphia: It is the inability to write, as a language disorder resulting from brain damage.
Anomia: In this, the word approximates the correct answer but it is phonetically inaccurate (plentiful for pencil)—phonemic paraphasia.
When the patient cannot say the appropriate name when an object is shown but can point to the object when the name is provided is known as one way or retrieval-based naming deficit.
The classification of aphasia
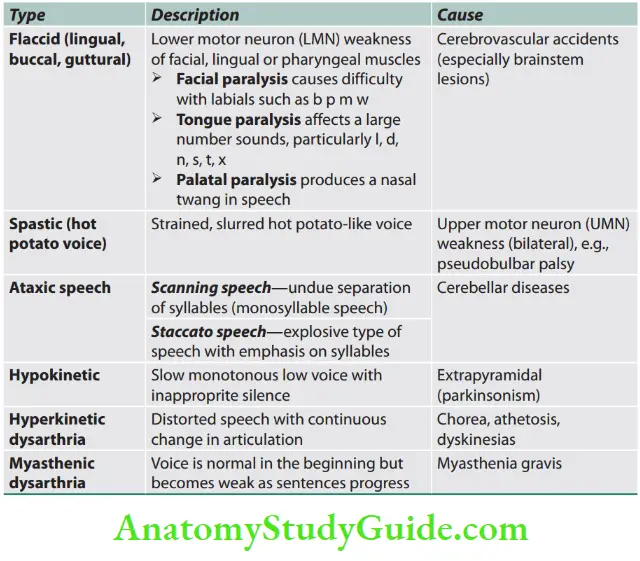
Dysarthrias
Dysarthrias involve the abnormal articulation of sounds or phonemes.
Types
Types of dysarthria are listed in
Apraxia
Question 10. Define apraxia. Give examples.
Answer: Apraxia is the impaired ability (inability) to carry out (perform) skilled, complex, organized motor activities in the presence of normal basic motor, sensory, and cerebella functions.
Examples of complex motor activities: are dressing, using cutlery, and geographical orientation.
Types
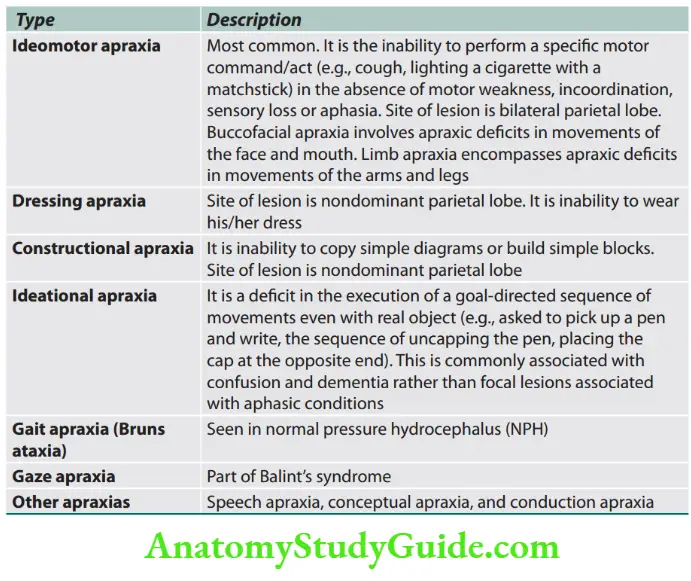
Gerstmann’s syndrome: The combination of acalculia (impairment of simple arithmetic), dysgraphia (impaired writing), finger anomia (an inability to name individual fingers such as the index or thumb), and right-left confusion (an inability to tell whether a hand, foot, or arm of the patient or examiner is one the right or left side of the body) is known as Gerstmann’s syndrome.
Site of lesion: Inferior parietal lobule (especially the angular gyrus) in the left (dominant) hemisphere.
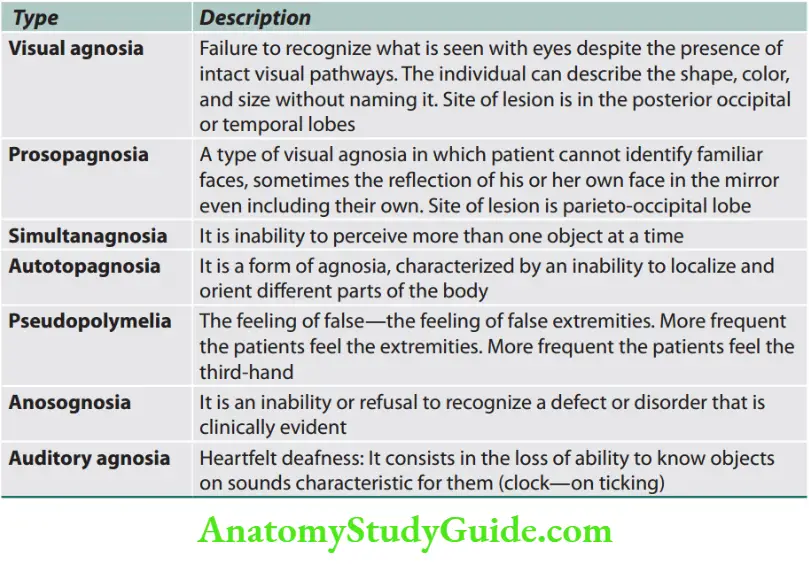
Agnosia
Question 11. Write a short note on agnosia.
Answer:
Agnosia is a failure to recognize objects (e.g., places, clothing, persons, sounds, shapes or smells), despite the presence of an intact sensory system.
Site of lesion: Contralateral parietal lobe.
Types
Types of agnosia.
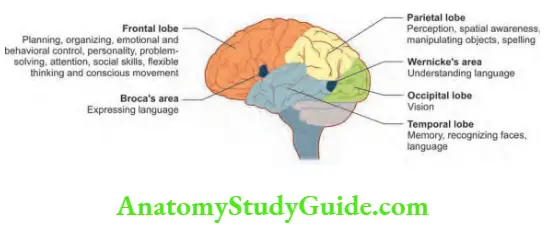
Functions of Cerebral Hemispheres
Question 12. Write a short note on the normal functions of different cortical lobes and their abnormalities. (or) Write a short note on the functions of the parietal lobe.
Answer:
Cerebral dominance aligns limb dominance with language function.
Right-handed individuals almost always (>95%) have the dominant left hemisphere, and about 7% of left-handers have a dominant right hemisphere.
Functions and effects of damage to various lobes of cerebral hemispheres

Headache
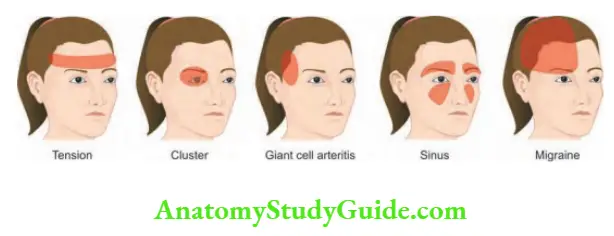
Headache is among the most common reasons, patients seek medical attention.
Classification of Headache
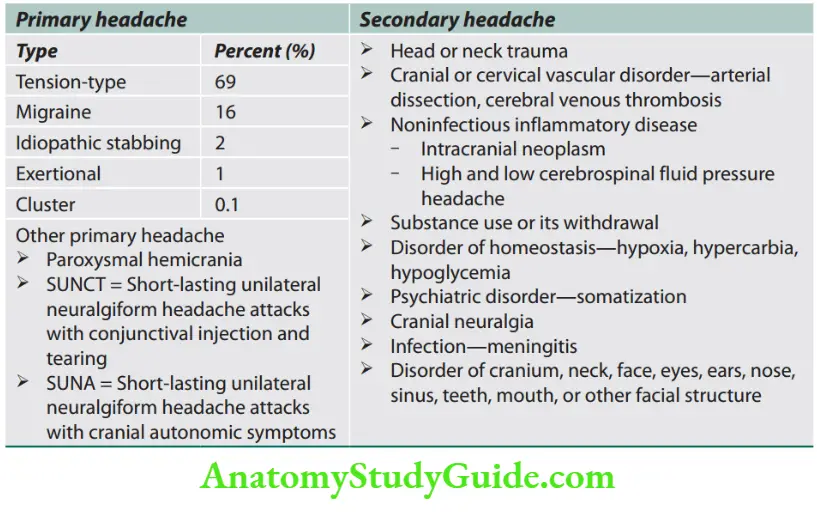
- Primary headaches: Benign, recurrent, no organic disease as their cause. It affects the quality of life of the patient.
- Secondary headaches: Underlying organic disease.
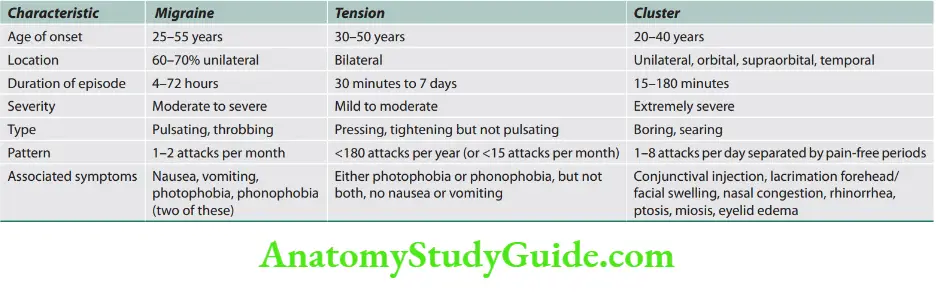
Migraine
Question 13. Write a short essay/note on classification, pathogenesis, clinical features, and management of migraine. Define and classify headaches and describe the presenting features, precipitating factors, and aggravating and relieving factors of various kinds of headaches.
Answer:
Migraine is a neurovascular disease caused by neurogenic inflammation and characterized by severe, recurring headaches.
- It is the second most common cause of headaches. It is usually characterized by episodic severe pain on one side of the head (headache) and is usually associated with certain features such as sensitivity to light, sound, or movement; nausea and vomiting often accompany the headache.
- Gender—Female: Male ratio is 5:1.
Classification
Question 14. Classify migraine and describe the distinguishing features between classical and nonclassical forms of migraine.
Answer:
Migraine without aura or common migraine: It does not give any warning signs before the onset of the headache. It occurs in about 70–80% of migraine patients.
Migraine with aura or classical migraine: It gives some warning signs called “aura” before the actual headache begins.
About 20–30% of migraine patients experience aura.
The most common aura is visual and may include both positive and negative (visual field defects) features.
Retinal migraine: It involves attacks of monocular scotoma or even blindness of one eye for less than an hour and is associated with headache.
Childhood periodic syndromes: These involve cyclical vomiting (occasional intense periods of vomiting), abdominal migraine (abdominal pain, usually accompanied by nausea), and benign paroxysmal vertigo of childhood (occasional attacks of vertigo).
They may be precursors or associated with migraine.
Complicated migraine: It describes migraine headaches and/or auras that are unusually long or unusually frequent, or associated with a seizure or brain lesion.
Basilar migraine: Occipital headache, preceded by vertigo, diplopia, and dysarthria, ±visual and sensory symptoms (brainstem symptoms).
Hemiplegic migraine: Rare autosomal dominant disorder characterized by prolonged headache lasting hours or days, followed by hemiparesis and/or coma that recovers slowly over days.
Ophthalmoplegic migraine: Migraine associated with transient IIrd nerve palsy with/without involvement of pupil; sometimes also affects IVth and VIth nerve.
Vestibular migraine (also called migrainous vertigo or migraine-associated vertigo).
Catamenial migraine: Migraine associated with menstruation-associated migraine.
Pathogenesis
- The cause of migraine is not known.
- Genetic factors: Play a role in causing neuronal hyperexcitability. Migraine is usually polygenic. Rarely, familial migraine is associated with mutations in the α1 subunit of the P/Q-type voltage-gated calcium channel or neuronal sodium channel (SCN1A) and a dominant loss of function mutation in a potassium channel gene (TRESK). Migraine is frequently associated with positive family history, and similar phenomena occur in disorders such as CADASIL.
Genes for familial hemiplegic migraine
-
- FHM1—CACNA1A: Neuronal P/Q calcium channel—increases neurotransmitter release
- FHM2—ATP1A2: Astrocyte sodium pump—dysfunction increases extracellular K+
- FHM3—SCN1A: Neuronal sodium channel—increased action potential firing
- Hormonal influences: Female preponderance and the frequency of migraine attacks at certain points in the menstrual cycle due to hormonal fluctuations. Estrogen-containing oral contraception can exacerbate migraine in a few patients.
- Right-to-left cardiac shunt: Migraine with aura has been associated with patent foramen ovale (PFO), atrial septal defect (ASD), and pulmonary arteriovenous malformations (AVMs) in hereditary hemorrhagic telangiectasia (Osler–WeberRendu syndrome).
Several theories have been proposed for the pathogenesis of migraine.
Vascular theory: Constriction of intracerebral blood vessels produces aura.
Vasodilatation of intracranial/extracranial blood vessels produces a headache phase.
Serotonin theory: Decreased serotonin levels linked with migraine and specific serotonin receptors found in blood vessels
of the brain.
Neurogenic theory: The aura (see clinical features below) is thought to be due to the spreading cortical depression wave of neuronal depolarization followed by depressed activity spreading slowly anteriorly across the cerebral cortex from the occipital region.
This spreading process occurs at a rate of about 3 mm/minute.
Dysfunction of activation of cells in the trigeminal nucleus releases vasoactive neuropeptides [e.g., calcitonin gene-related peptide (CGRP), substance P, and other vasoactive peptides including 5-HT] by activated trigeminovascular neurons.
They produce painful meningeal inflammation and vasodilation.
Dopamine plays a role and most migraine symptoms can be induced by dopaminergic stimulation.
There is dopamine receptor hypersensitivity in patients with migraine.


Cortical Spreading Depression
- A wave of activation is followed by a reduced activity that spreads across the surface of the brain.
- Initial dilation is conducted with intrinsic velocity ahead of cortical spreading depression (CSD), subsequent constriction, and eventual dilation.
Astrocyte Calcium Waves
- Slowly propagated waves evoked by a wide variety of stimuli
- Associated with the active release of ATP, glutamate, K+, lactate, prostanoids, and interleukins (ILs)
Precipitating factors: Anything can initiate or precipitate or amplify an attack.
Common triggers are excess stress, glare, exposure to bright light, loud noises/sounds, smoke or strong scents, menstruation, lack or excess of sleep, cheese, caffeine, alcohol, chocolate, citrus fruit, food additives such as monosodium glutamate, vasodilators, hunger, physical exertion, stormy weather or barometric pressure changes, contraceptive pills, etc.
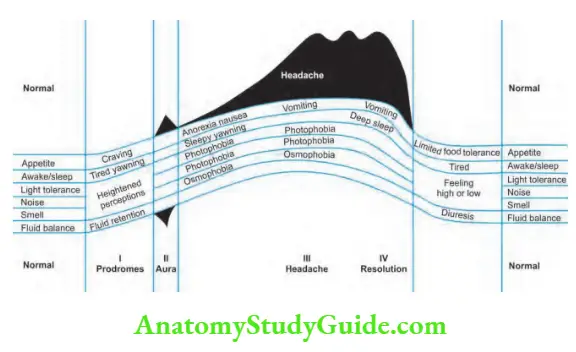
Clinical Features
Headache is usually hemicranial, throbbing, and associated with nausea and vomiting.
Migraine without aura (previously called “common” migraine).
About 70–80% of patients with migraine have a characteristic headache but without aura.
Typically attacks are episodic and start at puberty and prevalence increases in 4th decade. It may show variable degrees of spontaneous remissions.
The scalp may be tender to touch during episodes (allodynia is the production of pain from normally nonpainful stimuli) and the patients prefer to be still in a dark and quiet environment.
Other symptoms associated with migraine headaches.

Migraine with aura (previously known as “classical” migraine)
Migraine aura: About 20–30% of patients with migraine experience malaise, irritability, behavioral change, or focal neurological symptoms for some hours or days immediately preceding the headache phase.
Type of aura:
- Visual Aura,
- Sensory Aura, Or
- Language Aura.
Visual aura: It is the most common type characterized by positive visual symptoms such as shimmering, teichopsia (silvery zigzag lines also called fortification spectra) flashing lights, or fragmentation of the image (like looking through a pane of broken glass) or scintillating spots across the visual fields for up to 40 minutes.
Sometimes there may be temporary patchy visual field loss which may move across the visual field (scotomas) and even evolve into hemianopia or tunnel vision.
- Sensory aura: It consists of positive sensory symptoms such as tingling followed by numbness, spreading over 20–30 minutes, from one part of the body to another.
- Language aura: Dominant hemisphere involvement may cause transient speech disturbance.
- Motor aura-transient weakness.
Duration of aura: Usually it evolves over 5–20 minutes with symptoms changing as the wave of spreading neuronal depression moves across the surface of the cortex.
It rarely lasts for >60 minutes and is followed immediately by the headache phase

Diagnostic Criteria

Strongest predictors of migraine
- Are you nauseated or sick to your stomach when you have a headache?
- Has a headache limited your activities for a day or more in the last 3 months?
- Does light bother you when you have a headache?
- Patients who answer positively to two out of three have a 93% chance of having migraines.
Complications of Migraine
- Status migrainous is a debilitating migraine attack lasting for >72 hours.
- Persistent aura without infarction is defined by aura symptoms persisting for 1 week or more with no evidence of infarction on neuroimaging.
- Migrainous infarction: Migraine attack, occurring in a patient with migraine with aura, in which one or more aura symptoms persist for >1 hour and neuroimaging shows an infarction in a relevant brain area.
- Migraine aura-triggered seizure is a seizure triggered by an attack of migraine with aura.
Red Flags of Headache

Question 15. Write a short essay/note on the management of migraine in young adults.
Answer:
Mention three drugs used and write a short note on prophylactic therapy.
RED flags of headache.
- “Worst” headache ever
- First severe headache[
- Subacute worsening over days or weeks
- Altered level of sensorium/consciousness
- Abnormal neurologic examination
- Fever or unexplained systemic signs
- Significant weight loss
- Vomiting that precedes the headache
- Pain induced by bending, lifting, and cough, worsens with Valsalva maneuvers
- Pain that disturbs sleep or presents immediately upon awakening
- Known systemic illness, history of trauma, cancer, or HIV
- New-onset headache in a patient >50 years of age
- Focal neurologic deficits, jaw claudication
- Morning headache associated with nausea and vomiting
- Pain associated with local tenderness (e.g., region of a temporal artery)
- Mnemonic SNNOOP10
- Systemic symptoms including fever
- Neoplasm history
- Neurological deficit (including decreased consciousness)
- Onset is sudden or abrupt
- Older age (onset after age 50 years)
- P10:
- Pattern change or recent onset of new headache
- Positional headache
- Precipitated by sneezing, coughing, or exercise
- Papilledema
- Progressive headache and atypical
- presentations
- Pregnancy or puerperium
- Painful eye with autonomic
- features
- Post-traumatic onset of headache
- Pathology of the immune system such as HIV
- Painkiller (analgesic) overuse (e.g., medication overuse headache) or new drug at onset of headache.
Management
Nonpharmacological treatment (General measures)
- The explanation is that migraine has no grave prognosis.
- Identification of triggers and avoidance of identified triggers or exacerbating factors to prevent attacks.
- Women with aura should avoid estrogen treatment (oral contraception or hormone replacement). Lifestyle modification wherever possible.
- Other measures: Meditation, relaxation techniques, and psychotherapy.
Pharmacological treatment
- Abortive treatment: Treatment of acute attack.
- Preventive treatment: Drug prophylaxis.
Treatment of an acute attack
- Analgesic: Simple analgesia such as aspirin, paracetamol or nonsteroidal anti-inflammatory agents.
- Nausea may be treated by an antiemetic (metoclopramide or domperidone).
- Severe attacks: If there is previously no relief with a nonsteroidal anti-inflammatory drug (NSAID), use “triptans”.
- Triptans (e.g., sumatriptan: 50–100 mg tablet/5–20 mg nasal spray/6 mg S/C; rizatriptan: 5–10 mg tablet; frovatriptan: 2.5 mg oral; naratriptan: 2.5 mg oral; almotriptan: 12.5 mg oral; eletriptan: 40–80 mg oral; zolmitriptan: 2.5 mg oral/5 mg intranasal spray):
- Mode of action: Potent 5-HT1B/1D agonists, inhibit the release of CGRP and substance P, inhibit activation of the trigeminal nerve, and inhibit vasodilation in the meninges.
- Administration: Triptans are available as oral preparations, nasal sprays, and subcutaneous injections.
- Contraindications: Ischemic heart disease or stroke, high risk for coronary artery disease, pregnancy, hemiplegic or basilar migraine
and use with ergots.
Calcitonin gene-related peptide antagonists (e.g., erenumab, fremanezumab, galcanezumab) are very effective for acute treatment of migraine.
Lasmiditan, a selective serotonin 1F receptor agonist has been tried. Single-pulse transcranial magnetic stimulation (TMS) has shown good benefits.
Drug prophylaxis
Indications for drug prophylaxis in migraine
Various drugs can be used and the most frequently used are:
Anticonvulsants [antiepileptic drugs (AEDs)]: Valproate (800 mg) or topiramate (100–200 mg daily) is the most effective option. β-adrenoceptor antagonists (β-blockers), e.g., propranolol slow release 80–160 mg daily.
Tricyclic antidepressants, e.g., amitriptyline 10 mg increasing weekly in 10 mg steps to 50–60 mg or Dosulepin (10–200 mg at night).
Methysergide 1–2 mg TID in resistant cases (prolonged use may produce retroperitoneal and mediastinal fibrosis).
Botulinum toxin has been tried as a treatment for chronic migraine.
Vasoactive drugs and calcium channel blockers: These include flunarizine (5–10 mg OD at bedtime), verapamil (80–160 mg 3 times a day), and methysergide and are used in refractory cases. Pizotifen is rarely used.
Memantine—N-methyl-D-aspartate (NMDA) receptor antagonist, blocks glutamate.
Nonprescription medications:
- Riboflavin (B2): 400 mg daily
- Magnesium oxide: 400 mg daily
- Coenzyme Q10: 150 mg daily
- Feverfew: 10–30 mg daily
- Butterbur (Petadolex): 50–75 mg BID
- Melatonin
Indications for drug prophylaxis in migraine.
- Patients who have very frequent headaches (>2–3 weeks)
- Attack duration > 48 hours
- Migraine-related disability >3 days/month
- Headache extremely severe
- Migraine accompanied by severe aura
- Contraindications to acute treatment
- Unacceptable adverse effects with acute migraine treatment
- Patients preference
Cluster Headache
Question 16. Write a short note on cluster headaches.
Answer:
Cluster headache (migrainous neuralgia) is distinct from migraine and is much less common than migraine.
Cluster Headaches Age and gender: Usually it occurs in young adults in the 3rd decade (20 and 40 years) with male predominance (M: F = 5:1).
Cluster Headaches Pathophysiology
- The cause and precise mechanism are unknown.
- It differs from migraine in its character, absence of genetic predisposition, lack of triggering dietary factors, male predominance, and different drug effects.
- Abnormal hypothalamic activity is observed in functional imaging studies during an attack.
- Patients are often smokers and consume more than average alcohol.
Cluster Headaches Clinical Features
- Cluster headache is periodic with recurrent bouts of identical headaches beginning at the same hour for weeks at a time (the eponymous “cluster”).
- Patients may develop either one or several attacks within a 24-hour period.
- Cluster headache causes severe (excruciating) and worst, stabbing/boring, unilateral periorbital/retro-orbital pain with parasympathetic autonomic features in the same eye (e.g., unilateral lacrimation, nasal congestion, and conjunctival redness/injection or even a transient Horner’s syndrome). The pain is so severe that they may commit suicide.
- Circadian periodicity: Usually cluster period lasts for a few weeks and is followed by remission for months to years.
They typically recur a year or more later often at the same time of year.
Diagnostic Criteria for Cluster Headache
Diagnostic criteria for cluster headache
Management of Cluster Headache
- Acute attacks: Analgesics are not useful and acute attacks are usually halted by:
- Subcutaneous injection of sumatriptan (6 mg) is the drug of choice for acute treatment.
- It works quickly and usually shortens an attack to 10–15 minutes.
- There is no evidence of tachyphylaxis. Oral sumatriptan is not effective.
- Sumatriptan (20 mg) and zolmitriptan (5 mg) nasal sprays are also effective for
- Inhalation of 100% oxygen at 10–12 L/minute for 15–20 minutes.
- Many respond very well.
- The brevity of the attack probably prevents other migraine therapies from being effective.
- Octreotide is effective in the treatment of acute cluster headaches.
Most prophylactic migraine drugs are often ineffective.
Attacks can be prevented in some patients by sodium valproate, lithium, verapamil, methysergide, and/or a short course of oral corticosteroids.
Tension-type Headache
Question 17. Write a short note on a tension headache.
Answer:
It is the most common type of headache.
Pathophysiology is incompletely understood, and few consider this as a milder version of migraine.
Clinical Features
Characteristic features of headache are:
Pain is “dull” “tight” or like a “pressure”, and it may be accompanied by a sensation of a band around the head or pressure at the vertex.
It is of constant character and generalized, but often radiates forward from the occipital region. It may be episodic or persistent.
Severity may vary and is not associated with vomiting or photophobia. The pain often progresses throughout the day.
Tenderness may be present over the skull vault or in the occiput.
Diagnostic criteria for cluster headache.
- At least five attacks fulfilling the following:
- Severe or very severe unilateral orbital, supraorbital, and/ or temporal pain lasting 15–180 minutes if untreated
- Headache is accompanied by at least one of the following:
- Autonomic features: Unilateral
- Conjunctival redness/injection and/or lacrimation or
- Nasal congestion and/or rhinorrhea or
- Edema of eyelid
- Sweating on the forehead and face or
- Miosis and/or ptosis or
- Restlessness or agitation
- Frequency of attacks: From one every other day to eight/day
Management
- Carefully assess, followed by a discussion of likely precipitants and reassurance that the prognosis is good.
- Physiotherapy (with muscle relaxation and stress management) may be helpful.
- Low-dose amitriptyline may be beneficial. Investigation is rarely required.
The differences between the most common primary headaches are presented.
Stroke And Cerebrovascular Disease
Question 18. Classify cerebrovascular accidents and describe the etiology, predisposing genetic and risk factors, and pathogenesis of hemorrhagic and non-hemorrhagic stroke.
(or)
Describe clinical features, diagnosis of cerebral stroke in young, and its management.
(or)
Describe the etiology, clinical features, and treatment of cerebral thrombosis.
(or)
Write a short essay/note on cerebrovascular accidents/strokes.
Answer:
Stroke
- A stroke (cerebrovascular accident is a vague term that should be avoided) is defined as a syndrome of rapid (abrupt) onset of a neurologic deficit that is attributable to a focal vascular cause.
- World Health Organization (WHO) definition: Stroke is defined as a clinical syndrome consisting of “rapidly developing clinical signs of focal (or global) disturbance of cerebral function, with symptoms lasting for 24 hours or longer or leading to death, with no apparent cause other than of vascular origin”.
- Progressing stroke (or stroke in evolution): It is a stroke in which the focal neurological deficit worsens after the patient first presents.
- It may be due to an increasing volume of infarction, secondary hemorrhage in the infarcted area, or increasing cerebral edema.
- Complete stroke: Rapid onset with a persistent focal neurological deficit that does not progress beyond 96 hours.
- Evolving stroke: Gradual stepwise development of neurological deficits.
- Focal cerebral deficits that develop slowly (over weeks to months) are unlikely to be due to stroke and are more suggestive of tumor or inflammatory or degenerative disease.
Terminologies
Several terms are used to classify strokes, mainly based on the duration and evolution of symptoms.
- Transient ischemic attack (TIA): Described later
- Reversible ischemic neurological deficit (RIND): In some cases, deficits last for longer than 24 hours but resolve completely or almost completely within a few days.
- Stuttering hemiplegia: Internal carotid lesions are characterized by repeated episodes of TIA followed by fully evolved stroke.
Types of Stroke
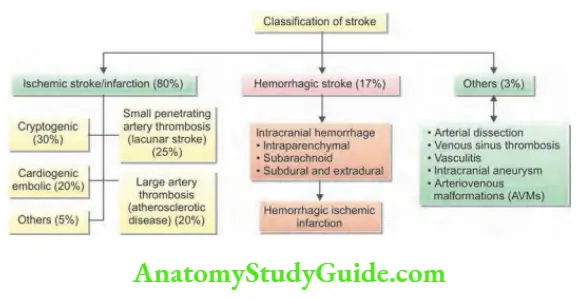
About 80% of patients develop cerebral infarction due to inadequate blood flow to part of the brain, and most of the remainder develop an intracerebral hemorrhage.
Ischemic stroke: Cerebral infarction is most commonly caused by thromboembolic disease secondary to atherosclerosis in the major extracranial arteries (carotid artery and aortic arch).
About 20% of infarctions are caused by emboli from the heart, and about 20% are caused by thrombosis in situ caused by intrinsic disease of small perforating vessels (lenticulostriate arteries), producing lacunar infarctions.
Hemorrhagic stroke: Intracranial hemorrhage (ICH) is caused by bleeding directly into or around the brain.
Neurological symptoms are produced by compression, toxic effects, or raised intracranial pressure (ICP).
Hemorrhagic conversion of ischemic stroke
- This may occur after thrombolytic drugs (alteplase) are administered to patients with an ischemic stroke or where a patient has a large cerebral clot, which tends to be more common with cardioembolic strokes.
- The major adverse effect of thrombolysis is symptomatic intracerebral hemorrhage, observed in around 6–7% of cases.
- The risk of symptomatic ICH increases with age, high blood pressure, very severe neurological deficits, severe hyperglycemia, and with early ischemic changes on computed tomography (CT) scans.
Cerebrovascular anomalies such as intracranial aneurysms and AVMs.
Risk (Predisposing) Factors for Stroke.
Risk factors for stroke.
Risk factors in patients of all age groups
High-risk
- Hypertension (including
- isolated systolic)
- Smoking
- Diabetes mellitus
- Atrial fibrillation
- Drugs: Cocaine, amphetamine
- Dilated cardiomyopathy
- Endocarditis
- High cholesterol
- Obesity
- Vasculitis: Systemic vasculitis [e.g., polyarteritis nodosa (PAN)], granulomatosis with polyangiitis (Wegener’s, etc.), primary CNS vasculitis, meningitis (syphilis, tuberculosis, fungal, bacterial, zoster)
Low-risk
- Migraine
- Oral contraceptives or alcohol
- Patent foramen ovale
- Recent myocardial infarction
- Prosthetic valve
- Sleep apnea
Additional risk factors that are more common in young patients
Hypercoagulable disorders
- Protein C and S deficiencies
- Antithrombin III deficiency
- Antiphospholipid syndrome
- Factor V Leiden mutation
- Prothrombin G20210A heterozygous mutation
- Sickle cell anemia
- Hyperhomocysteinemia
- Thrombotic
- thrombocytopenic purpura
- Arterial dissection
- Infections (e.g., syphilis, HIV)
- Systemic malignancy
Question 19. Write a short essay/note on risk factors for stroke.
Answer:
Pathophysiology

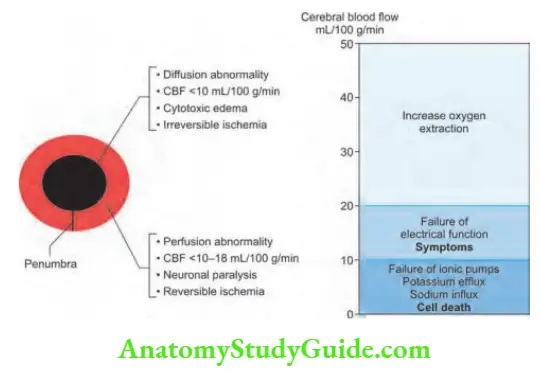
A complete stop of cerebral blood flow (CBF) causes the death of brain tissue within 4–10 minutes.
The CBF to the ischemic core is <10 mL/100 g/minute.
The ischemic penumbra is the region surrounding the central core.
The tissue of the penumbra is functionally impaired and is at risk of infarction.
CBF is 10–18 mL/100 g/minute. It has the potential to be salvaged by revascularization.
The damage to the penumbra is coupled with inflammation and excitotoxicity mediated by glutamine and sodium.
Also, hyperglycemia and fever worsen the penumbra, so need to be controlled.
Etiology of Ischemic Stroke
Thrombotic occlusion
- Small vessel (lacunar) stroke
- Large vessel thrombosis
Embolic occlusion
- Artery-to-artery: Carotid bifurcation, aortic arch, arterial dissection
- Cardioembolic: Atrial fibrillation, mural thrombus, myocardial infarction, dilated cardiomyopathy, valvular lesions, mitral stenosis, mechanical valve, bacterial endocarditis
- Paradoxical embolus: Atrial septal defect, patent foramen ovale Venous sinus thrombosis Subarachnoid hemorrhage due to vasospasm
Small Vessel (Lacunar) Stroke
Embolic occlusion
Question 20. Write a short essay/note on embolic stroke, its causes, and management.
Answer:
Artery-to-artery embolic stroke
- Any vessel may be the source of emboli, including the aortic arch, common and internal carotid, vertebral and basilar arteries.
- Atherosclerosis within the carotid artery: Carotid bifurcation atherosclerosis is the most common source of artery-to-artery embolus.
- In young (age <60 years) patients: Intracranial atherosclerosis, dissection of the internal carotid or vertebral arteries or even vessels beyond the circle of Willis is a common source of embolic stroke.
Cardioembolic stroke
- The heart is a common source of emboli. Emboli from the heart most often lodge in the middle cerebral artery (MCA), the posterior cerebral artery (PCA), or one of their branches. Infrequently, the anterior cerebral artery (ACA) territory is involved.
- Nonrheumatic atrial fibrillation (and other arrhythmias) causing thrombosis in a dilated left atrium is the most common cause of cerebral embolism.
- Other causes include cardiac valvular disease such as congenital valve disorders, and infective vegetations; rheumatic and degenerative calcific changes may cause embolization.
- Simultaneous infarcts in different vascular territories are suggestive that the heart or aorta is the source of emboli.
Paradoxical embolus
Atrial septal defect or PFO may occasionally allow passage of fragments of thromboembolic [e.g., from a lower limb deep vein thrombosis (DVT)] from the right atrium to the left.
This is believed to be the most common cause of cryptogenic strokes.
Other embolisms like fat embolism, air embolism, amniotic fluid embolism, and tumor embolism are rarely associated with neurological deficits.
Thrombosis occlusion
Large vessel thrombosis
Thrombosis developing at the site of ruptured mural atheromatous plaque may produce occlusion of the involved vessel or maybe a source of artery-to-artery embolism.
Small Vessel (lacunar stroke)
Question 21. Write a short essay/note on lacunar infarct.
Answer:
- Small penetrating arterial branches of 200–800 μm in diameter, supply the deep brain parenchyma.
- Each of these small branches can be occluded either by atherothrombotic disease at its origin or by the development of occlusive vasculopathy—lipohyalinotic thickening (a consequence of hypertension).
- Thrombosis of these vessels causes small infarcts that are referred to as lacunes.
- These infarcts range in size from 0.2 to 15 mm in diameter.
- Risk factors include hypertension and age.
- Small vessel strokes account for 20% of all strokes.
- Symptoms: Lacunar strokes present with fluctuating symptoms “capsular warning syndrome”.
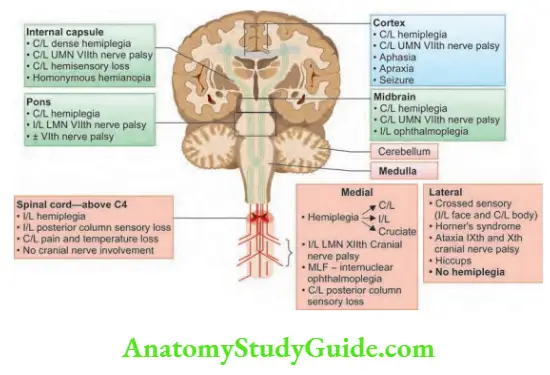
Clinicoanatomic Corralation
Stroke Localization -Clinical Features

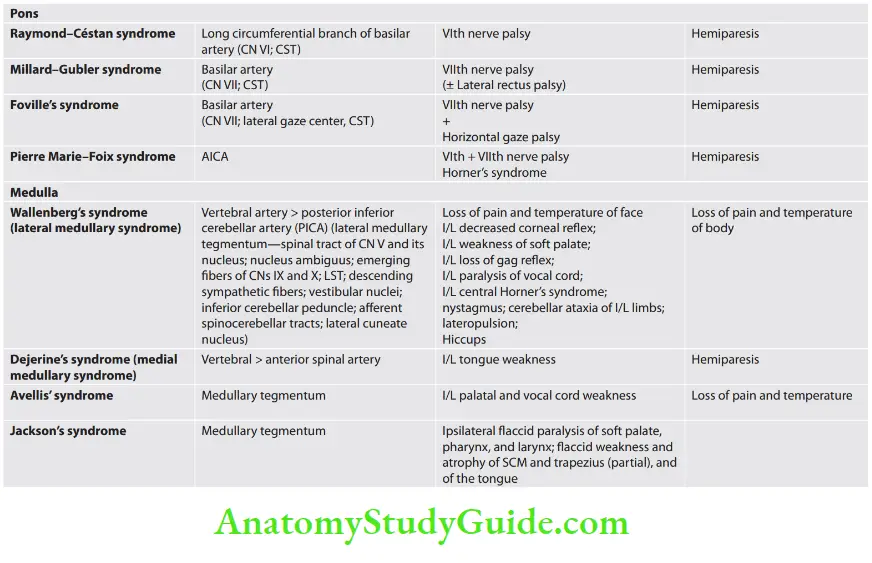
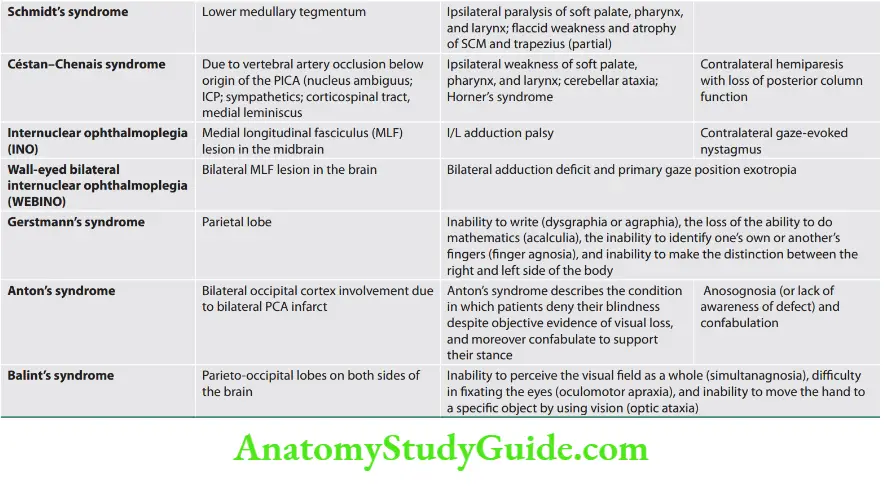

Lateral medullary syndrome
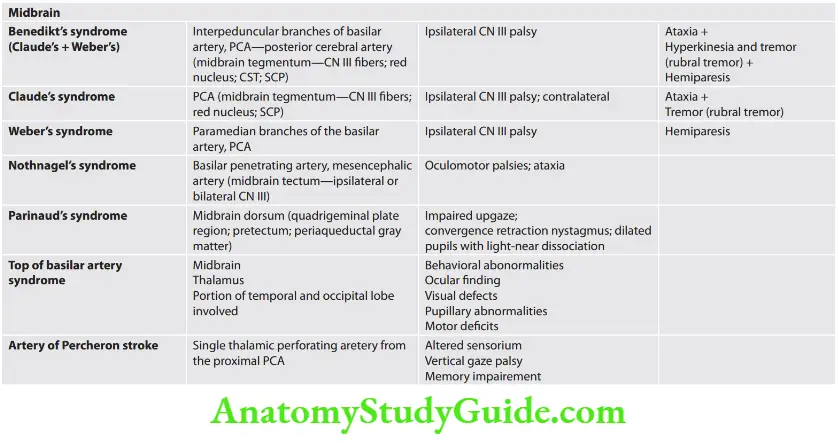
Question 22. Write a short note on the lateral medullary syndrome.
Answer:
It is also known as Wallenberg’s syndrome and is due to occlusion of the posterior inferior cerebellar artery or vertebral artery.
Localization/structures involved and associated symptoms are presented.

Oxfordshire Community Stroke Project (OCSP)—Bamford classification:
- TACS (Total anterior circulation syndrome)
- Hemianopia, hemiparesis, and higher cortical dysfunction
- PACS (Partial anterior circulation syndrome)
- Any two of the TACS criteria or isolated higher cortical dysfunction
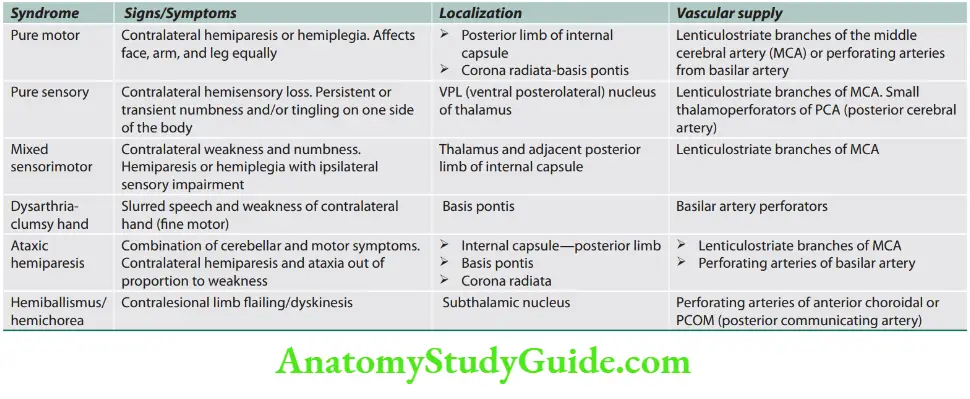
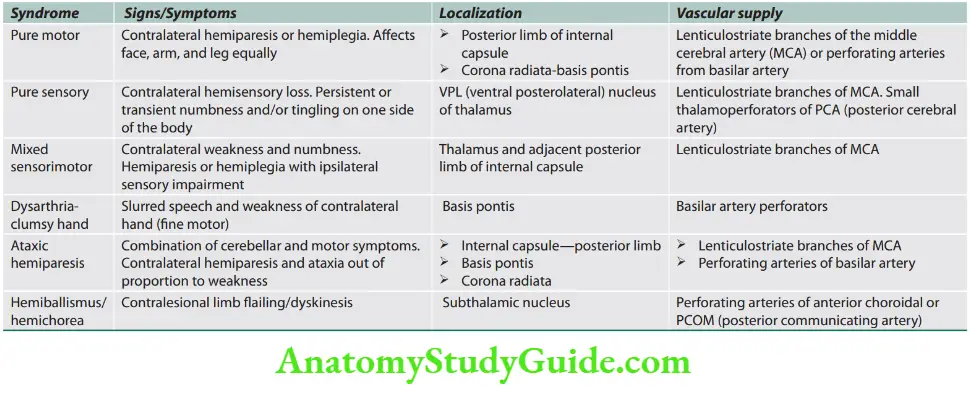
- LACS (Lacunar syndrome)
- Pure motor, pure sensory, sensorimotor strokes, dysarthria-clumsy hand syndrome, or ataxic hemiparesis
- POCS (Posterior circulation syndrome)
- Isolated hemianopia or brainstem or cerebellar signs
TOAST (the Trial of ORG 10172 in Acute Stroke Treatment) classification of subtypes of acute ischemic stroke.
TOAST classification of subtypes of acute ischemic stroke.
- Large artery atherosclerosis
- Cardioembolism
- Small vessel occlusion
- Stroke of other determined etiology
- Stroke of undetermined etiology
- Hypoperfusion and its Consequences
- A generalized reduction in CBF due to systemic hypotension (e.g., cardiac arrhythmia, cardiac arrest, myocardial infarction, or hemorrhagic shock) usually produces syncope.
- If low CBF persists for a longer duration, then infarction in the border zones (watershed areas) between the major and PCA distributions may develop (particularly if there are severe
stenosis of proximal carotid vessels). - In more severe instances, global hypoxia-ischemia causes widespread brain injury, the constellation of cognitive sequelae that ensues is called hypoxic-ischemic encephalopathy.
- Focal ischemia or infarction, conversely, is usually by thrombosis of the cerebral vessels themselves or by emboli from a proximal arterial source of the heart.
Carotid and vertebral artery dissection
- About 20% of cases of young (below the age of 40 years) stroke are due to dissection of the carotid or vertebral artery.
- It may develop sometimes as a sequel of trivial neck trauma or hyperextension (e.g., after whiplash, hair washing in a salon, or exercise).
- Predisposing factors include subtle collagen disorders (e.g., partial forms of Marfan’s syndrome).
- Most dissections occur in large extracranial neck vessels and are characterized by penetration of blood into the subintimal region of the vessel wall and forming a false lumen.
- However, it is the thrombosis within the true lumen due to tissue thromboplastin release which results in embolization from the site of dissection. This type of stroke sometimes develops days after the initial event.
- Symptoms: Pain in the neck or face is the clue for diagnosis.
Venous stroke
- Only 1% of strokes are due to venous thrombosis (within intracranial venous sinuses).
- Predisposing factors: It may occur in pregnancy, hypercoagulable states, and thrombotic disorders or with dehydration or malignancy.
- Consequences: Cortical infarction, seizures, and raised ICP.
Investigations/Diagnosis
Neuroimaging
- CT scans: To identify or exclude hemorrhage as the cause of stroke.
- They identify intraparenchymal hemorrhage, neoplasms, abscesses, and other conditions mimicking stroke.
- Brain CT scans in the first several hours after an infarction generally do not show any abnormality, and the infarct may not be detected reliably for 24–48 hours.
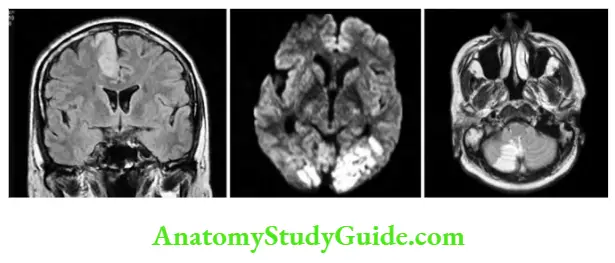
- CT may fail to show small ischemic strokes in the posterior fossa because of bone artifacts; small infarcts on the cortical surface may also be missed.
- Magnetic resonance imaging (MRI): Reliably documents the extent and location of infarction in all areas of the brain, including the posterior fossa and cortical surface. MRI is less sensitive than CT in detecting acute bleeding.
- Diffusion-weighted imaging and FLAIR (fluid-attenuated inversion recovery) imaging are more sensitive for early brain infarction than standard MR sequences or CT. It can identify ischemic penumbra and patients showing large regions of mismatch may be better candidates for acute revascularization.
Vascular imaging
- Many ischemic strokes are due to atherosclerotic thromboembolic disease of the major extracranial vessels.
- Detection of extracranial vascular disease can establish the diagnosis of ischemic stroke and, it may also help for specific treatments (e.g., carotid endarterectomy to reduce the risk of further stroke).
- The extracranial arterial disease can be noninvasively detected by duplex ultrasound, magnetic resonance angiography (MRA)
or CT angiography, or occasionally intra-arterial contrast radiography.
Cardiac investigations
Cardioembolism is responsible for 20% of ischemic strokes.
- The source of emboli includes atrial fibrillation, prosthetic heart valves, other valvular abnormalities, and recent myocardial infarction.
- Clinical examination and ECG help in identifying the source of cardiac emboli.
- However, it may be necessary to perform a transthoracic or transesophageal echocardiogram to confirm the cardiac source or for detecting an unsuspected source (e.g., endocarditis, atrial myxoma, intracardiac thrombus or PFO).
- Such findings will help in providing specific cardiac treatment.
Blood investigations
- Complete blood count, erythrocyte sedimentation rate (ESR), glucose, renal function test (RFT), liver function test (LFT), serum electrolytes, prothrombin time-international normalized ratio (PT-INR), activated partial thromboplastin time (aPTT)
- Workup for hypercoagulable states.
- CBC with differential and platelets
- PT/aPTT
- Fibrinogen
- Factor VIII
- Factor VII
- C-reactive protein
- Antithrombin III
- Protein C
- Protein S (total and free)
- Lipoprotein (a)
- Activated protein C resistance (APCR)
- Leiden factor V mutation if APCR negative
- Prothrombin G20210A mutation
- Antiphospholipid antibodies (Abs)
- Lupus anticoagulant
- Anticardiolipin Abs
- Anti-β-2-glycoprotein I Abs
- Antiphosphatidylserine Abs
- Methyltetrahydrofolate reductase
- MTHFR C677T and A1298C mutations
- Sickle cell screen
Paramedical personnel and the general public are taught how to make the diagnosis of stroke on a simple history and examination—FAST
Complications of Acute Stroke.
- Cerebral edema
- Cardiac arrhythmias, myocardial infarction, and neurogenic
- cardiac injury
- Chest infection
- Seizures
- Deep venous thrombosis/pulmonary embolism
- Pressure sores
- Urinary infection
- GI bleed: Stress ulcers, drug-induced
- Constipation
- Painful shoulder/contractures
- Depression and anxiety
- Increased ICT and coning
Question 23. Write a short essay/note on the complications of a stroke.
Answer:
Differential Diagnosis of Stroke
- Structural stroke mimickers
- Tumors: Primary and
- metastatic cerebral tumors
- Demyelinating disorders:
- Multiple sclerosis
- Subdural hematoma Peripheral nerve lesions
- Cerebral abscess (vascular or compressive)
- Functional stroke mimickers
- Seizure and postictal state Ménière’s disease/other vestibular disorders
- Focal seizures Encephalitis
- Syncope Wernicke’s encephalopathy
- Hypoglycemia Transient global amnesia
- Migrainous aura Metabolic encephalopathy
- Conversion disorders and
- other psychiatric disorders
- Treatment of Ischemic Stroke—“Time is Brain”
Treatments designed to reverse or lessen the amount of tissue infarction and improve clinical outcomes fall into six categories:
- IV thrombolysis,
- Endovascular techniques,
- Antithrombotic treatment,
- Medical support,
- Neuroprotection, and
- Stroke centers and rehabilitation.
1. Intravenous thrombolysis
- Intravenous (IV) administration for recombinant tissue plasminogen activator (r-tPA) within 3 hours of the onset of symptoms reduces disability and mortality from ischemic stroke.
- Indications and contraindications for r-tPA
Administration
- Administer at the rate of 0.9 mg/kg intravenously (maximum 90 mg) as 10% of the total dose as an IV bolus and the remainder of the dose as a continuous IV infusion over 60 minutes.
Diagnosis of stroke on a simple history and examination—FAST.
- Face: Sudden weakness of the face
- Arm: Sudden weakness of one or both arms
- Speech: Difficulty in speaking, slurred speech
- Time: Sooner the start of treatment, the better the outcome

- No other antithrombotic treatment for 24 hours.
- Avoid urethral catheterization for 2 hours.
- For a decline in neurologic status or uncontrolled blood pressure, stop an infusion, give cryoprecipitate, and reimage the brain emergently.
2. Endovascular mechanical thrombectomy: It is an alternative or adjunctive treatment of acute stroke in patients who are ineligible for, or have contraindications to, thrombolytics or in those who have failed to have vascular recanalization with IV thrombolytics?
3. Antiplatelet therapy
Platelet inhibition: Aspirin 150 mg + clopidogrel 75 mg daily. Note: Glycoprotein IIb/IIIa receptor inhibitor abciximab causes excess ICH and should be avoided in acute stroke.
Randomized studies of unfractionated heparin, low-molecular-weight heparins, or heparinoids have shown no proven benefits in the reduction of stroke-related mortality, stroke-related morbidity, early stroke recurrence, or stroke prognosis except in the case of cerebral venous thrombosis (CVT).
4. Anticoagulation for acute ischemic stroke:
Routine use of anticoagulation for acute ischemic stroke is not recommended.
The only indications for anticoagulants are:
Conditions with a potentially high risk of early cardiogenic re-embolization (e.g., atrial fibrillation) Symptomatic arteriosclerotic stenosis with crescendo TIAs Known hypercoagulable states Cerebral venous sinus thrombosis.
5. Medical support
Prevention of the common complications of bedridden patients:
Infections (pneumonia, urinary, and skin): Prophylactic antibiotic.
Deep venous thrombosis with pulmonary embolism: Subcutaneous heparin and pneumatic compression stockings to prevent DVT.
Others: Catheterization, Ryle’s tube (RT) feeding, maintenance of hygiene, etc.
Maintenance of blood pressure: Collateral blood flow within the ischemic brain is dependent on the blood pressure; hence, it is to be maintained.
- Treat if BP >220/120 mm Hg.
BP goals in the first 24–48 hours post-stroke:
- No IV tPA: <220/120 mm Hg
- IV tPA: <185/110 mm Hg.
First-line agents: labetalol, nicardipine, and clevidipine; second-line agent: nitroprusside [if diastolic blood pressure (DBP) >120 mm Hg].
Avoid excessive lowering of BP just to give t-PA—“Do not kill the penumbra to save the penumbra”.
Do not use antihypertensive drugs except—for left ventricular failure, aortic dissection, acute myocardial infarction, acute renal failure, and hypertensive encephalopathy.
Fever: It is detrimental and should be treated with antipyretics and surface cooling.
Serum glucose should be monitored and kept at <6.1 mmol/L (110 mg/dL) using an insulin infusion if necessary.
Decreasing intracranial tension (ICT): By water restriction and IV mannitol, oral glycerol, head end elevation.
Hemicraniectomy (craniotomy and temporary removal of part of the skull) markedly reduces mortality.
People with MCA infarction who meet all of the criteria below should be considered for decompressive hemicraniectomy.
They should be referred within 24 hours of the onset of symptoms and treated within a maximum of 48 hours:
Aged 60 years or under
Clinical deficits suggestive of infarction in the territory of the MCA with a score on the National Institute of Health Stroke Scale (NIHSS) of above 15
Decrease in the level of consciousness to give a score of 1 or more on item 1a of the NIHSS
Signs on MRI of an infarct of at least 50% of the MCA territory, with or without additional
Indications and contraindications for recombinant tissue plasminogen activator (r-tPA).
Indications for r-Tpa
- Clinical diagnosis of stroke
- The onset of symptoms to time of drug administration ≤3 hours (time window: 3–4.5 hours)
- Age ≥18 years
Contraindications for r-tpa
- CT scan showing no hemorrhage or infarct size >1/3rd of the middle cerebral artery (MCA) territory.
- Consent by patient or surrogate
Contraindications for r-tPA
- Sustained BP >185/110 mm of Hg despite treatment
- Platelets <100,000/μL
- Hematocrit <25%
- Glucose <50 or >400 mg%
- Use of heparin/warfarin within 48 hours and prolonged activated partial thromboplastin time (aPTT) or international normalized ratio (INR) Prior intracranial hemorrhage Prior stroke or head injury within 3 months
- Major surgery in the preceding 14 days
- Minor stroke symptoms
- GI bleeding in the preceding 21 days
- Recent myocardial infarction (within 3 months)
- Coma or stupor
- Pregnancy
- Age >80 years
Infarction in the territory of the anterior or PCA on the same side, or Infarct volume >145 cm 3 as shown on diffusion-weighted MRI.
Control of intracranial pressure: In cerebellar strokes, even small amounts of cerebellar edema can acutely increase ICP or directly compress the brainstem.
Prophylactic suboccipital decompression of large cerebellar infarcts before brainstem compression is very useful.
Endovascular treatment approaches:
- EKOS (ultrasound-enhanced thrombolysis)
- Neuroflo (perfusion augmentation)
- Mechanical endovascular thrombectomy
6. Statins: Atorvastatin, rosuvastatin—10–20 mg
7. Neuroprotection: Drugs that block the excitatory amino acid pathways (nimodipine, magnesium, lubeluzole, basic fibroblast growth factor, and citicoline) protect neurons and glia in animals, but they have not yet been proven to be beneficial in humans.
8. Rehabilitation:
Proper rehabilitation of the stroke patient includes early physical, occupational, and speech therapy.
It is directed toward educating the patient and family about the patient’s neurologic deficit, preventing the complications of immobility (e.g., pneumonia, DVT and pulmonary embolism, pressure sores of the skin, and muscle contractures), and providing encouragement and instructions in overcoming the deficit.
The goal of rehabilitation: It is to return the patient to home and to maximize recovery.
The use of restraint therapy (immobilizing the unaffected side) has been shown to improve hemiparesis following stroke, even years following the stroke.
Secondary Prevention of Stroke and Transient Ischemic Attack
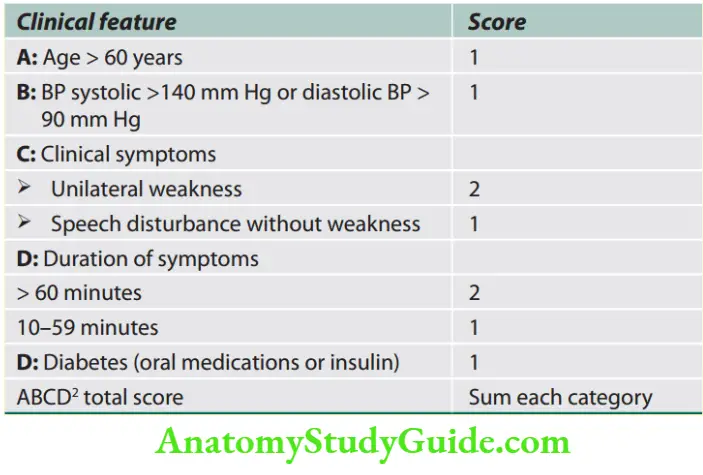
ABCD2 score can help in identifying stroke risk.
A score of <4 is associated with a minimal risk whereas >6 is a high risk for a stroke
Treatment of Atherosclerosis
Risk factors: Treatment of hypertension, diabetes, dyslipidemia, and cessation of smoking.
Patients with higher risk scores need to be on anticoagulation while for those with lower scores antiplatelet agents would suffice.
Atorvastatin: At the dose of 80 mg/day is useful both in secondary and primary prevention even with normal lipid levels.
Antiplatelet agents: Aspirin, clopidogrel or dipyridamole are used. Anticoagulation therapy for embolic stroke: Anticoagulation (INR range, 2–3) in patients with chronic nonvalvular (nonrheumatic) atrial fibrillation prevents cerebral embolism and is safe.
Warfarin, apixaban, dabigatran, rivaroxaban, and edoxaban are equally effective.
Calculation of CHADS2 score: One point for age > 75 years, one point for hypertension, one point for congestive heart failure (CHF), one point for diabetes, and two points for stroke or TIA; the sum of points is the total CHADS2 score.
Treatment of Carotid Atherosclerosis
High-grade stenosis of the carotid artery: Patient with recent symptomatic (within 2 weeks of symptoms) hemisphere ischemia, high-grade stenosis (>70%) in the appropriate internal carotid artery, and an institutional perioperative morbidity and mortality rate of 6% generally should undergo carotid endarterectomy.
Low-grade stenosis of the carotid artery: For asymptomatic stenosis and symptomatic low-grade stenosis (50–70%), medical therapy for the reduction of atherosclerosis risk factors, including cholesterol-lowering agents and antiplatelet medications, is generally recommended.
Endovascular therapy: Balloon angioplasty coupled with stenting to open stenotic carotid arteries and maintain their patency.
Treatment of Intracranial Atherosclerosis
- Symptomatic lesions with intracranial angioplasty and stenting.
- Dural sinus thrombosis: By short-term anticoagulants, regardless of the presence of ICH, for venous infarction following sinus thrombosis.
Causes for young stroke.
- Cardiac
- Congenital heart disease, patent foramen ovale
- Atrial myxoma
- Atrial fibrillation and other arrhythmia
- Cardiomyopathy, myocarditis, myocardial infarction
- Cardiac surgery, cardiac catheterization
- Endocarditis, rheumatic heart disease
- Prosthetic valve
Hematologic
- Sickle cell disease, iron deficiency anemias, polycythemia vera
Hypercoagulable states
- Inherited prothrombotic states, protein C and S deficiency, antithrombin III deficiency, factor V Leiden gene mutation, prothrombin gene mutation
- Antiphospholipid syndrome
- Hyperhomocysteinemia
- Myeloproliferative disorders (e.g., leukemia, lymphoma)
- Pregnancy
- Exposure to hormonal treatments such as anabolic steroids and erythropoietin
- Nephrotic syndrome
Vascular
Noninflammatory
- Arterial dissection
- Secondary to connective tissue disease (Ehlers–Danlos, Marfan)
- Moyamoya disease
- Hypertension
- Radiation vasculopathy
- Vasculitis and postinfectious vasculopathy
- Migraine
- Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) Fibromuscular dysplasia, Susac’s syndrome, Sneddon’s syndrome, Fabry’s disease
Inflammatory
- Takayasu arteritis
- Giant cell arteritis
- Kawasaki disease
- Polyarteritis nodosa
- Human immunodeficiency virus (HIV)
- Bacterial meningitis
- Intracranial Hemorrhage
Question 24. Describe the risk factors, etiopathogenesis, clinical features, investigations, diagnosis, and management of cerebral/intracerebral hemorrhage. (or) Describe the etiology, clinical features, investigations, and management of hemorrhagic stroke.
Answer:
This includes:
- intracerebral and cerebellar hemorrhage,
- subarachnoid hemorrhage (SAH), and
- subdural and extradural hemorrhage/hematoma.
- It causes about 10% of acute stroke events.
It is usually due to the rupture of a blood vessel within the brain parenchyma but may also occur in association with a SAH if the artery ruptures into the brain substance as well as into the subarachnoid space.
Hypertensive intraparenchymal hemorrhage (hypertensive hemorrhage or hypertensive intracerebral hemorrhage) usually results from spontaneous rupture of a small penetrating artery deep in the brain.
The most common sites are the basal ganglia (especially the putamen), thalamus, cerebellum, and pons.
Cortical bleeds are rare.
Pontine hemorrhages: 3Ps namely pinpoint pupil, hyperpyrexia, paralysis (quadriplegia).
There are prominent decerebrate rigidity and “pinpoint” (1 mm) pupils that react to light.
There is impairment of reflex horizontal eye movements evoked by head turning (doll’s head or oculocephalic maneuver) or by irrigation of the ears with ice water.
Primary versus secondary hemorrhage (hemorrhage that frequently occurs also into an area of brain infarction)—may be difficult to distinguish from primary intracerebral hemorrhage both clinically and radiologically.
Consequences of Hemorrhage
Immediate cessation of function: The sudden entry of blood into the brain parenchyma causes immediate cessation of function in that area as neurons are structurally disrupted with the splitting of white matter fiber tracts.
Mass Effect: The hemorrhage may expand over the first minutes or hours, or it may produce a rim of cerebral edema.
Progression of neurological deficit occurs due to the mass effect.
If large, they can cause a shift of the intracranial contents, producing transtentorial coning and sometimes rapid death.
Resorption of hematoma: If the patient survives, the hematoma is gradually absorbed, and produces a hemosiderin lined slit in the brain parenchyma.
Causes of Intracranial Hemorrhage
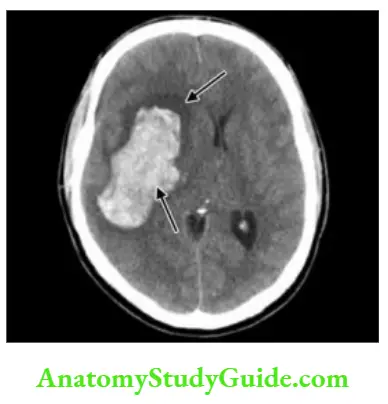
Clinical Features
- Clinical presentation depends upon which arterial territory is involved and the size of the lesion.
- Rapid-onset: Occurs over minutes.
- Most common presentation: Weakness of the face or arm, or disturbance of speech.
- Focal deficit of brain function: Affects an identifiable area of the brain and is “negative” in character (i.e., abrupt loss of function without positive features such as abnormal movement).
Features that suggest the location of the lesion are:
- The lesion in the cerebral hemisphere: Usually characterized by unilateral motor deficit, a higher cerebral function deficit such as aphasia or neglect, or a visual field defect.
- The lesion in the brainstem or cerebellum: Ataxia, diplopia, vertigo, and/or bilateral weakness.
- Intracerebral hemorrhage: Combination of severe headache and vomiting at the onset of the focal deficit.
Management/Treatment
Any identified coagulopathy should be reversed as soon as possible.
- For patients taking VKAs (vitamin K antagonists), rapid reversal of coagulopathy can be achieved by infusing prothrombin complex concentrates which can be administered quickly, followed by fresh frozen plasma and vitamin K.
- When ICH is associated with thrombocytopenia (platelet count < 50,000/μL), transfusion of fresh platelets is indicated.
Control of BP
- BP >185/110 increases the size of the hematoma by more bleeding. Hence, maintain a mean arterial pressure (MAP) < 130 mm Hg, unless an increase in ICP is suspected.
- In patients who have ICP monitors in place, keep the cerebral perfusion pressure (MAP-ICP) > 60 mm Hg (i.e., one should lower MAP to this target if blood pressure is elevated).
- Blood pressure should be lowered with non vasodilating IV drugs such as nicardipine, labetalol, or esmolol.
Hyperosmolar therapy
- Mannitol: Bolus of 0.25 g/kg to 1 g/kg body weight.
- Hypertonic saline, concentrations ranging from 3 to 23.4%.
Hyperventilation decreases PaCO2, which can induce constriction of cerebral arteries by alkalinizing the cerebrospinal fluid (CSF).
Barbiturate coma
Pentobarbital is given in a loading dose of 10 mg/kg body weight followed by 5 mg/kg body weight each hour for three doses.
Hypothermia
Steroids are used for primary and metastatic brain tumors, to decrease vasogenic cerebral edema.
Treatment of raised intracranial tension
If the hematoma causes a marked midline shift of structures with consequent obtundation, coma, or hydrocephalus, osmotic agents coupled with induced hyperventilation can be instituted to lower ICP.
Once ICP is recorded, further hyperventilation and osmotic therapy can be tailored to the individual patient to keep cerebral perfusion pressure (MAP-ICP) above 60 mm Hg.
If ICP is still found to be high, CSF can be drained from the ventricular space and osmotic therapy continued.
Persistent or progressive elevation in ICP may prompt surgical evacuation of the clot.
Glucocorticoids are not helpful for edema from intracerebral hematoma.
Most cerebellar hematomas >3 cm in diameter will require surgical evacuation.
Transient Ischemic Attack
Question 25. Write a short essay/note on TIA including its definition.
Answer:
A transient ischemic attack is characterized by a brief episode of neurological dysfunction (sudden loss of function) in which symptoms and signs resolve completely after a brief period of 24 hours (usually within 30 minutes).
A transient ischemic attack is defined as a transient episode of neurologic dysfunction caused by the focal brain, spinal cord, or retinal ischemia, without acute infarction.
However, TIAs may herald a stroke.
It may be better to describe as symptoms <1 hour with no evidence of infarction.
It may have an infarct even with symptoms lasting a few hours (~50% of TIA patients have MRI evidence of ischemia).
Newer, neuroimaging-informed, operational definitions of TIA—“a brief episode of neurological dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than one hour, and without evidence of acute infarction”
Clinical Features
Hemiparesis and aphasia are the most common. Other features include
amaurosis fugax (sudden transient loss of vision in one eye), chemosensory loss, hemianopia visual loss, diplopia, vertigo, vomiting, choking, dysarthria, ataxia, etc.
Types of transient ischemic attack (TIA).
- Embolic TIA—recurrent, short-lasting episodes of stereotyped symptoms (shotgun TIA)
- Large artery TIA—longer-lasting less frequent episodes with varied symptoms
- Lacunar TIA
- Recurrent same side, same territory TIA is suggestive of vessel disease
- TIA in both anterior and posterior circulation is suggestive of cardioembolism
Diagnosis
Diagnosis of TIA often depends on its description. There may be clinical evidence of a source of embolus (e.g., carotid arterial bruit/stenosis, atrial fibrillation or other dysrhythmia, valvular heart disease/endocarditis, or recent myocardial infarction.
An underlying risk factor/condition may be present.
These include atheroma, hypertension, postural hypotension, bradycardia or low cardiac output, diabetes mellitus, etc.
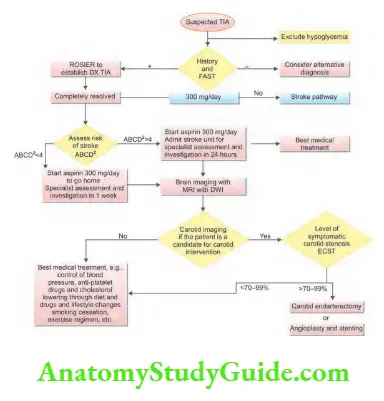
Treatment
Medical therapy and surgical treatment if appropriate. If patients have ABCD 2 score >4, or have had two recent TIAs (especially within the same vascular territory), they should be investigated and advised for secondary prevention of stroke.
Subarachnoid Hemorrhage
Question 26. Discuss the clinical manifestations and treatment of primary SAH. (or) Write a short essay/note on SAH.
Answer:
Subarachnoid hemorrhage is a pathologic condition in which the blood enters the subarachnoid space. SAH is less common than ischemic stroke or intracerebral hemorrhage.
It accounts for about 5% of strokes.
Etiology
- The most common cause of SAH is trauma.
- The most common cause of spontaneous SAH is an aneurysmal (saccular or “berry” aneurysm) bleed (65– 80%).
- SAH causes intense reductions in CBF, reduced cerebral autoregulation, and acute cerebral ischemia.
- The overall case fatality varied from 32 to 67%.
- Risk factors for SAH
Circle of Willis
- Circle of Willis is a circulatory anastomosis that supplies blood to the brain and surrounding structures. This anastomotic pathway is important for the preservation of brain function when major blood flow is disrupted in one of the major feeding vessels.
- Constituents’ arteries of the circle of Willis
- The MCAs, supplying the brain, do not form part of the circle of Willis.
Saccular (“berry”) aneurysm
Saccular aneurysms develop within the circle of Willis and adjacent arteries.
The three most common locations are—
- The terminal internal carotid artery,
- MCA bifurcation, and
- Top of the basilar artery.
- It can undergo spontaneous rupture and cause SAH.
- If the patient survives, but the aneurysm is not obliterated, the rate of rebleeding is about 20% in the first 2 weeks, 30% in the first month, and about 3% per year afterward.
- The annual risk of rupture for aneurysms <10 mm in size is 0.1%, and for aneurysms >10 mm in size is 0.5–1%.
Clinical Features of Subarachnoid Hemorrhage
Headache: Most common symptom (97%). Usually severe (the worst headache of my life) and sudden (thunderclap) in onset.
Few patients may have milder warning headaches (sentinel headaches) in 2–8 weeks preceding the major hemorrhage.
Headache may pulsate toward the occiput and sometimes may be felt as neck pain.
Occipital and posterior cervical pain may signal a posterior inferior cerebellar artery or anterior inferior cerebellar artery aneurysm.
If there is expanding MCA aneurysm, pain may be observed in or behind the eye and in the low temple.
Headache commonly occurs on physical exertion, straining, and sexual excitement.
- Vomiting may be present.
- Other symptoms: Decreased consciousness and alertness, seizure (10%) and stiff neck, etc.
- Rerupture/rebleed: The rupture of an untreated aneurysm in the first month following SAH is ~30%, with the peak in the first 7 days.
Risk factors for subarachnoid hemorrhage.
- Hypertension
- Cigarette smoking
- Oral contraceptives
- Diurnal variations in blood pressure
- Pregnancy and parturition
- Slight increased risk during lumbar puncture and/or cerebral angiography in patients with cerebral aneurysm
- Slightly increased risk with advancing age
- Alcohol consumption (debatable)
- Following cocaine abuse
- Increased incidence of polycystic kidneys, fibromuscular dysplasia of extracranial arteries, Ehlers–Danlos syndrome, moyamoya, AV malformations, and coarctation of aorta.
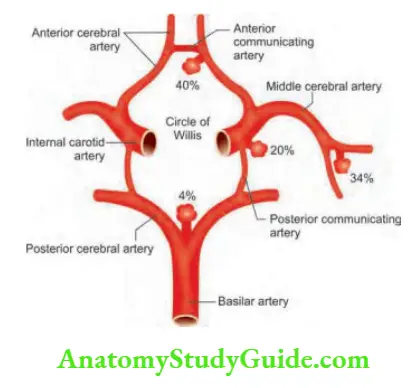
- Constituents arteries of the circle of Willis.
- Two internal carotid arteries (left and right)
- Anterior cerebral artery (left and right)
- Anterior communicating artery
- Posterior cerebral artery (left and right)
- Posterior communicating artery (left and right)
- The basilar artery is formed by the joining of two vertebral arteries
Physical examination:
- The patient is usually distressed and irritable, with photophobia.
- Neck stiffness (and a positive Kernig’s sign) due to subarachnoid blood may be present but this may take a few hours to develop.
- Focal hemisphere signs, e.g., intracerebral hematoma may cause hemiparesis or aphasia.
- Rarely, IIIrd nerve palsy may be developing due to local pressure from an aneurysm of the posterior communicating artery.
Fundoscopy: It may show subhyaloid hemorrhage which is canoe-shaped, (SAH plus subhyaloid hemorrhage = Terson’s syndrome) produced due to tracking of blood along the subarachnoid space around the optic nerve.
Investigations
Computed tomography scan: It shows blood in the subarachnoid space (if performed in the first few days), intracerebral hematoma, hydrocephalus, associated brain ischemia, and occasionally aneurysmal location.
Question 27. Write a short essay/note on CSF findings in SAH.
Answer:
Lumbar puncture: It should be performed when clinical suspicion of SAH is high, the CT scan does not reveal subarachnoid blood, and there is no mass effect.
- In the initial few hours, CSF will be uniformly blood-stained.
- Xanthochromia appears within 6 hours of SAH and persists for nearly 2 weeks.
- Naked inspection of supernatant CSF is usually sufficiently reliable for diagnosis of SAH.
- Spectrophotometry to estimate bilirubin in the CSF released from lysed cells confirm the diagnosis.
Angiography: It is required to locate the site of the bled aneurysm and details that are needed to ligate the aneurysm by the neurosurgeon.
- It should be done in all patients fit for surgery (i.e., aged < 65 years and who are not in a coma).
- Magnetic resonance angiography: It will accurately identify aneurysms > 5 mm in size.
CT angiography: It is a rapid readily available, less invasive alternative to catheter angiography.
It is equivalent to conventional angiography for detecting large aneurysms.
ECG changes: Prolonged QRS complex, increased QT interval, and prominent “peaked” or deeply inverted symmetric T waves are usually secondary to the ICH (cerebral T waves)
Differential Diagnosis of Subarachnoid Hemorrhage
- Migraine
- Acute bacterial meningitis
- Cervical arterial dissection
Complications
- Obstructive hydrocephalus: Due to blood in the subarachnoid space. It may be asymptomatic but may cause deteriorating consciousness following SAH. Shunting may be required.
- Arterial spasm (cause of coma or hemiparesis): It is a serious complication and is a poor prognostic feature.
- Hyponatremia.
Question 28. Write a short essay/note on the treatment of SAH.
Answer:
Management
- Immediate treatment is absolute bed rest (for 4 weeks) and supportive measures.
- These include protecting the airway, managing blood pressure and raised ICP, pain management, and sedation.
- It should be discussed urgently with a neurosurgical team. The patient is advised to gradually resumption of physical activities after recovery.
Control of hypertension: In conscious patients without raised ICP, active treatment of hypertension is necessary. A target MAP of 130mm, Hg is recommended.
Drugs include labetalol, esmolol, or nicardipine. Sodium nitroprusside should be avoided because may cause an increase in ICP.
Avoid strictly: Hypovolemia, hypotension, hyperthermia, hyperglycemia, and hyponatremia.
Calcium channel blocker: Nimodipine (30–60 mg IV for 5–14 days, followed by 360 mg orally for a further 7 days) provides a modest but significant improvement in outcome by reducing cerebral arterial vasospasm.
It is given for 3 weeks to prevent delayed ischemia in the acute phase and decreases mortality.
Prophylactic anticonvulsants
Interventional management: Early aneurysm repair prevents re-rupture/rebleeding and improves blood flow.
- An aneurysm can be “clipped” by a neurosurgeon or “coiled” by an endovascular surgeon.
- Insertion of platinum coils into an aneurysm: If angiography shows an aneurysm (the most common cause of SAH), endovascular treatment by inserting platinum coils via a catheter in the aneurysm sac to promote thrombosis and ablation of the aneurysm, is the first-line treatment.
- Surgical clipping of the aneurysm neck also helps in reducing the risk of recurrence (both early and late).
- Endovascular coiling has fewer perioperative complications and better outcomes than surgery.
- For unruptured aneurysms > 8 mm in diameter, the risk of treatment is less than the risk of hemorrhage if not treated.
Treatment of complications: Managing delayed cerebral ischemia due to vasospasm (may be treated with vasodilators), treating obstructive hydrocephalus (may require drainage via a shunt), treating hyponatremia (managed by fluid restriction), and systemic complications associated with immobility, such as chest infection venous thrombosis and preventing pulmonary embolus.
Question 29. Identify the nature of the cerebrovascular accident based on the temporal evolution and resolution of the illness.
Answer:
The differences between hemorrhagic, thrombotic, and embolic strokes.
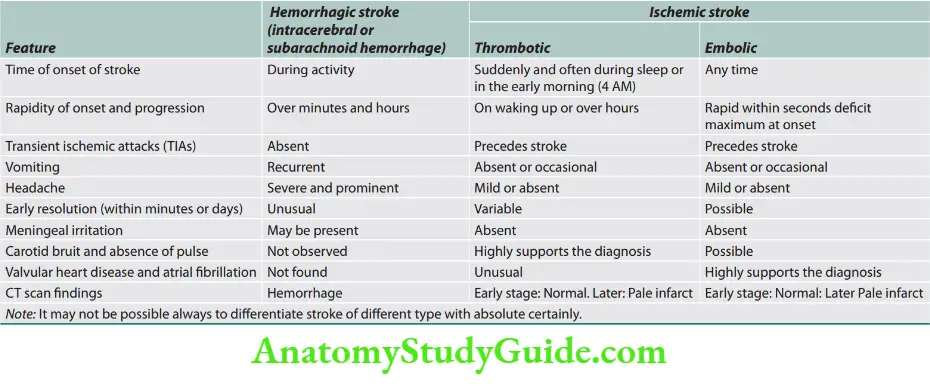
Cerebral Venous Thrombosis
Question 30. Write a short note about cerebral venous thrombosis.
Answer:
- It is the thrombosis of the draining venous sinuses of the cerebral cortex and the least common of all other types of cerebrovascular accidents (1–2% of strokes in young adults).
- Thrombosis most commonly involves superior sagittal sinus and it may extend into transverse and sigmoid sinuses.
- Thrombosis of the deep venous system (e.g., internal cerebral veins, a vein of Galen, or straight sinus) is less common.
Age and gender: It frequently affects young adults and children. More common in women than men with an F: M ratio of 3:1.
It may be due to the presence of gender-specific risk factors such as pregnancy, puerperium, oral contraceptive pills, and hormone replacement therapy (HRT).
One of the leading causes of maternal mortality and morbidity with cases of puerperal CVT occurring 10–12 times more frequently in India than the West.
Causes of Cerebral Venous Thrombosis

Pathophysiology
Two basic mechanisms are responsible for the development of the clinical features in CVT.
- Thrombosis of cerebral veins or sinuses leading to cerebral parenchymal lesions or dysfunctions.
- Occlusion of the dural venous sinus causes reduced absorption of CSF and raised ICP.
Obstruction of venous structures → increased venous pressure → decreased capillary perfusion → increased cerebral blood volume → disruption of blood-brain barrier → plasma leakage into interstitial space → cerebral edema, parenchymal changes, and venous hemorrhage.
Clinical Features
The clinical presentation of CVT is highly variable and it may present as acute/ subacute/ chronic.
Symptoms and signs: Depends on the sinuses involved, the place of occlusion, involvement of cortical veins and presence of collaterals, parenchymal changes, and the patient’s age.
Symptoms are grouped under three major clinical syndromes:
- Isolated intracranial hypertension syndrome (headache with or without vomiting, papilledema, and visual problems)
- Focal syndrome (focal deficits or seizures or both)
- Encephalopathy (multifocal signs, mental status changes, stupor or coma).
Diagnosis
CT or MR venography: Direct signs of CVT in CT include:
Dense triangle sign—seen as hyperdensity with the triangular shape at the posterior end of the superior sagittal sinus caused by venous thrombus.
Empty delta sign—seen in head CT with contrast as a triangular pattern of contrast enhancement surrounding a central region lacking contrast enhancement.
Cord sign—as linear or curvilinear hyperdensity over the cerebral cortex caused by thrombosed cerebral veins.
CT or MRI to detect secondary parenchymal lesions.
CSF examination: To rule out meningitis or SAH.
Workup for the prothrombotic state.
Treatment
- Treatment should be started as soon as the diagnosis is confirmed and consists of:
- Treatment of the underlying cause when known.
- Control of seizures and intracranial hypertension.
- Antithrombotic therapy.
- The main treatment option to achieve these goals is anticoagulation using heparin or LMWH (IV/SC).
- It is recommended that anticoagulant therapy with warfarin should be given for a minimum of 3 months after acute CVT aiming at an INR target of 2.5 (2–3).
Demyelinating Diseases
Question 31. What are the common demyelinating diseases?
Answer:

Multiple Sclerosis
Question 32. Write a short essay on multiple sclerosis (MS) and its clinical features.
Answer:
Chronic autoimmune T-cell-mediated inflammatory disorder with selective destruction of myelin of the central nervous system (CNS). It has a relapsing-remitting or progressive course.
The neurological deficits are disseminated in time and space.
It is termed multiple sclerosis because of the multiple scarred areas (well-demarcated gray areas termed plaques) of demyelination throughout the brain and spinal cord observed on macroscopic examination.
The plaques are crucial for diagnosis. The peripheral nervous system (PNS) is generally spared.
Epidemiology
- Gender: Female preponderance and female to male ratio is 2:1.
- Age: Usually present between 20 and 40 years of age and is rare after 60 years of age.
Pathophysiology
Both genetic and environmental factors play a causative role.
- Genetic factors: The risk of familial recurrence in MS is 15%, with the highest risk in first-degree relatives. Multiple genes interact and it has a complex polygenic inheritance pattern. HLA-DR2 association. Other autoimmune disorders occur in patients with MS and their relatives, indicating a genetic predisposition to autoimmunity.
- Environmental factors: Epidemiologic evidence supports the role of environmental exposure in MS but these factors are still largely unknown. For example, sunlight exposure, vitamin D, and exposure to Epstein–Barr virus (EBV). Viral infections can precipitate MS relapses in genetically susceptible individuals.
Etiology and Pathogenesis
- The etiology is not known.
- It is probably a T-cell-mediated autoimmune disease causing an inflammatory process within the white matter of the brain and spinal cord.
MS risk correlates with high socioeconomic status, which may reflect improved sanitation and delayed initial exposures to infectious agents.
Clinical Manifestations
- No single group of signs or symptoms is diagnostic of MS.
- Onset: May be abrupt/insidious, vary from trivial to severe form.
- Presentation varies and depends on the anatomical site of lesions and severity.
- It has been called the modern “great imitator”. MS commonly involves the pyramidal tract, optic nerve, posterior cord, cerebellum, and medial longitudinal fasciculus (MLF).
- Common symptoms in MS: Disability and neurological impairments gradually accumulate over the years.
Clinical course: Main clinical patterns are:
Relapsing-remitting MS (RRMS) (85–90%): Discrete attack and complete recovery.
Secondary progressive MS (SPMS): Begins as RRMS, with clinical steady deterioration at some stage.
Primary progressive MS (PPMS) (10–15%): Steady function decline from onset.
Progressive relapsing MS (PRMS)
Malignant MS/Aggressive MS refers to a disease with a rapidly progressive course, leading to significant disability in multiple neurologic systems in a relatively short time after disease onset.
Tumefactive MS is an acute tumor-like MS variant in which some patients with demyelinating disease present with large (>2 cm) acute lesions, often associated with edema or ring enhancement.
Diagnosis
The diagnosis of MS is usually easily made in a young adult with relapsing and remitting symptoms referable to different areas of CNS white matter.
No single diagnostic test for a definitive diagnosis of MS.
Magnetic resonance imaging
- Characteristic abnormalities are found in >95% of patients.
- The lesion appears as a focal area of hyperintensity on T2-weighted.
- Typical lesions are oval up to 2 cm in diameter and frequently orientated perpendicular to the lateral ventricular surface, corresponding to the pathologic pattern of perivenous demyelination (Dawson’s fingers).
- Lesions are multifocal (periventricular, juxtacortical, infratentorial, or spinal cord).
Evoked potential (EP) or evoked responses
Assesses function in afferent (visual, auditory, and somatosensory) or efferent (motor) CNS pathways, abnormalities on one or more EP modalities occur in 80–90% of MS patients.
However, they are less important than MRIs.
Cerebrospinal flid examination
- CSF cell count may be raised (5–60 mononuclear cells/mm)— mononuclear cell pleocytosis.
- Increased immunoglobulin G (IgG), total CSF protein, CSF IgG index.
- Protein electrophoresis: Oligoclonal IgG bands detected by agarose gel, but these are not specific.
McDonald’s criteria for MS are presented.
Treatment of Multiple Sclerosis
- Treatment of acute attack
Intravenous methylprednisolone 500–1,000 mg/day for 3–5 days, followed by a course of oral prednisone beginning at a dose of 60–80 mg/day and gradually tapered over 2 weeks.
High-dose cyclophosphamide has been used for induction therapy to stabilize aggressive MS.
Plasma exchange: 5–7 changes are done with 40–60 mL/kg per exchange alternate days for 14 days for cases that are unresponsive to glucocorticoids.
Common presentations of multiple sclerosis.
- Optic neuropathy (neuritis)
- Relapsing/remitting sensory symptoms, e.g. Lhermitte’s sign (barber chair phenomenon, is an electrical sensation that runs down the back and into the limb on neck flexion)
- Subacute painless spinal cord lesion
- Acute brainstem syndrome: Sudden diplopia, and vertigo with nystagmus
Subacute loss of function of upper limb (dorsal column deficit):
- Clumsy/useless hand or limb
- Fatigue, cognitive impairment
- Sixth cranial nerve palsy, wall-eyed bilateral
- internuclear ophthalmoplegia (WEBINO)
- Unsteadiness or ataxia. Charcot’s triad of dysarthria, ataxia, and tremor
- Urinary symptoms—bladder hyperreflexia
- Neuropathic pain, fatigue, spasticity, depression, sexual dysfunction
Uhthoff’s phenomenon: Worsening of neurologic symptoms (especially vision) when the body gets overheated

Disease-modifying drugs: For prophylaxis of RRMS and SPMS after the acute stage has passed. No effective treatment for PPMS. These include interferon (IFN)-β (1a-1b), glatiramer acetate, natalizumab, mitoxantrone hydrochloride, rituximab, siponimod, fingolimod, and cladribine.
Ocrelizumab, a recombinant human anti-CD20 monoclonal antibody designed to optimize B-cell depletion, is the first drug to significantly reduce the risk of disability progression among patients with PPMS.
Symptomatic therapy
- Weakness (heat-induced): K+ channel blockers—4-aminopyridine, 3,4-diaminopyridine
- Spasticity/spasm: Physiotherapy, avoid trigger factors. Drugs like baclofen, diazepam, tizanidine, and dantrolene can be used.
- Pain:
- Anticonvulsant: Carbamazepine, phenytoin, gabapentin, pregabalin
- Antidepressants: Amitriptyline/nortriptyline, desipramine, venlafaxine
- Antiarrhythmic: Mexiletine
- Bladder care.
The differences between MS and neuromyelitis optica (NMO) are listed. It is important to differentiate as IFN is contraindicated in NMO.
Seizures And Epilepsy
Question 33. Classify seizure/epilepsy. Discuss the evaluation of adult seizures and management of seizures.
(or)
Describe the types, etiology, clinical features, investigations (evaluation), and management of epilepsy. Enumerate antiepileptic agents.
(or)
Classify epilepsy. Describe the clinical features and treatment of grand mal (generalized tonic-clonic) epilepsy.
(or)
Write a short essay/note on:
- Enumerate the causes and investigation of a seizure. Mention drugs used to treat grand mal epilepsy.
- Temporal lobe epilepsy.
- Management of complex partial seizures.
- Complications of status epilepticus (SE).
- Causes of convulsion.
- Primary epileptic disorders.
- Complications of seizures.
- Define complex partial seizure.
Answer:
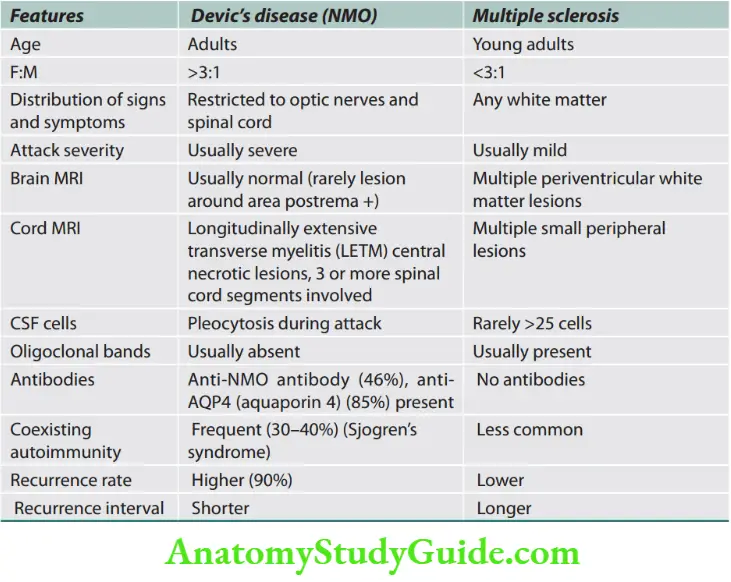
Seizure: It is a paroxysmal event due to abnormal, excessive, hypersynchronous discharges from an aggregate of CNS neurons.
Epilepsy is a chronic neurological disorder defined as the occurrence of 2 or more unprovoked or reflex seizures at least 24 hours apart, the occurrence of a single unprovoked or reflex seizure in an individual with an underlying condition that increases the risk of subsequent seizures (e.g., a brain tumor), or the presence of an epilepsy syndrome.
An individual with a single seizure or recurrent seizures due to correctable or avoidable circumstances does not necessarily have epilepsy.
Classification of Seizures
Question 34. Classify seizure/epilepsy.
Answer:
Seizures are classified as “simple” if there is no impairment of consciousness or as “complex” if an alteration in consciousness occurs. Seizures may be either focal or generalized.
- Focal seizures: They originate within networks limited to one cerebral hemisphere across both cerebral hemispheres. They are usually associated with structural abnormalities of the brain.
- Generalized seizures: In contrast to focal, they may result from cellular, biochemical, or structural abnormalities that have a more widespread distribution. However, there are clear exceptions in both cases.
- Most seizures last seconds to minutes.
- Status epilepticus (discussed later).
Focal Seizures
Focal Seizures without Dyscognitive Features
- In this type, consciousness is fully preserved during the seizure and the clinical manifestation is relatively simple.
- Focal seizures can cause motor, sensory, autonomic, or psychic symptoms without an obvious alteration in consciousness.
- A focal motor seizure arising from the right primary motor cortex near the area controlling hand movement will note the onset of involuntary movements of the contralateral, left hand.
Three additional features of these seizures are:
Question 35. Write a short note on Jacksonian epilepsy.
Answer:
Jacksonian march: Representing the spread of seizure activity over a progressively larger region of the motor cortex.
In Jacksonian seizures, the march of symptoms occurs where if it is motor, clonic jerking starts at a point.
For example, it may start at the face, spread to the upper limb, then to the lower limb, and then on to the opposite side to become a generalized fit.
This denotes the path of the spread of the epileptic activity and is called Jacksonian spread/march.
Question 36. Write a short note on Todd’s paralysis.
Answer:
Todd’s paralysis or localized paresis: It is a condition characterized by brief, temporary paralysis that follows a seizure.
It may occur for minutes to hours in the involved region following the seizure.
Epilepsia partialis continua: Analogous to partial SE. It is often quite refractory to medical therapy.
Other manifestations:
Changes in somatic sensation (e.g., paresthesia), vision (flashing lights or formed hallucinations), equilibrium (sensation of ailing or vertigo), and autonomic function (flushing, sweating, piloerection).
Focal Seizures with Dyscognitive Features
Focal seizure activity may be accompanied by a transient impairment of the patient’s ability to maintain normal contact with the environment.
The patient is unable to respond to visual or verbal commands during the seizure and has impaired recollection or awareness of the ictal phase.
The seizures frequently begin with an aura (i.e., focal seizures without dyscognitive features) that is stereotypic for the patient.
The start of the ictal phase is often a sudden behavioral arrest or motionless stare, and this marks the onset of the event for which the patient will be amnesic.
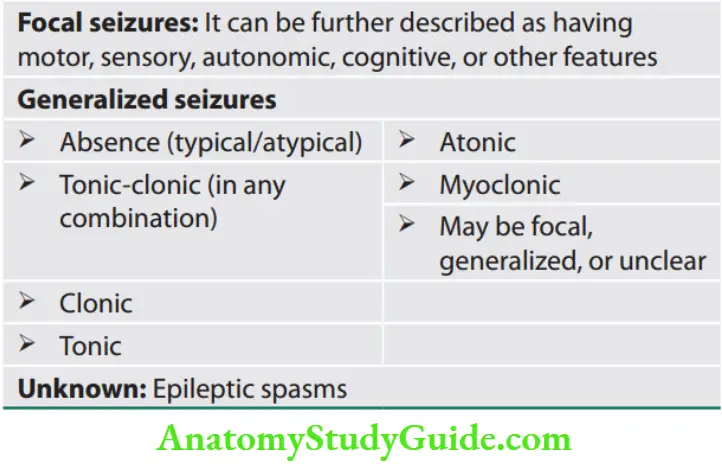

Behavioral arrest is usually accompanied by oromandibular or hand automatisms, which are involuntary, automatic behaviors that have a wide range of manifestations.
Automatisms may consist of very basic behaviors such as chewing, lip smacking, swallowing, or picking movements of the hands or more elaborate behaviors such as a display of emotion or running.
Complex Partial Seizures (Temporal Lobe Seizures)
Focal seizures arising from the temporal or frontal cortex may also cause alterations in hearing, olfaction, or higher cortical function (psychic symptoms-like to a sensation of unusual intense odors (e.g., burning rubber or kerosene) or sounds (crude or highly complex sounds) or illusions that objects are growing (metamorphopsia) smaller (micropsia) or larger (macropsia).
When such symptoms precede focal seizures with dyscognitive features or secondarily generalized seizures, these seizures serve as a warning or aura.
Generalized Seizures
- Generalized seizures arise from both cerebral hemispheres simultaneously.
- It may be practically defined as bilateral clinical and electrographic events without any detectable focal onset.
- Though they arise at some point in the brain they rapidly engage neuronal networks in both cerebral hemispheres.
Absence Seizures (Petit Mal Seizures)
- Sudden, brief lapses of consciousness without loss of postural control.
- It lasts for only a few seconds and consciousness returns rapidly and there is no postictal confusion.
- Usually accompanied by subtle, bilateral motor signs such as rapid blinking of the eyelids, chewing movements, or small amplitude, clonic movements of the hands.
- Absence seizures can be typical or atypical.
- Onset is usually in childhood (ages 4–8 years) or early adolescence and is the main seizure type in 15–20% of children with epilepsy.
- May occur hundreds of times in a day without the knowledge of parents or the child.
- The first clue may be often unexplained “daydreaming” and a decline in school performance recognized by a teacher.
- Electroencephalography (EEG): Generalized, symmetric, 3-Hspike-and-wave discharge that begins and ends suddenly, superimposed on a normal EEG background which can be provoked by hyperventilation.
Generalized Tonic-Clonic Seizures (Grand Mal Seizures)
Question 37. Write a short essay/note on clinical features and treatment of grand mal (generalized tonic-clonic) epilepsy.
Answer:
- The main type of seizure is in 10% of all individuals with epilepsy.
- Usually begins abruptly without warning. Some may develop vague premonitory symptoms which are distinct from the stereotypic auras associated with focal seizures that generalize.
- Initially, there is a tonic contraction of muscles throughout the body leading to a loud moan or ictal cry, cyanosis, biting of the tongue, etc.
- Marked enhancement of sympathetic tone leads to increases in heart rate, blood pressure, and pupillary size.
- After 10–20 seconds, the clonic phase starts with superimposed relaxation which progressively increases until the end ofictal period.
- The postictal phase is characterized by unresponsiveness, muscular flaccidity, and excessive salivation, bladder or bowel incontinence.
- Patients gradually regain consciousness over minutes to hours with accompanying postictal confusion, headache, and muscle ache.
- EEG: Generalized high-amplitude, polyspike discharges in tonic phases which in the clonic phase are typically interrupted by slow waves to create a spike-and-wave pattern.
- Other variants include pure tonic and pure clonic types.
Atonic Seizures
- They are characterized by sudden loss of postural muscle tone lasting 1–2 seconds.
- Consciousness is briefly impaired, but there is usually no postictal confusion.
- They may cause only a quick head drop or nodding movement, while a longer seizure will cause the patient to collapse.
- They are rarely seen in isolation and are usually seen in association with known epileptic syndromes.
- EEG: Brief, generalized spike-and-wave discharges followed immediately by diffuse slow waves that correlate with the loss of muscle tone.
Myoclonic Seizures
- Sudden, brief jerky muscle contraction that may involve one part of the body or the entire body.
- Normal, common physiologic forms of myoclonus are the sudden jerking movement observed while falling asleep and hiccups.
- Most pathologic myoclonus are commonly seen in association with metabolic disorders, degenerative CNS diseases, or anoxic brain injury.
- Myoclonic seizures are the predominant feature of juvenile myoclonic epilepsy (JME).
- EEG: May show bilaterally synchronous spike-and-wave discharges synchronized with the myoclonus.
Epilepsy Syndromes
Epilepsy syndromes are disorders in which epilepsy is a predominant feature, and there is sufficient evidence to suggest a common underlying mechanism.
Juvenile Myoclonic Epilepsy
- Juvenile myoclonic epilepsy is a generalized seizure disorder of unknown cause that appears in early adolescence and is usually characterized by bilateral myoclonic jerks that may be single or repetitive.
- Myoclonic seizures are most frequent in the morning after awakening and can be provoked by sleep deprivation.
- Unless severe consciousness is preserved.
- Many patients also experience generalized tonic-clonic seizures (GTCSs), and up to one-third have absence seizures.
- The condition is otherwise benign, and although complete remission is uncommon, the seizures respond well to appropriate anticonvulsant medication.
- Lifelong treatment is necessary and sodium valproate is the drug of choice.
Lennox–Gastaut Syndrome
Lennox–Gastaut syndrome is seen in children between the ages of 1–8 and is characterized by the following triad.
- Multiple seizure types (usually including generalized tonic-clonic, atonic, and atypical absence seizures).
- EEG: Shows slow (<3 Hz) spike and wave discharges and a variety of other abnormalities.
- Impaired cognitive function in most but not all patients.
Lennox–Gastaut syndrome is an epileptic encephalopathy associated with CNS disease or dysfunction from a variety of causes, including developmental abnormalities, perinatal hypoxia/ischemia, trauma, infection, and other acquired lesions.
- The multifactorial nature of this syndrome suggests that it is a nonspecific response of the brain to diffuse neural injury.
- A similar syndrome in infancy that often evolves into Lennox–Gastaut syndrome is West syndrome, characterized by infantile spasms, Salaam attacks, other findings of cerebral dysfunction, and abnormal EEG pattern (hypsarrhythmia).
Mesial Temporal Lobe Epilepsy Syndrome
- Mesial temporal lobe epilepsy (MTLE) is the most common syndrome associated with complex partial seizures and is an example of symptomatic, partial epilepsy.
- Characteristic hippocampal sclerosis on MRI is an essential element in the pathophysiology of MTLE for many patients.
- Recognition of this syndrome is especially important because it tends to be refractory to treatment with anticonvulsants but responds extremely well to surgical intervention.
Febrile Seizures
- Usually occur between 3 months and 5 years of age and has a peak incidence between 18 and 24 months.
- It is GTCS in children during a febrile illness in the setting of common childhood infections. The seizure is likely to occur during the rising phase of the temperature curve (i.e., during the first day).
- A simple febrile seizure is a single, isolated event, brief and symmetric in appearance. Complex febrile seizures are characterized by repeated seizure activity, duration >15 minutes, or by focal features.
- Simple febrile seizures are not associated with an increase in the risk of developing epilepsy; while complex febrile seizures have a risk of 2–5%. Other risk factors include the presence of pre-existing neurologic deficits and a family history of nonfebrile seizures.
Causes of Seizures According to Age
Neonates (<1 month)
- Perinatal hypoxia and ischemia
- Intracranial hemorrhage and trauma
- CNS infections
- Metabolic
Infants and children (>1 month and <12 years)
- Febrile seizures
- Genetic disorders (metabolic, degenerative, primary epilepsy syndromes)
- CNS infections
Adolescents (12–18 years)
- Trauma
- Genetic disorders
- Infection
Young adults (18–35 years)
- Trauma
- Alcohol withdrawal
Older adults (>35 years)
- Cerebrovascular disease
- Trauma (including subdural hematoma)
- Brain tumor
- Alcohol withdrawal
- Degenerative diseases
Evaluation of the Patient with a Seizure
- When a patient is seen shortly after a seizure, the first priorities are attention to vital signs, respiratory and cardiovascular support, and treatment of seizures if they resume.
- When the patient is not acutely ill, the evaluation will initially focus on whether or not there is a history of earlier seizures.
First Seizure
If this is the patient’s first seizure, then the emphasis will be to establish whether the reported episode was a seizure rather than another paroxysmal event.
In the case of GTCS, features supporting organicity are postictal confusion, tongue bite, history of fall, or having sustained injury, etc.
- Determine the cause of the seizure by identifying risk factors and precipitating events. Precipitating factors such as sleep deprivation, systemic diseases, electrolyte or metabolic derangements (like hypoglycemia, hyper and hypocalcemia, hyponatremia hypomagnesemia) acute infection, drugs that lower the seizure (like penicillins, quinolones, antipsychotics,
lithium, amphetamine, barbiturates, cocaine) threshold or alcohol or illicit drug use should be identified. - General physical examination: Look for signs of system illnesses or infection. Careful examination of the skin may reveal signs of neurocutaneous disorders such as tuberous sclerosis or neurofibromatosis, or chronic liver or renal disease.
- Organomegaly may indicate a metabolic storage disease and limb asymmetry may provide a clue to brain injury early in development.
Finally, decide whether anticonvulsant therapy is required in addition to treatment for any underlying illness.
It is usually not required in case of metabolic or electrolyte derangements and seizures due to alcohol withdrawal.
If no metabolic or infectious causes are found, then look for
- Focal features of seizure
- Any focal neurological deficits
- Any other neurological dysfunction/mental retardation
Unusual features such as prolonged duration of seizures (>6 hours), more than six seizures, SE, or a prolonged postictal state.
- If present, usually antiepileptic therapy is required. Then MRI and EEG are done to find any mass lesion/stroke/ degenerative lesion. Treatment of underlying cause besides antiepileptic therapy.
- If absent, it is idiopathic epilepsy and antiepileptic therapy continued.
Recurrence of a Seizure
In the patient with prior seizures or a known history of epilepsy, the evaluation is directed toward:
Identification of the underlying cause and precipitating factors. Common precipitating factors are—sleep deprivation, fever, hypoglycemia, and alcohol.
Determination of the adequacy of the patient’s current therapy.
If no precipitating factors are found, measure the plasma concentration of AED.
If subtherapeutic concentration: Appropriate increase in drug dosage is done.
If the therapeutic concentration is normal: Either drug is increased to the maximum tolerated dose or alternate therapy is started by gradually tapering the first drug.
Laboratory Studies
Routine investigations: These include serum glucose, calcium, electrolytes, and renal and hepatic functions.
Lumbar puncture: If indicated.
EEG: This may help establish the diagnosis of epilepsy, classify the seizure type, and provide evidence for the existence of a particular epilepsy syndrome.
The presence of electrographic seizure activity, i.e., of abnormal, repetitive, rhythmic activity having an abrupt onset and termination, clearly establishes the diagnosis.
The EEG findings may also be helpful in the interictal period by showing certain abnormalities that are strongly supportive of epilepsy.
Such epileptiform activity consists of bursts of abnormal discharges containing spikes or sharp waves.
- EEG is normal in 40% of epileptic patients.
- Neuroimaging studies (MRI preferred over CT).
- The differences between seizures and syncope.
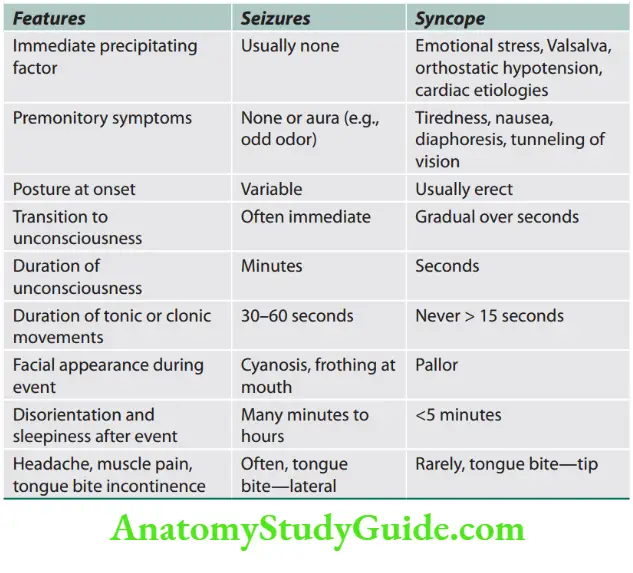
Treatment
Indications to Initiate Antiepileptic Drug Therapy
Antiepileptic drug therapy should be started in any patient with recurrent seizures of unknown etiology or a known cause that cannot be reversed within a short time.
Risk factors associated with recurrent seizures
- An abnormal neurologic examination
- Seizures presenting as status epilepticus
- Postictal Todd’s paralysis
- Strong family history of seizures
- An abnormal EEG
- Abnormal CT or MRI
Selection of AEDs. Certain AEDs like phenytoin and carbamazepine can worsen myoclonic seizures. Hence proper AED for proper seizure must be given.
Treatment Modification
- If a treatment is modified recently, one should wait for at least four half-lives of the drug before further modifying dosage.
- If one drug is unable to control seizures then another drug should be considered when either the maximum dose of the first drug is reached or the patient starts showing intolerable side effects.
- Whenever a new drug is added, the first drug is continued till the second drug controls seizures. Only after achieving adequate seizure control, the first drug should be gradually withdrawn.
Indications for Discontinue Therapy
- Complete medical control of seizures for 1–5 years.
- Single seizure type, either focal or generalized.
- Normal neurologic examination (including intelligence)
- Normal EEG
- In most cases, it is preferable to reduce the dose of the drug gradually over 2–3 months.
- Most recurrences occur in the first 3 months after discontinuing therapy, and patients should be advised to avoid potentially dangerous situations such as driving or swimming during this period.
Treatment of Refractory Epilepsy
- The therapy combines first-line drugs, i.e., carbamazepine, phenytoin, valproic acid, and lamotrigine.
- If these drugs are unsuccessful, then the addition of a newer drug such as levetiracetam and topiramate is indicated.
- Patients with myoclonic seizures resistant to valproic acid may respond to a combination of valproic acid and ethosuximide.
Surgical Treatment of Refractory Epilepsy
- About 20–30% of patients with epilepsy are resistant to medical therapy. The ketogenic diet—has been advised to decrease seizure recurrence. Low carbohydrates, adequate protein, and high fat have been advised.
- Anteromedial temporal lobe (temporal lobectomy) or a more limited removal of the underlying hippocampus and amygdala (amygdalohippocampectomy).
- Focal seizures arising from extratemporal regions may be abolished by a focal neocortical resection with precise removal of an identified lesion (laminectomy).
- Others: Hemispherectomy, corpus callosotomy, etc.
- Vagus nerve stimulation (VNS) may be used—in some of these cases, although the benefit for most patients seems to be very limited.

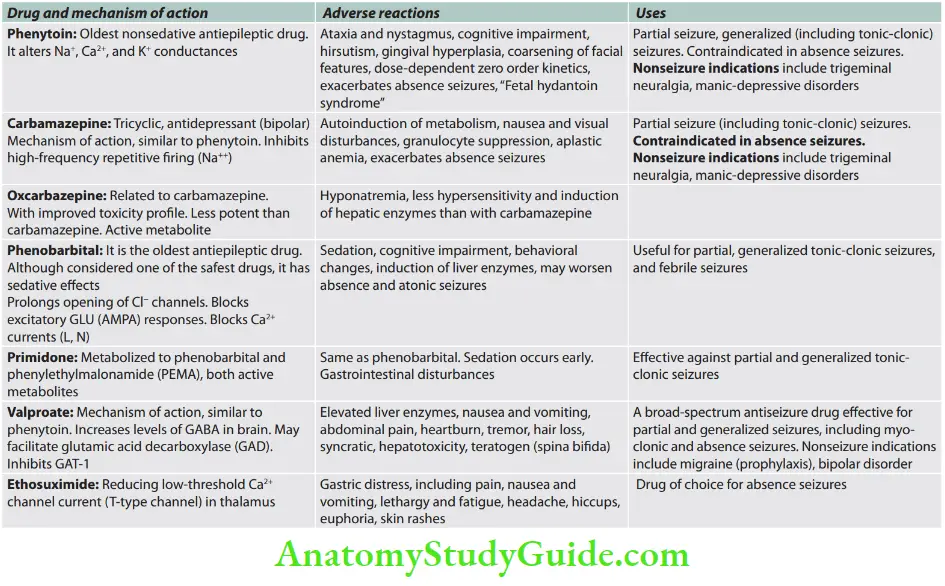
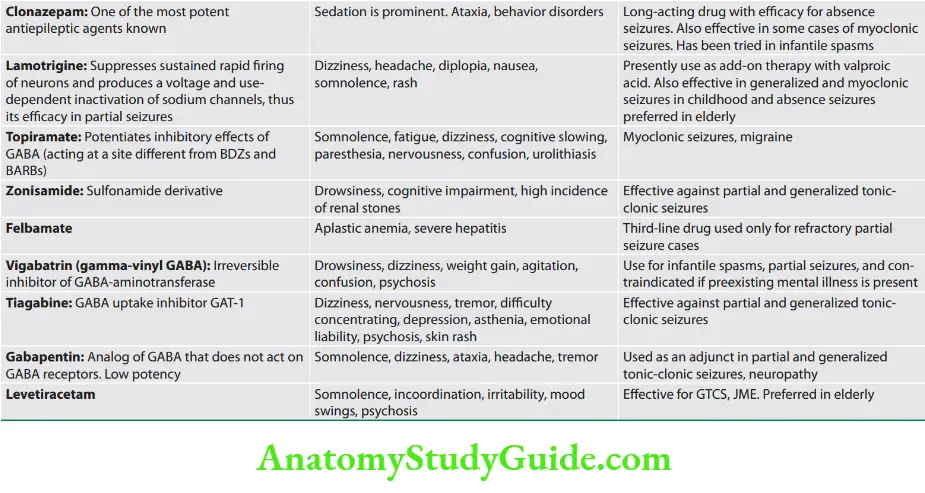
Newer AEDs (third-generation AEDs): Carisbamate, esclicarbazepine, brivaracetam, carabersat, ganaxalone, huperzine, lacosamide, losigamone, remacemide, retigabine, rufinamide, safinamide, perampanel.
Drug-resistant epilepsy/Intractable epilepsy may be defined as the failure of adequate trials of two tolerated and appropriately chosen and used antiseizure drug schedules to achieve sustained seizure freedom.
Nonmedical management can be tried.
- Surgical treatments—lobar and multilobar resections, hemispherectomy, corpus callosotomy, multiple subpial transections
- Vagus nerve stimulation
- Responsive cortical stimulation
- Subcortical deep brain stimulation
- Transcranial magnetic stimulation
- Trigeminal nerve stimulation
- A ketogenic diet (high-fat, low-protein) diet has demonstrated efficacy in children
Status Epilepticus
Question 38. Write a short essay/note on the definition, complications of SE, and its emergency management.
Answer:
- Status epilepticus refers to continuous seizures or repetitive, discrete seizures with impaired consciousness in the interictal period.
- Status epilepticus is an epileptic seizure of >5 minutes or more than one seizure within a 5-minute period without the patient returning to normal between them. Previous definitions used a 30-minute time limit.
Subtypes
Generalized convulsive status epilepticus (GCSE), e.g., persistent, generalized electrographic seizures, coma, and tonic-clonic movements.
Nonconvulsive status epilepticus, e.g., persistent absence seizures or focal seizures, confusion or partially impaired consciousness, and minimal motor abnormalities.
Etiology of status epilepticus.
- Stroke, including hemorrhagic
- Low AED levels
- Alcohol withdrawal
- Anoxic brain injury
- Metabolic disturbances
- Remote brain injury/congenital
- malformations
- Infections
- Brain neoplasms
- Idiopathic
Clinical Features
- Self-perpetuating, GTCS or
- Series of GTCSs
- Without a return to consciousness in between seizures.
Phases
- Initial compensatory phase: Sympathetic overdrive, increased CO, increased BP.
- Decompensation → homeostatic failure
- Reduced → CO/sugar/lactate/O2 levels leading to:
Cardiorespiratory collapse
- Electrolyte imbalance
- Rhabdomyolysis and delayed tubular necrosis
- Hyperthermia
- Multiorgan failure (MOF)
- Raised ICP and cerebral edema.
Complications
These include aspiration, hypotension, cardiac arrhythmias, and renal or hepatic failure.
Diagnosis
- Diagnosis of nonconvulsive SE in critically ill patients.
- Correlate with poorer outcomes.
- EEG patterns are difficult to interpret (equivocal patterns)—criteria are not validated.
- A trial of rapidly acting IV AED is used to observe improvement in both clinical → EEG by several hours.
Management of Status Epilepticus
- Convulsive/nonconvulsive.
- Other available modalities:
- Brain imaging (perfusion/metabolic imaging)
- Intracranial monitoring with intracortical EEG
- Brain tissue O2 monitoring
- Cerebral microdialysis.
Pregnancy and Epilepsy
- In approximately 50% of epileptic women, the frequency of epilepsy remains the same. In about 30% it increases and in about 20% it decreases in frequency.
- Uncontrolled seizure in the mother causes harm to both mother and fetus greater than the teratogenic effects of AEDs. Hence, pregnant women should be maintained on effective drug therapy.
- Due to the numerous side effects, phenytoin is not used in pregnancy. Neural tube defects are associated with valproic acid and carbamazepine.
Idiopathic
Management of status epilepticus (SE).
First 5 minutes
- Check emergency ABCs
- Give O2
- Obtain IV access
- Begin ECG monitoring
- Check finger stick glucose
- Draw blood for serum electrolytes.
- RFT, magnesium, calcium, phosphate, CBC, LFTs, AED levels, ABG, troponin
- Toxicology screen (urine and blood)
6–10 minutes
- Thiamine 100 mg IV; 50 mL of D50 IV unless adequate glucose knew Lorazepam 4 mg IV over 2 minutes; if still seizing, repeat × 1 in 5 minutes
- If no rapid IV access give diazepam 20 mg PR or midazolam 10 mg intranasally, buccally, or IM for
10–20 minutes
- If seizures persist, begin fosphenytoin 20 mg/kg IV at 150 mg/min, with blood pressure and ECG monitoring Or
- Phenytoin 15–20 mg/kg at 30–50 mg/min Reasonable to bypass this step, or perform subsequent steps simultaneous with fosphenytoin loading
10–60 minutes: One (or more) of the following four options (intubation usually necessary except for valproate):
- Continuous IV midazolam. Load: 0.2 mg/kg; repeat 0.2–0.4 mg/kg boluses every 5 minutes until seizures stop, up to a maximum total loading dose of 2 mg/kg.
- Initial rate: 0.1 mg/kg/h. Continuous IV dose range: 0.05–2.9 mg/kg/h
- Continuous IV propofol. Load: 1 mg/kg; repeat 1–2 mg/kg boluses every 3–5 minutes until seizures stop, up to a maximum total loading dose of 10 mg/kg.
- Initial continuous IV rate: 2 mg/kg/h. Continuous IV dose range: 1–15 mg/kg/h. Avoid > 48 hours of >5 mg/kg/h (increased risk of propofol infusion syndrome) Or
- IV valproate: 40 mg/kg over ~10 minutes. If still seizing, an additional 20 mg/kg over ~5 minutes Or
- IV phenobarbital: 20 mg/kg IV at 50–100 mg/min
60 minutes
Continuous IV pentobarbital Load: 5 mg/kg at up to 50 mg/min; repeat 5 mg/kg boluses until seizures stop. Initial continuous IV rate: 1 mg/kg/h.
Continuous IV-dose range: 0.5–10 mg/kg/h; traditionally titrated to suppression-burst on EEG
Perform neuroimaging when convulsive activity is controlled
Begin continuous EEG, if the patient does not awaken rapidly or if continuous IV Rx is used
Treat metabolic abnormalities and hypothermia
Lumbar puncture and antibiotics can be considered if the infection is suspected
SE not controlled even with anesthetic agents is called super refractory SE, for which IVIg/pulse steroid can be tried as a last resort
Carbamazepine is considered relatively safe in pregnancy, because of its low teratogenic potential. Levetiracetam is the safest drug.
If the mother is on phenobarbital, she should be given oral vitamin K (20 mg daily) in the last 2 weeks of pregnancy, and the the infant should be given an intramuscular injection of vitamin K (1 mg) at birth.
This is because phenobarbital may cause a transient and reversible deficiency of vitamin K-dependent clotting factors in up to 50% of newborn infants.
Neuroinfections
Question 39. Classify meningitis. List the causes of meningitis.
Answer:
Classifications of Meningitis
Infective Noninfective (sterile)
1. Bacterial meningitis
- Common organisms: Streptococcus pneumoniae, Neisseria meningitides, Haemophilus influenzae
- Uncommon organisms: Staphylococcus aureus, Staphylococcus epidermidis, Group B streptococci, Escherichia coli, Klebsiella, Proteus spp., Listeria monocytogenes.
- Rare organisms: Salmonella, Shigella, Clostridium perfringens, Neisseria gonorrhoeae
2. Tuberculous meningitis (TBM): Mycobacterium tuberculosis
3. Viral meningitis (aseptic meningitis)
- Enteroviruses (coxsackie virus, poliovirus)
- Mumps virus
- Arboviruses
- Human immunodeficiency virus (HIV)
- Herpes simplex-2
4. Spirochetal: Leptospirosis, Lyme disease, syphilis
5. Rickettsial: Typhus fever
6. Protozoal: Cysticerci, amoeba, Naegleria
7. Fungal: Cryptococcus neoformans, Candida, Histoplasma,
Blastomyces, Coccidioides, Sporothrix
1. Malignant disease
- Breast cancer
- Bronchial cancer
- Leukemia (leukemic meningitis)
- Lymphoma
2. Subarachnoid hemorrhage (SAH) (causes meningismus)
3. Inflammatory disease (may be recurrent)
- Sarcoidosis
- Systemic lupus erythematosus, rheumatoid arthritis
- Behçet’s disease
- Vasculitis
Neurosyphilis
Question 40. Write a short essay on the clinical features and management of neurosyphilis.
Answer:
- Treponema pallidum invades the nervous system within 3–18 months (may take years to develop) after primary infection.
- Neurosyphilis may be asymptomatic or symptomatic. The initial event is usually in the form of asymptomatic meningitis.
- Later it may produce more damage. All forms of neurosyphilis have meningitis of variable severity.
- Secondary to meningitis, the blood vessels show endarteritis obliterans.
Asymptomatic Neurosyphilis
- Asymptomatic invasion of CNS by treponema is common and occurs within a few months of primary infection by Treponema pallidum.
- Neurosyphilis may develop in 25% of cases of latent syphilis. Many of them may develop symptomatic neurosyphilis.
- Cerebrospinal fluid shows lymphocytosis with increased protein and low glucose.
- Antibodies in the CSF are the most specific test for neurosyphilis. Venereal disease research laboratory test is positive.
- Treatment: Penicillin.
Symptomatic Neurosyphilis
It takes one of several forms, although mixed features are common (Box 15.25).
Meningeal syphilis
Symptoms of meningitis may develop at any time after infection but usually occur within 2 years after primary infection.
Symptoms:
- These include headache, neck stiffness, seizures, altered sensorium, and cranial nerve palsies.
- It may show skin rash on palms and soles.
- Papilledema with symptoms of increased ICP may develop. The patient is afebrile and CSF is abnormal.
Meningovascular syphilis
- Chronic meningitis involves the base of the brain, cerebral convexities, and spinal leptomeninges.
- Usually presents 6–7 years after primary infection. However, it can develop as early as 6 months and as late as 10–12 years.
- It should be suspected when a young patient develops a stroke (generally subacute) which results in hemiparesis, aphasia, visual loss, etc. Other features include headache, vertigo, insomnia, and psychological abnormalities.
Cerebrospinal fluid findings in neurosyphilis
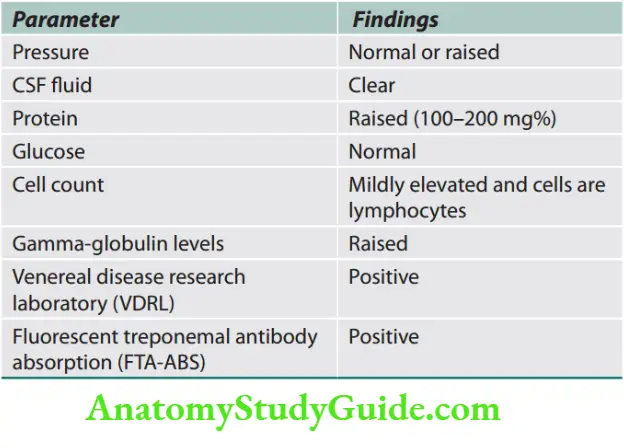
General paralysis of insane
It develops about 20 years after primary infection.
Shows generalized/diffuse brain parenchymal disease with dementia; hence called general paresis of insane.
Clinical manifestation includes personality changes, illusions, delusions, hallucinations, dementia (reduced memory), hyperactive reflexes, and Argyll Robertson pupils.
Major categories of symptomatic neurosyphilis.
- Meningeal syphilis
- Chronic meningovascular disease
- Parenchymatous syphilis
- General paresis of insane
- Tabes dorsalis
Tabes dorsalis
- Tabes dorsalis is the parenchymal form of neurosyphilis characterized by demyelination of the posterior column, dorsal root, and dorsal root ganglia in the spinal cord.
- Usually develops 20–25 years after primary infection.
- Symptoms: Severe lightening pains in the trunk and extremities, ataxia, and urinary incontinence.
- Signs: Patchy tactile sensory loss and severe impairment of proprioception with sensory ataxia.
- Muscular strength is normal and tendon jerks are absent. Complications include trophic lesions such as perforating ulcers of feet and Charcot’s joints.
- Argyll Robertson pupils may also be observed in tabes dorsalis.
- Visceral crisis: It consists of sudden epigastric pain with vomiting that Abadie’s sign (Pinching of, or the application of firm pressure to, the Achilles tendon does not result in pain) lasts for hours.
- Barium studies show pylorospasm (gastric crisis).
- Other crisis includes an intestinal crisis with diarrhea, a rectal crisis with tenesmus, a genitourinary crisis with strangury, and a pharyngeal-laryngeal crisis with gulping movements and dyspnea
- CSF findings:
Treatment
- Penicillin: Drug of choice and given in the dose of 18–24 million units/ day for 15–20 days.
- If the patient is sensitive to penicillin: Erythromycin and tetracycline 0.5 g 6 hourly for 20–30 days.
Follow-up
- Re-examine the patient every 3 months and CSF examination at 6 months intervals. If CSF is normal and VDRL titers are reduced, no further treatment is necessary.
- However, if CSF remains abnormal, the patient should be treated with another full course of penicillin.
Cerebrospinal Fluid
- Cerebrospinal fluid is formed within the ventricles and circulates in the subarachnoid space (between the arachnoid and pia matter) and in the ventricles. The total volume of CSF in adults ranges from 90 to 150 mL. It is a medium for the transfer of substances from the brain and spinal cord into the blood.
- Importance of CSF examination: Analysis of the CSF is of diagnostic importance in conditions like meningitis or primary/ metastatic tumor of CNS with CSF involvement.
- Collection of CSF: CSF is usually obtained by lumbar puncture using a lumbar puncture needle under strict aseptic conditions.
Lumbar Puncture
Question 41. Write a short essay on the procedure indications, contraindications, and complications of lumbar puncture.
Answer:
- Lumbar puncture is the technique done to obtain CSF samples and also provides an indirect measure of ICP.
- After local anesthetic injection, a lumbar puncture needle is inserted in the midline between lumbar spinous processes usually between L3 and L4 (3rd lumbar space) through the dura
and into the spinal canal. - In children, it is collected from the 4th lumbar space.
- Intracranial pressure can be assessed (if patients are lying on their side) and CSF obtained for analysis.
- CSF pressure is important in the diagnosis and monitoring of idiopathic intracranial hypertension.
Indications and contraindications for lumbar puncture
Complications of Lumbar Puncture
Most common complication is post spinal headache. Herniation of the cerebellum through the foramen magnum due to raised ICP.
- Indications and contraindications for lumbar puncture.
- Indications for lumbar puncture
- Diagnostic indications
Infection:
Meningeal infection: Bacterial (pyogenic, tuberculosis, syphilitic (to differentiate general paresis of insane, tabes dorsalis, and meningeal syphilis), viral, fungal
- Encephalitis
- Subarachnoid hemorrhage
- Primary or metastatic malignancy (e.g., acute leukemia, lymphoma)
Demyelinating diseases: Multiple sclerosis and subacute sclerosing panencephalitis (SSPE)
- Guillain–Barré syndrome
- Spinal canal blockage leading to elevated
- intracranial tension (spinal cord tumors)
- Injecting the radiopaque dye for myelography
- Therapeutic indications
- Spinal anesthesia, epidural analgesia
- Intrathecal injection of chemotherapeutic drugs
- for CNS prophylaxis/relapse of ALL, lymphomas
- Therapeutic CSF drainage in cases of normal
- pressure hydrocephalus
- Contraindications for lumbar puncture
- Raised intracranial pressure, coagulopathy
- Local infective lesion
- Bony deformities at the site of puncture
- Hematoma, either extradural or subdural.
- Introduction of infection by the lumbar puncture needle through the infected skin or subcutaneous tissue.

CSF osmolality and sodium levels are the same as that of serum
Constituents that are in higher concentration in CSF than in blood include magnesium, chloride, H+ concentration, and lactate.
Question 42. Write a short note on the causes of elevated CSF proteins.
Answer:
Causes of low sugar in CSF: Pyogenic, tuberculous, fungal, and carcinomatous meningitis.
Causes of elevated CSF proteins.
- Meningitis
- Brain abscess
- Brain or spinal cord tumors
- Multiple sclerosis
- Guillain–Barré syndrome
- Syphilis
- Hemorrhage
- Froin’s syndrome
Question 43. Write a short note on xanthochromia in CSF analysis. Xanthochromia is the yellowish appearance of CSF.
Causes: Xanthochromia is always pathological and conditions associated with it are:
Old SAH: The yellow appearance is due to RBCs which may leak into the CSF during the hemorrhage. RBCs are breakdown and liberate hemoglobin which is converted into yellow bilirubin.
Other causes are high protein in the CSF, jaundice, and Froin’s syndrome.
Meningitis and Encephalitis
Question 44. What are the causes of meningitis? Discuss the clinical features, investigations, diagnosis, complications, and management of acute pyogenic (bacterial/meningococcal) meningitis.
Answer:
Acute Bacterial (Pyogenic/Purulent) Meningitis
Question 45. Write a short essay/note on the common organisms causing pyogenic meningitis.
Answer:
The relative frequency of various bacterial species causing meningitis varies with age.
Neonatal period: Major causative agents include gram-negative bacilli (principally Escherichia coli), Listeria monocytogenes, and group B streptococci.
Infants and children: Major causes in children beyond 1 month of age are Haemophilus influenzae and Neisseria meningitidis.
Adolescents and young adults: Meningococcus (N. meningitidis) is the most common pathogen.
Extremes of life: Streptococcus pneumoniae and L. monocytogenes.
Causes of community-acquired bacterial meningitis: Important causes are:
- S. pneumoniae (50%): Most common.
- N. meningitides (25%): It is the only major cause of epidemics of bacterial meningitis.
- Group B streptococci (15%)
- L. monocytogenes (10%)
- H. influenza (10%)
Predisposing conditions
For Pneumococci: Other Pneumococcal infections (pneumonia, sinusitis, otitis media), splenectomy, hypogammaglobulinemia, complement deficiency.
For Neisseria meningitidis: Complement deficiency (including properdin), B-serotype (not protected by the vaccine is responsible for one-third of cases).
Usually associated with petechial or purpuric skin lesions.
Listeria monocytogenes: Important cause in neonates (<1 month), pregnant women, >60 years, and immunocompromised persons.
Clinical manifestations
Question 46. Write a short essay/note on the physical signs of meningitis.
Answer:
Classic triad:
- Fever,
- headache, and
- nuchal rigidity.
Consciousness: Vary from lethargy to coma (>75%).
Nausea, vomiting, and photophobia are common Seizures/convulsions (especially in children) may be an initial presentation or present during the course (20–40%).
Focal: Due to arterial ischemia, infarction, cortical venous thrombosis, or focal edema.
Generalized or status: Due to hyponatremia, cerebral anoxia, or toxic effects of antimicrobial drugs.
- Occasionally cranial nerve palsies, with focal neurologic deficits such as visual field defects, dysphasia, and hemiparesis may occur.
- In meningococcus: The rash begins as diffuse erythematous maculopapular that becomes petechial, found on the trunk, lower limbs, mucous membrane, conjunctiva, and rarely on the palm and sole.
- Raised ICP: One of the complications of bacterial meningitis
and CSF pressure may be raised to 180–400 mm H2O.
A disastrous complication of ICP is cerebral herniation.
Other signs: Reduced level of consciousness, papilledema, dilated poorly reactive pupil, VIth cranial palsy, decerebrate posture, Cushing reflex (bradycardia, hypertension, and irregular respiration).
Diagnosis
- By examination of the CSF: It is usually obtained by lumbar puncture. Lumbar puncture should be postponed if there is papilledema and or focal neurologic findings suggestive of an intracranial mass lesion. It can be done only after ruling out the same by CT or MRI.
- Blood culture: If meningitis seems likely, blood should be sent for culture, and sensitivity and empirical antimicrobial therapy should be started while the neuroimaging study is being carried out.
CSF findings in bacterial meningitis
Question 47. Write a short essay/note on CSF findings on pyogenic/bacterial meningitis.
(or)
Write a short essay/note on the treatment of pyogenic meningitis.
Answer:
Treatment
Antibiotics used in empirical therapy of bacterial meningitis and focal CNS infections.
Empirical therapy
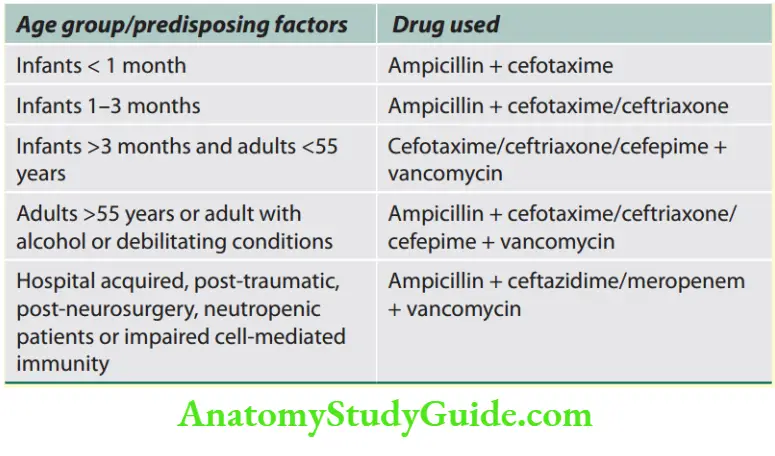
Streptococcus pneumoniae and Neisseria meningitidis are common organisms.
Due to the emergence of penicillin and cephalosporin resistance S.
pneumoniae a combination therapy of 3rd or 4th generation cephalosporin (ceftriaxone, cefotaxime, cefepime) and vancomycin, plus acyclovir (HSV is a differential diagnosis) and doxycycline (tick infection) is given as empirical therapy.
Specific antimicrobial therapy
- Neisseria meningitidis
- Penicillin sensitive: Penicillin G—250,000–300,000 U/kg/ day in divided doses, Ampicillin (3 g IV TID/QID).
- Penicillin resistant: Ceftriaxone 2 g IV BID/Cefotaxime 2 g IV BID.
- 7 days IV doses are adequate.
- All close contacts should be given chemoprophylaxis with rifampicin for 2 days or azithromycin (500 mg once) or one IM ceftriaxone (250 mg).
- Streptococcus pneumoniae
- Should be tested for penicillin and cephalosporin sensitivity
- Penicillin sensitive: Penicillin G
- Penicillin-intermediate: Ceftriaxone/cefotaxime/cefepime
- Penicillin-resistant: Ceftriaxone/cefotaxime/cefepime + vancomycin 2 weeks IV is adequate.
- Lumbar puncture is to be repeated 24–36 hours after initiation to see the response.
- Intraventricular vancomycin may be more effective than IV or intrathecal.
- Gram-negative bacilli (except Pseudomonas): Ceftriaxone/cefotaxime for 3 weeks.
- Ampicillin + ceftazidime/meropenem + vancomycin
Classical signs of meningitis.
- Cervical rigidity/neck stiffness
- Kernig’s sign: Knee pain with hip flexion
- Brudzinski’s sign: Knee/hip flexion when the neck is flexed
Complications of bacterial meningitis.
Obstructive hydrocephalus
Thrombophlebitis of leptomeningeal veins may lead to venous thrombosis, cerebral infarction, focal infection of the underlying brain parenchyma
- Chronic adhesive arachnoiditis
- Cerebral abscess
- Subdural empyema
- Focal neurologic deficits (e.g., cranial nerve palsy, hemiparesis)
- Sensorineural hearing loss
- Vasculitis of cranial vessel
- Epilepsy
Waterhouse–Friderichsen syndrome: It results from meningitis-associated septicemia with hemorrhagic infarction of the adrenal glands and cutaneous petechiae.
It occurs most often with meningococcal and pneumococcal meningitis
Pseudomonas aeruginosa: Ceftazidime/cefepime/meropenem
Staphylococci spp:
Methicillin sensitive: Nafcillin
Methicillin resistant: Vancomycin (1 g IV TID) (IV or intraventricular)
Listeria monocytogenes: Ampicillin for 3 weeks. Gentamicin may be added in critically ill cases.
- Trimethoprim and sulfamethoxazole are alternative
- influenzae-intermediate: Ceftriaxone/cefotaxime/cefepime
- Streptococcus agalactiae: Penicillin G/ampicillin
- Bacteroides fragilis: Metronidazole
- Fusobacterium spp: Metronidazole
Adjunctive therapy
Dexamethasone inhibits the synthesis of IL-1 β and TNF-α, decreases CSF outflow resistance, and stabilizes the blood-brain barrier (BBB).
It is to be given 20 minutes before antimicrobial therapy. It is less effective if given 6 hours after antibiotic therapy.
Treatment of raised ICP
Elevation of the head end of the bed to 30–45°, hyperventilation, and administration of mannitol.
Duration of therapy: 7 days for N. meningitides, 7–10 days for H. influenzae, 10–14 days for S. pneumoniae, and 3 weeks for gram-negative bacilli.
Tubercular Meningitis
Question 48. Describe the pathology, clinical features, investigation, complications, and treatment/management of tuberculous meningitis.
Answer:
In India, tubercular meningitis (TBM) remains the most common form of meningitis.
Risk factors
- Previous history of exposure to tuberculosis or illness.
- Immunocompromised state of acquired immunodeficiency syndrome (AIDS) Young children.
Pathology
- The main neuropathologic finding is basal meningeal exudates containing mainly mononuclear cells.
- Tubercles may be seen on the meninges and on the surfaces of the brain.
- The ventricles may be dilated as a result of hydrocephalus, and the ependymal surface may be covered by exudates.
- Hydrocephalus is common in children and most develop symptoms in 2–3 weeks.
- Communicating type: Common, due to blockage of the basal cistern by exudates in the acute phase or adhesive leptomeningitis in the chronic phase.
- Obstructive type: Less common, due to narrowing or occlusion of the aqueduct by ependymal inflammation, tuberculomas, or obstruction of the outlet of the IVth ventricle.
- Arteritis can cause cerebral infarction and basal inflammation, and fibrosis can compress cranial nerves.
Clinical features
Onset: Usually subacute/chronic. Acute: children—50%, adult—14%.
Past history of TB: Children—50%, adults—10%.
Prodromal symptoms: Two to three weeks, vague ill health, apathy, irritability, anorexia, changes in behavior.
Features of meningitis: Headache, vomiting, fever, and focal neurological deficit.
Features of raised ICP:
- Convulsions (focal/generalized—20–30%).
Cranial nerve palsy: 20–30% (VIth cranial nerve).
Loss of vision: Partial/complete. Due to opticochiasmatic exudate, and arteritis.
Other presentations: Hemiplegia, facial nerve palsy, optic atrophy, abnormal movement, oculomotor palsy, choroid tubercle, etc.
In untreated cases: Consciousness deteriorates, pupillary abnormality, pyramidal signs due to hydrocephalus and tentorial herniation.
Complications of tubercular meningitis
- Raised intracranial pressure (ICP), cerebral edema
- Basal meningitis with cranial nerve palsy—II, III, IV, VI, and VII
- Focal neurologic deficit and seizure
- Hydrocephalus
- Tuberculoma
- Opticochiasmatic pachymeningitis: Visual loss
- Endocrine abnormality: Growth hormone and gonadotropin
- Hypothalamic disorder: Loss of control of blood pressure (BP)
and temperature, delayed or precocious sexual development - Diabetes insipidus, syndrome of inappropriate ADH secretion
(SIADH)
Investigations
Question 49. Write a short essay/note on laboratory diagnosis and CSF findings in tuberculous meningitis.
Answer:
1. CSF study
- Cytological study
- Leukocyte count: 100–500 cells/μL, rarely >1,000/cells/μL.
- Cell type: Lymphocytes predominant, polymorphonuclear cells in the acute stage. Hemorrhagic due to fibrinoid necrosis of vessels. No malignant cells were seen.
Biochemical study
- Protein: Usually 100–200 mg/100 mL. In spinal block >1 g/100 mL and xanthochromic.
- On standing a pellicle/cobweb formed indicating fibrinogen—high suggestion of TBM.
- Glucose: Reduced to 20–50 mg/100 mL. In most cases <40% of corresponding blood sugar, but unlike pyogenic never undetectable.
- Chloride: Low value 450–600 mg/100 mL and is nonspecific indicating hypochloremia.
It may be seen in bacterial, viral meningitis also.
Adenosine deaminase (ADA) is produced by T lymphocytes elevated in CSF (60–100%).
Microbiological study
- Negative with Gram stain, Indian ink stain, and culture is sterile.
- Acid-fast bacilli (AFB): AFB in smear and culture-confirmatory but the number of bacteria should be >104/ml. Stained by Ziehl–Neelsen and auramine (4–40% positive).
Centrifuged CSF: The thick smear from the pellicle and repeated culture enhance detection.
CSF culture: In Lowenstein–Jensen media in 4–8 weeks.
It may be enhanced by liquid media like the Septi-Chek AFB system or Middlebrook 7H9.
Isolation is better from cisternal/ventricular CSF.
2. Radiological investigations
- Chest X-ray: It may reveal military mottling in the lung.
- CT scan:
- Thickening of meninges in the basal cistern (60%)
- Hydrocephalus in 50–80% depending on duration
- Cerebral infarct (28%) in MCA, edema (periventricular), tuberculoma (10%).
3. Immunological methods
- Antibody detection: CSF shows antibodies against various antigens that are sensitive and are detected by ELISA and RIA.
- Antigen detection: More specific. The antigen is detected by Latex agglutination, ELISA, etc.
- Molecular methods: Amplification of specific DNA sequence by PCR used for rapid diagnosis. PCR is confirmatory and not affected by other organisms.
Management
- Antituberculous treatment (ATT) for one and a half year in uncomplicated cases is usually sufficient.
- Steroids: It is recommended to give steroids during the initial 6 weeks to decrease the possibility of adhesion formation.
- Steroids prevent complications. There is no definite duration for which treatment might be continued but should be judged on the basis of neuroimaging findings.
- Surgical intervention: If hydrocephalus, tuberculoma, or abscess develops. The tubercular abscess needs drainage.
Viral Meningitis/Aseptic Meningitis
Viral infections of the meninges (meningitis) or brain parenchyma (encephalitis) often present as acute confusional states.
Children and young adults are frequently affected.
Etiology
Common viruses include:
- Enteroviruses (most common, i.e., coxsackieviruses, echoviruses, and human enteroviruses): At least two-thirds of the cases of CSF culture-negative aseptic meningitis are due to enteroviruses.
- Herpes simplex virus-2 (HSV-2) meningitis: It may occur during the initial episode of genital herpes.
- Most cases of benign recurrent lymphocytic meningitis (previously called Mollaret’s meningitis) appear to be due to HSV.
- Arthropod-borne viruses: These are transmitted through infected insect vectors.
- Human immunodeficiency virus (HIV): Aseptic meningitis is a common manifestation of primary exposure to HIV.
- Cranial nerve palsies, most commonly involving cranial nerves V, VII, and VIII are more common in HIV meningitis than in other viral infections.
- Mumps can also cause meningitis.
Clinical Manifestations
Fever, headache, and meningeal irritation. Other features include malaise, myalgia, anorexia, nausea and vomiting, abdominal pain, and/or diarrhea.
Laboratory Diagnosis
Question 50. Write a short answer on CSF changes in viral meningitis.
Answer:
CSF examination:
- Reveals lymphocytosis, mildly elevated protein, and normal glucose.
- As a rule, a lymphocytic pleocytosis with a low glucose level (<25 mg/dL) should suggest the presence of fungal, listerial, or tuberculous meningitis or of noninfectious disorders (e.g., neoplastic meningitis)
- Polymerase chain reaction (PCR): Amplification of viral-specific DNA or RNA from CSF by PCR important method for the diagnosis of CNS viral infection.
Treatment
- Symptomatic and hospitalization are not required.
- Oral or IV acyclovir may be of benefit in patients with meningitis caused by HSV-1 or HSV-2 and in cases of severe EBV or VZV infection.
- Patients with HIV should receive highly active antiretroviral therapy.
Mollaret’s Meningitis
Question 51. Write a short note on Mollaret’s meningitis.
Answer:
Definition: It is a syndrome characterized by recurrent aseptic, self-limiting meningitis and the presence of large typical monocytes (Mollaret cells) in the CSF.
Cause: Not known, but may be due to some virus infections.
Herpes simplex-2 infection must be excluded in these patients.
Clinical features: Repeated self-limiting episodes of fever, meningismus, and severe headache (of 2–5 days duration) separated by symptom-free intervals.
Noninfective causes of meningitis.

CSF Findings in Meningitis
Question 52. Write a short note comparing the CSF findings of pyogenic, aseptic, and tuberculous meningitis.
Answer:
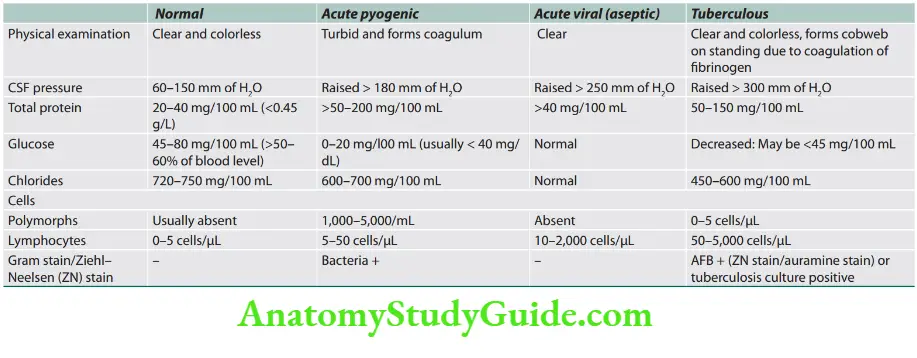
Causes of Neck Stiffness
Question 53. List the causes of neck stiffness.
Answer:
Encephalitis
Viral Encephalitis
In meningitis, the infectious process and associated inflammatory response are limited largely to the meninges, whereas in encephalitis, the brain parenchyma is also involved.
Encephalitis is characterized by nonsuppurative inflammation of the brain by an inflammatory process.
Causes of neck stiffness.
Meningism:
- Meningitis
- Subarachnoid hemorrhage
Other conditions that mimic meningism (also resist cervical rotation):
- Cervical spondylosis
- After cervical fusion
- Parkinson’s disease
- Raised intracranial pressure especially if there is impending tonsillar herniation
- Acute dystonic reaction
- Tetanus
- Strychnine poisoning
- Intermittent neck stiffness is characteristic of Arnold–Chiari malformation
Viruses causing encephalitis.
- Epidemics of encephalitis are caused by arboviruses
- Acute encephalitis
- Herpesvirus
- Herpes simplex virus I (MC)
- Varicella zoster virus
- Epstein–Barr virus
- Arthropod-borne viruses
- West Nile virus
- Japanese encephalitis Colorado tick fever
- Others: Rabies, enteroviruses, mumps, cytomegalovirus
Etiology
Clinical manifestations
- Acute febrile illness with evidence of meningitis and encephalitis.
- Altered level of consciousness (ranging from mild lethargy to coma), an abnormal mental state, and evidence of either focal or diffuse neurologic signs or symptoms.
- It may have hallucinations, agitation, personality change, behavioral disorders, and at times a frankly psychotic state.
- Focal or generalized seizures occur in many patients with encephalitis.
- Most common focal neurological findings: Aphasia, ataxia, upper or lower motor neuron patterns of weakness, involuntary movements (e.g., myoclonic jerks, tremors), and cranial nerve
deficits (e.g., ocular palsies, facial weakness). - Involvement of the hypothalamic-pituitary axis may result in temperature dysregulation, diabetes insipidus, or the development of the syndrome of inappropriate secretion of antidiuretic hormone (SIADH).
Laboratory diagnosis
1. Cerebrospinal fluid
CSF examination: Indistinguishable from that of viral meningitis and typically consists of lymphocytic pleocytosis, a mildly elevated protein concentration, and a normal glucose
concentration.
CSF PCR: Primary diagnostic test for CNS infections caused by CMV, EBV, HHV-6, and enteroviruses.
CSF culture: Limited utility.
2. Serologic studies and antigen defection: Demonstration of antibodies or antigens.
3. Brain biopsy: Reserved for patients in whom CSF PCR studies fail to lead to a specific diagnosis, who have focal abnormalities on MRI, and who continue to show progressive clinical deterioration despite treatment with acyclovir and supportive therapy.
4. MRI, CT, and EEG.
Management
- General measures: Care of the unconscious patient. Anticonvulsants may be needed. Brain edema is managed with dexamethasone 4 mg 6 hourly.
- Herpes simplex encephalitis: Acyclovir (10 mg/kg IV 8 hourly for 14–21 days), if instituted early.
Froin’s Syndrome
Question 54. Write a short note on Froin’s syndrome.
Answer:
Froin’s syndrome describes the CSF findings in cases of complete spinal (subarachnoid) block (below the block).
CSF below the block shows the following features:
- CSF pressure: Reduced
- Queckenstedt’s test: It is an outdated clinical test, formerly used for diagnosing spinal block.
- Physical examination: Yellowish discoloration (xanthochromia) coagulum may form due to high protein content.
- Chemical examination: Very much elevation of protein levels, sugar levels are normal or occasionally reduced if the obstruction is due to tuberculous meningitis.
- Cytology: Normal cell count. Increased cells if the obstruction is due to tuberculous meningitis.
Causes of total block:
- Intraspinal tumors
- Vertebral disease with compression.
- Chronic spinal arachnoiditis.
Diseases of the spinal cord
Features Suggestive of Spinal Cord Involvement
- Presence of sensory deficit and/or motor weakness in both lower limbs and/or upper limbs
- Bladder and bowel involvement
- Brown–Sequard type of clinical picture
- Presence of definite sensory level
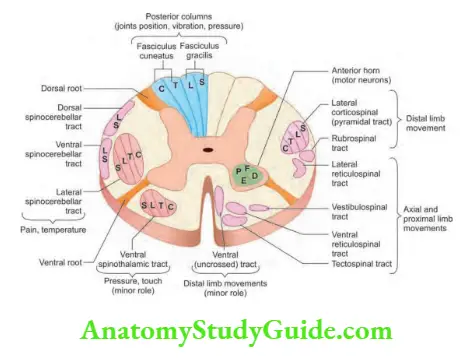
Question 55. List the spinal cord syndrome with examples.
Answer:
Patterns of Spinal Cord Disease
- Complete cord transection syndrome (trauma)
- Brown–Sequard syndrome (bullet injury, multiple sclerosis)
- Central cord syndrome (syringomyelia)
- Posterior column syndrome (tabes dorsalis, Friedreich’s ataxia, HIV myelopathy)
- Posterolateral cord syndrome—subacute combined degeneration of the cord (SACDC)
- Combined anterior horn cell (AHC) and pyramidal tract syndrome [amyotrophic lateral sclerosis (ALS)]
- Anterior horn cell syndrome (polio)
- Anterior cord syndrome (anterior spinal artery occlusion)
- Conus medullaris and cauda equina syndrome
Causes and Clinical Features of Spinal Cord Syndromes





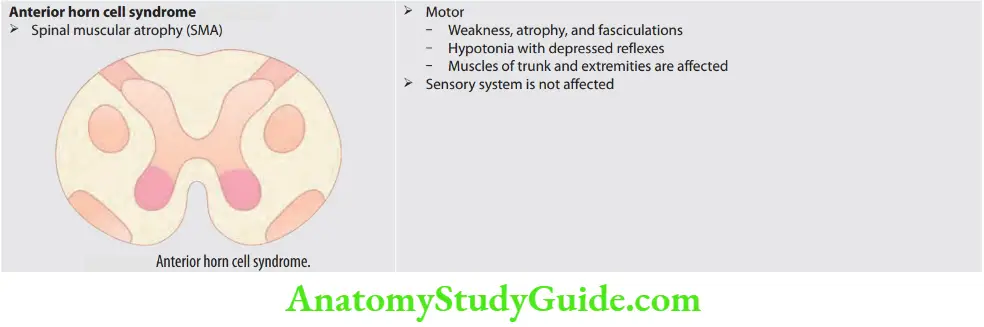
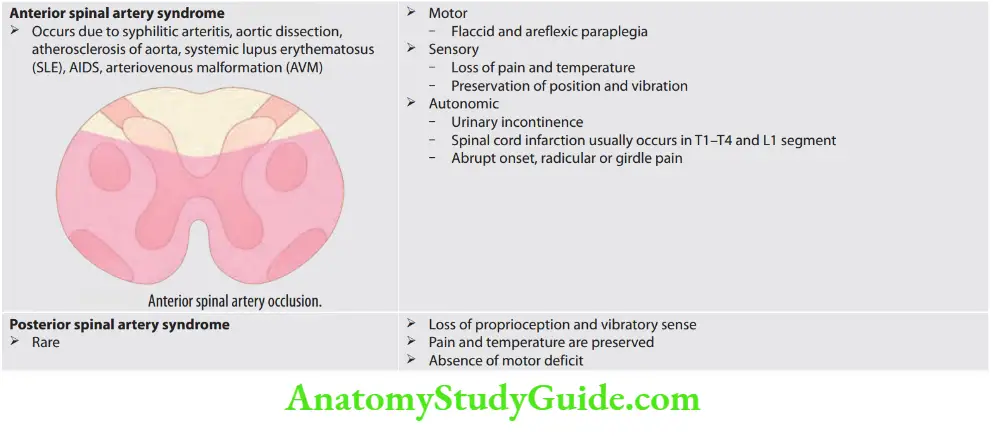

Question 56. List the differences between compressive and noncompressive myelopathy.
Answer: The differences between compressive and noncompressive myelopathy

Question 57. Write a short essay on the differences between extramedullary and intramedullary lesions of the spinal cord.
Answer:
The differences between extramedullary and intramedullary lesions of the spinal cord.
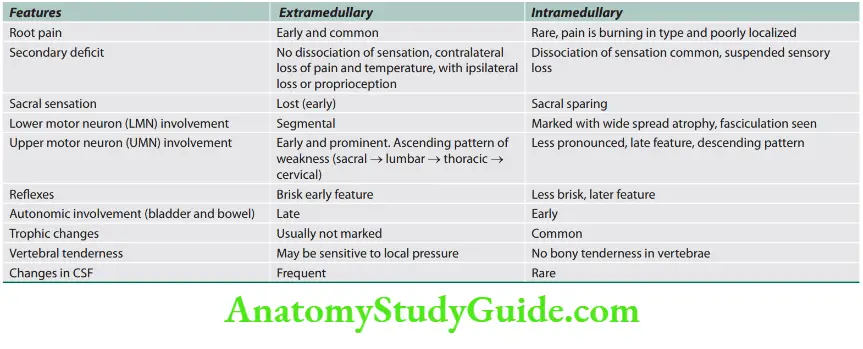
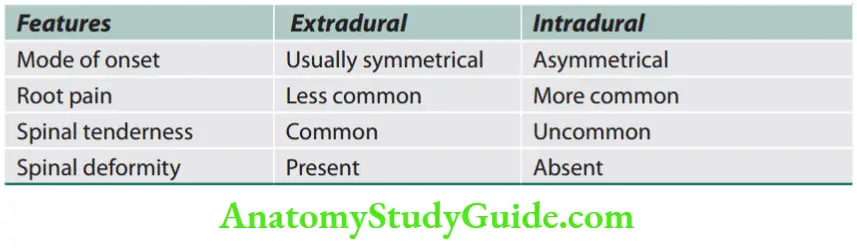
Causes of Compressive Myelopathies
Question 58. Write a short note on common causes of compressive myelopathies.
Answer:
Localization of the level of lesion in a compressive myelopathy
- Distribution root pain: Ask for specific dermatomes involved.
- The upper border of sensory loss: Examine the patient from below upward for a demonstration of the upper border of sensory loss (spinothalamic tract).
- Girdle-like sensation or sense or constriction at the level of lesion (involvement of posterior column).
- Zone of hyperesthesia or hyperalgesia (zone of hyperesthesia is present just above the level of girdle-like sensation and is due to compression of posterior nerve roots.
- Analysis of abdominal reflex: If the upper abdominal reflex is intact with loss of the middle and lower one, the site of the lesion is probably at T10 spinal segment.
- Atrophy of the muscles in a segmental distribution (due to anterior horn cells).
- Loss of deep reflexes: If the particular segment is involved. The reflexes will be brisk below the involved segment.
- Deformity or any swelling in the vertebra
- Tenderness in the vertebra
- The area of sweating may help (lack of sweating below the level) in localizing the level of the lesion.
- The level can also be localized by X-ray of the spine, myelography, CT scan, or MRI.
Beevor’s Sign
Analysis of Beevor’s sign: It is a medical sign seen in the selective weakness of the lower abdominal muscles.
Rectus abdominis is innervated by the terminal branches lower six or seven thoracic spinal nerves via the lower intercostal and subcostal nerves.
If a lesion lies above T6, the entire rectus abdominis is weak so there is no contraction of the muscle.
If it is at or below T10, the upper abdominal muscular function is preserved, whereas the lower abdominal muscles are weak.
Therefore, when the head is flexed against resistance (patient supine), the intact upper abdominal muscles pull the umbilicus upward and the shift of the umbilicus 3 cm when the head flexed is considered significant.
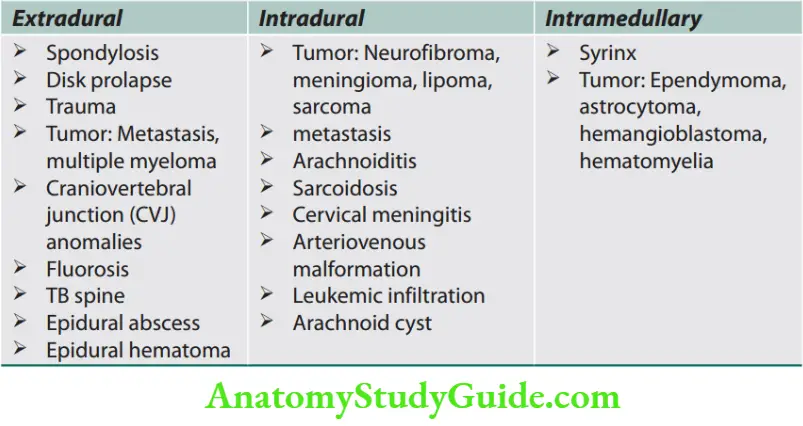
Question 59. List the differences between paraplegia in flexion and paraplegia in extension.
Answer:
The differences between paraplegia in flexion and paraplegia in extension.
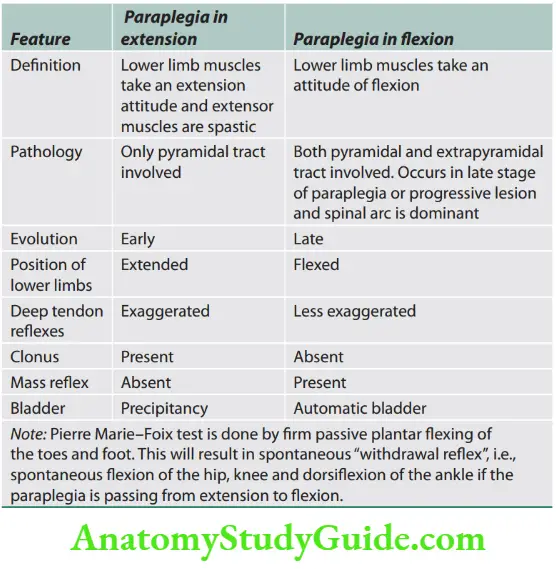
Flexor Spasms
- After recovery from spinal shock, many types of innocuous or noxious cutaneous or muscle stimuli to the lower limb can elicit a prolonged, coordinated pattern of hip flexion and ankle dorsiflexion, similar to flexion withdrawal.
- It is attributed to increased hyperexcitability of the spinal cord circuitry and leads to flexor spasms.
- Spinal cord lesions are associated with flexor spasms except for incomplete and high spinal cord lesions that usually have dominant extensor tone.
Acute and Subacute Spinal Cord Diseases
Spinal Cord Compression
Question 60. Write a short note on the causes of extramedullary spinal cord compression.
Answer:
- Spinal cord compression is one of the common neurological emergencies in clinical practice.
- Mechanism of damage: A space-occupying lesion within the spinal cord may damage nerve tissue either by direct pressure or indirectly by interfering with blood supply.
- Edema due to venous obstruction impairs neuronal function, and ischemia due to arterial obstruction may cause necrosis of the spinal cord.
- Consequences: During early stages, the damage is reversible but severely damaged neurons cannot recover.
- Hence, it is important to diagnose and treat early.
Various causes of spinal cord compression
Causes of spinal cord compression.
Vertebral (80%)
- Vertebral body destruction by bone metastases, e.g., breast, prostate, bronchus, myeloma
- Disk and vertebral lesions Trauma (extradural), chronic degenerative, and acute central (intervertebral) disk prolapse.
- Inflammatory: Tuberculosis, Staphylococcus aureus, melioidosis
Meninges (intradural, extramedullary) (15%)
- Tumors (e.g., meningioma, neurofibroma, ependymoma, metastasis, lymphoma, leukemia)
- Inflammatory: Epidural abscess, epidural hemorrhage/hematoma
Spinal cord (intradural, intramedullary) tumors (5%)
- Extramedullary, e.g., meningioma or neurofibroma, metastasis
- Intramedullary, e.g., glioma or ependymoma
Investigations
Patients with acute or subacute spinal cord syndrome should be investigated urgently.
- MRI of the spine: MRI is the investigation of choice. MRI can define the extent of compression and associated abnormality in the soft tissue.
- Plain X-rays of the spine: They may show destruction of bone and soft tissue abnormalities.
- Chest X-ray: It may show evidence of systemic disease
- Myelography
- CSF: If there is a complete spinal block, CSF shows a normal cell count with a raised protein causing yellow discoloration of the fluid (Froin’s syndrome).
- Serum vitamin B12
- Biopsy: If a secondary tumor is causing the cord compression, a needle biopsy may be an established diagnosis.
Symptoms and signs of spinal cord compression
Symptoms of spinal cord compression
- Pain: Occurs early. Localized over the spine or in root distribution.
- May be aggravated by coughing, sneezing, or straining
- Sensory: Occurs early. Paresthesia, numbness or cold sensations (especially in the lower limbs), spread proximally to a level on the trunk
- Motor: Occurs late. Weakness, heaviness, or stiffness of the limbs (commonly legs)
- Sphincters: Occurs late. Urgency or hesitancy of micturition, and retention of urine
Signs of spinal cord compression: Vary according to the level of the cord compression and the structures involved
Cervical, above C5: Frequently life-threatening
- Upper motor neuron signs and sensory loss in all four limbs (quadriplegia)
- Weakness of the diaphragm (phrenic nerve)
Cervical, C4–C5: Quadriplegia with preserved respiratory function
Cervical, C5–T1
- Lower motor neuron (LMN) signs and segmental sensory loss in the arms; upper motor neuron (UMN) signs in the legs
- Weakness of respiratory (intercostal) muscle
Thoracic cord
- Spastic paraplegia with a sensory level on the trunk
- Weakness of legs, sacral loss of sensation, and extensor plantar responses
- Midline back pain is a useful localizing sign
Lumbar cord and Cauda equina
- The spinal cord ends at the T12/L1 spinal level.
- Spinal lesions below this level can cause only LMN signs by affecting the cauda equina
- L1–L2—cremasteric reflex is a cutaneous reflex useful in the localization of lumbar cord disease
- L2–L4 paralyzes flexion and abduction of the thigh, weakens leg extension at the knee, and abolishes the patellar reflex
- L5–S1 paralyzes movements of the foot and ankle, flexion at the knee, and extension of the thigh, and abolishes the ankle jerk (S1)
Brown–Séquard Syndrome
Question 61. Write a short note on the causes and clinical manifestations of Brown–Séquard syndrome.
Answer:
It refers to findings seen when the damage is confined to one side (lateral half ) of the spinal cord.
With compressive lesions, there is a band of pain at the level of the lesion in the distribution of the nerve roots involved by compression.
Causes: Due to extramedullary lesions, usually caused by penetrating injuries (gunshot) or tumors.
Clinical Features
- Sensory
- Ipsilateral loss of proprioception due to posterior column involvement
- Contralateral loss of pain and temperature due to involvement of lateral spinothalamic tract.
- Motor
- Ipsilateral spastic weakness due to descending corticospinal tract involvement
- Lower motor neuron signs at the level of the lesion.
Chronic Myelopathies
Syringomyelia
Question 62. Write a short essay on the pathogenesis of syringomyelia.
Answer:
- A syrinx is a fluid-filled cavity within the spinal cord. Syringomyelia is a cavitary expansion of the spinal cord.
- Syrinxes commonly develop in the lower cervical and high thoracic regions or in the high cervical region.
- They may extend proximally to the medulla or pons (syringobulbia).
- Pathogenesis: More than 50% are associated with Arnold–Chiari type I malformation characterized by herniation of cerebellar tonsils through the foramen magnum.
- This abnormality at the foramen magnum probably allows normal pulsatile CSF pressure waves to be transmitted to fragile tissues of the cervical cord and brainstem.
- This results in secondary cavity formation. There is gradual destruction of spinothalamic neurons, anterior horn cells, and lateral corticospinal tracts.
- It leads to progressive myelopathy.
- The classic presentation is a central cord syndrome with dissociated sensory loss and areflexic weakness in the upper limbs.
- Sensory loss: Most cases begin asymmetrically with unilateral sensory loss. The sensory loss has a distribution that is “suspended” over the nape of the neck, shoulders, and upper arms (cape distribution) or in the hands.
- Neurogenic arthropathies (Charcot’s joint): Most common in the shoulder. Thoracic kyphoscoliosis is common.
Management
- Treatment depends on the underlying lesion.
- Benign tumors should be surgically excised.
- Extradural compression due to malignancy has a poor prognosis.
- Useful function can be regained if treatment (e.g., radiotherapy), is started within 24 hours of the onset of severe weakness or sphincter dysfunction.
- Spinal cord compression due to tuberculosis may require surgical treatment and antituberculous chemotherapy.
- Traumatic lesions of the vertebral column needed treatment by neurosurgeon.La main succulent of syringomoyelia (subcutaneous thickening and swollen fingers)
Morvan’s syndrome: Progressive loss of pain sensation, ulceration, loss of soft tissues, and resorption of phalanges.
MRI scans: Accurately identify syrinx cavities and associated spinal cord enlargement.
Treatment is surgery: Posterior decompression of Arnold– Chiari malformation and the peritoneal shunt to drain the syrinx cavity.
Syringobulbia
It is characterized by dysphagia, nystagmus, pharyngeal and palatal weakness, asymmetric weakness, and atrophy of the tongue, and loss of pain, the temperature in the distribution of the
trigeminal nerve (facial sensory loss resembling distribution a balaclava helmet), involving the outer parts of the face but sparing the nose and mouth.
This pattern of facial sensory impairment may also be known as the onion-peel or onion-skin pattern.
Subacute Combined Degeneration of Cord
Question 63. Write a short essay on the etiology, clinical features/neurological signs, and management of subacute combined degeneration (SACD).
Answer:
It is myelopathy that develops due to nutritional deficiency of vitamin B12 deficiency, including pernicious anemia.
- This treatable myelopathy presents with paresthesia in the hand and feet, early loss of vibration and position sensation, and progressive spastic and ataxic weakness.
- Loss of reflexes due to a superimposed peripheral neuropathy in a patient who also has Babinski’s sign is an important diagnostic clue.
- The myelopathy of SACD tends to be diffuse rather than focal; signs are generally symmetric and reflect predominant involvement of the posterior and lateral tracts.
- Optic atrophy and irritability are prominent in advanced cases.
- Low vitamin B
- 12 levels confirm the diagnosis.
- MRI spinal cord: Inverted V sign or rabbit ear sign due to T2 hyperintensity along the posterolateral column of the spinal cord.
Treatment is by replacement therapy, beginning with 100 mg of intramuscular vitamin B 12 repeated at regular intervals or by subsequent oral treatment.
Causes of posterolateral column disease.
- Vitamin B12 deficiency
- AIDS
- HTLV-associated myelopathy
- Cervical spondylosis, hypocupremia, vitamin E deficiency
Paraplegia
Question 64. Write a short note on the causes and differential diagnosis of spastic paraplegia.
Answer:
Paraplegia is a weakness or paralysis of both lower limbs, sparing the upper limbs.
Causes: It can occur in disorders of the cerebrum, spinal cord, spinal roots, peripheral nerves, or muscles. It is usually due to disorders of the spinal cord.
Spastic Paraplegia
Spasticity is due to an upper motor neuron lesion. It is usually produced in subacute or chronic lesions. Acute lesions usually cause flaccid paralysis.
Causes of spastic paraplegia
![Neurology Causes of spastic paraplegia [upper motor neuron (UMN) type lesion]](https://anatomystudyguide.com/wp-content/uploads/2023/08/Neurology-Causes-of-spastic-paraplegia-upper-motor-neuron-UMN-type-lesion.png)
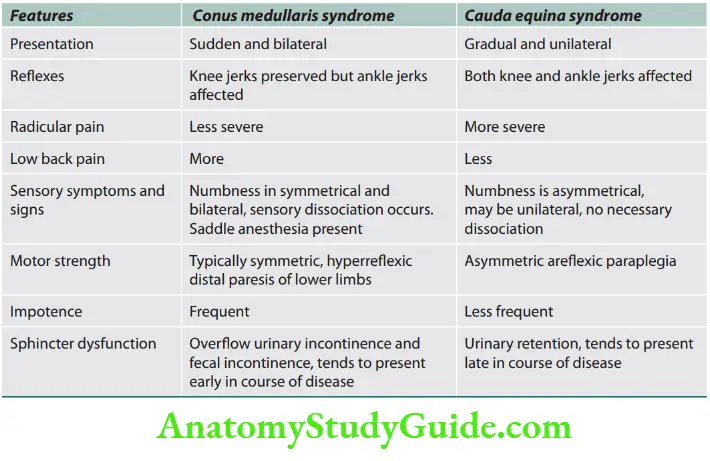
Cauda Equina Syndrome
- The epiconus comprises the cord segment between L4 and S1, corresponding to the T12 and L1 vertebrae.
- The most distal bulbous part of the spinal cord (L1–L2) is called the conus medullaris.
- The conus medullaris consists of the cord segment between S2 and S5 as well as the coccygeal segments.
- Distal to this end of the spinal cord is a collection of nerve roots (L2–L3 onward to coccygeal), which are horsetail-like in appearance called the cauda equina
- MRI showing cervical syringomyelia.
![Neurology Causes of spastic paraplegia [upper motor neuron (UMN) type lesion]](https://anatomystudyguide.com/wp-content/uploads/2023/08/Neurology-Causes-of-spastic-paraplegia-upper-motor-neuron-UMN-type-lesion-1.png)
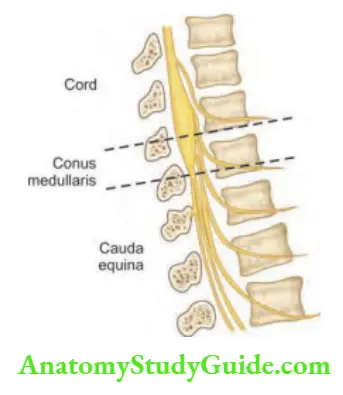
Causes of posterolateral column disease.
- Vitamin B12 deficiency
- AIDS
- HTLV-associated myelopathy
- Cervical spondylosis, hypocupremia, vitamin E deficiency
Complications of paraplegia.
- Pressure/bed sore
- Urinary infection and renal stones
- Fecal impaction with intestinal obstruction
- Contracture of limbs
- It may lead to death
Question 65. Write a short note on the differences between conus medullaris lesions and cauda equina lesions.
Answer: The differences between conus medullaris and cauda equina syndromes.
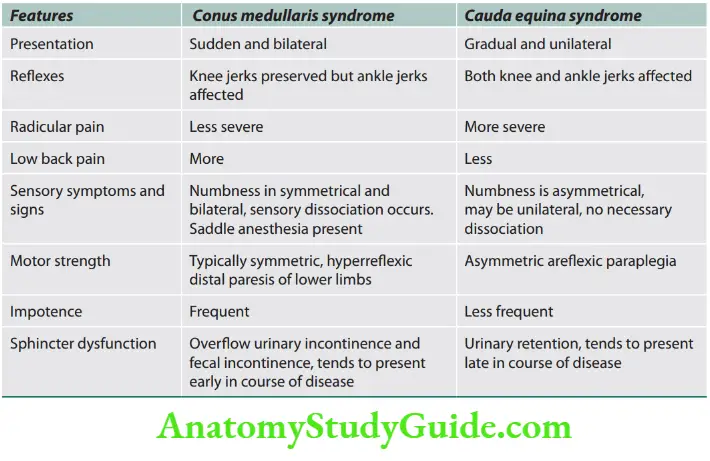
Complications of Paraplegia
Management of Paraplegia
- Skincare: Prevention of pressure sores. They develop due to loss of sensation and reduced blood supply. The following measures will be helpful.
- Turn the patient every 2–4 hours to avoid pressure over bony prominences.
- Keep the skin dry and clean.
- Specially designed mattresses like water or air-cushioned bed will be useful.
- The patient should be prevented from lying on the side of the pressure sores.
- Needs aseptic care and may require skin grafting.
- Bladder
- Aseptic intermittent catheterization.
- An indwelling catheter is not advisable as it predisposes to infection, reduces bladder capacity, and promotes calculus formation.
- Prompt treatment of urinary infections and maintenance of adequate fluid intake.
- Bowel: Laxative to prevent constipation. If the fecal matter becomes hard, manual evacuation is done.
- Paralysis
- Spasticity can lead to contractures and flexor spasms. Hence, regular passive movements of the limbs should be encouraged.
- Posture should be such that flexion is prevented at joints.
- Drug treatment of spasticity: Baclofen, diazepam, and tizanidine. In severe spasticity, intrathecal baclofen is via a pump or sectioning of the anterior roots (rhizotomy).
- Rehabilitation: Use a caliper or wheelchair and physiotherapy.
Spinal Pain
Question 66. Discuss the different types of spinal pain.
Answer:
Radicular pain:
- It is unilateral, lancinating, dermatomal pain often exacerbated by cough, sneeze, or Valsalva maneuver.
- Common with extradural growths (e.g., neurilemmoma which is intradural extramedullary) and rare with intramedullary lesions.
Vertebral pain:
- It is an aching pain localized to the point of the spine involved in the compressive process and often accompanied by point tenderness.
- Common with neoplastic or inflammatory extradural lesions and infrequent with intramedullary or intradural extramedullary lesions.
Funicular (central) pain:
-
- It is deep, ill-defined painful dysesthesia, usually distant from the affected spinal cord level (and therefore of poor localizing value), probably related to dysfunction of the spinothalamic tract or posterior columns.
- It is common with intramedullary lesions and very unusual with extradural lesions.
- With dysfunction of the posterior columns in the cervical region, neck flexion may elicit a sudden “electric-like” sensation down the back or into the arms (Lhermitte’s sign or Barber’s chair syndrome).
- More reliable band-like radicular pain or segmental paresthesia may occur at the level of the lesion and may be of localizing value for the appropriate spinal level.
Causes of Flaccid Paraplegia (LMN type)
- Upper motor neuron lesion in shock stage, i.e. sudden onset or history of long duration as in extradural transverse myelitis and spinal injury
- Lesion involving anterior horn cells
- Acute anterior poliomyelitis
- Progressive muscular atrophy (a variety of motor neuron diseases)
- Diseases affecting nerve root: tabes dorsalis, radiculitis, GBS
- Diseases affecting peripheral nerves
- Acute infective polyneuropathy (GBS)
- High cauda equina syndrome
- The disease of peripheral nerves involving both lower limbs
- Lumbar plexus injury (psoas abscess or hematoma)
- Diseases affecting myoneural junction
- Myasthenia gravis (MG), Lambert–Eaton syndrome
- Periodic paralysis due to hypo- or hyperkalemia
Diseases affecting muscles: Myopathy.
Causes of Pure Motor Paraplegia
Causes of Quadriplegia
Weakness in all 4 limbs can occur in the lesions from the cortex to the C5 level of the spinal cord and various lower motor neuron lesions affecting anterior horn cells, roots, peripheral nerve, neuromuscular junction, and muscles.
Craniovertebral Junction Anomalies
Question 67. Discuss craniovertebral junction (CVJ) anomalies.
Answer:
- Abnormal general physical appearance: Head may be cocked to one side, short neck, and scoliosis.
- Neurological: Most common posterior occipital headache that worsens with neck flexion or extension. Others include myelopathy and brainstem and lower cranial nerve deficits.
- Vascular symptoms: Intermittent attacks of altered consciousness, confusion, transient loss of visual fields, and vertigo.
Classification of CVJ anomalies Causes of pure motor paraplegia.
- Hereditary spastic paraplegia
- Lathyrism
- Guillain–Barré syndrome
- Amyotrophic lateral sclerosis [motor neuron disease (MND)]
- Fluorosis
- Erb’s spastic paraplegia (syphilitic)
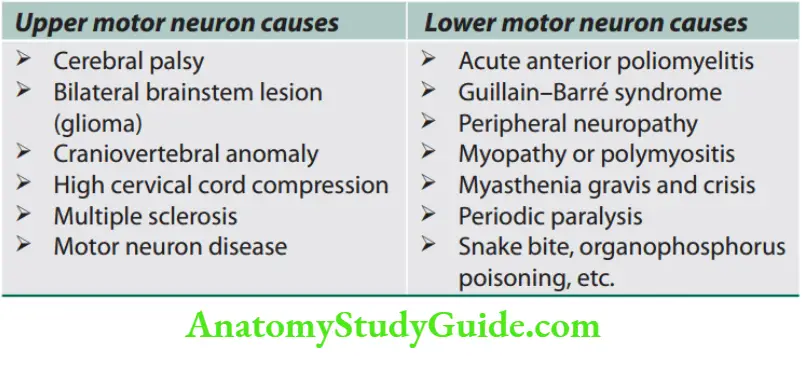
Hemiplegia
Question 68. Discuss hemiplegia in an elderly male. Give the differential diagnosis, investigations, and treatment.
Answer:
Hemiplegia is paralysis of one side of the body. Hemiparesis is the weakness of one side of the body. For differential diagnosis, investigations, and treatment, refer to individual diseases.
Causes of Hemiplegia
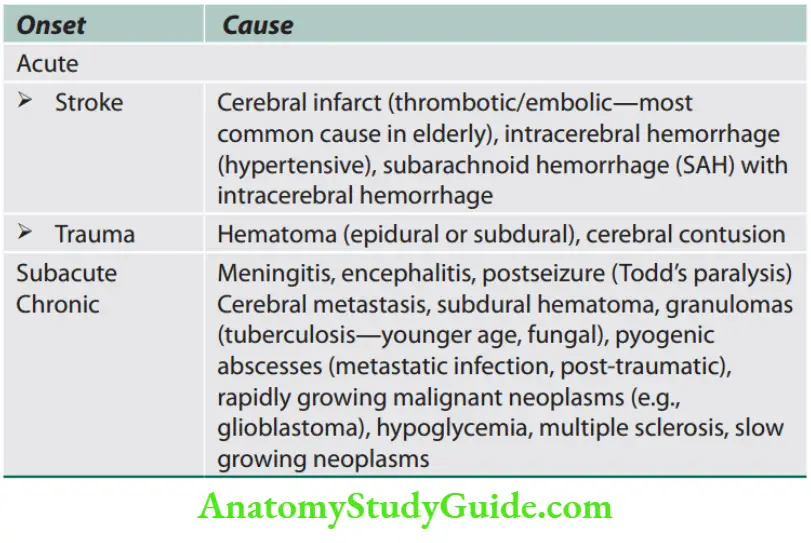
Small Muscle Wasting of the Hand
Question 69. Write a short note on the wasting of small muscles of the hand.
Answer:
Causes of small muscle wasting of the hand.
- Lesions of vertebra: Craniovertebral anomalies, vertebral metastasis
- Lesions of spinal cord: Syringomyelia, cord compression by tumor
- Lesion of anterior horn cell: Motor neuron disease (MND), poliomyelitis
- Lesions of spinal root: Cervical cord tumor, Pancoast tumor, cervical disk prolapse
- Lesion of brachial plexus: Cervical rib
- Lesions of peripheral nerve: Hansen’s disease, carpal tunnel syndrome, lead poisoning
- Diseases of muscle: Distal muscular dystrophy, polymyositis
- Disuse atrophy: Therapeutic immobilization (e.g., fracture), rheumatoid arthritis, postparalytic (hemiplegia)
Motor Neuron Diseases
Question 70. Write a short essay/note on motor neuron disease.
Answer:
Motor neuron disease (MND) is a devastating, progressive, heterogeneous group of neurodegenerative conditions caused by the loss of upper and lower motor neurons in the spinal cord, cranial nerve nuclei, and motor cortex.
There is no involvement of sensory or other nonmotor tracts. It causes progressive weakness and eventually death (usually as a result of respiratory failure or aspiration).
Various types of MND are
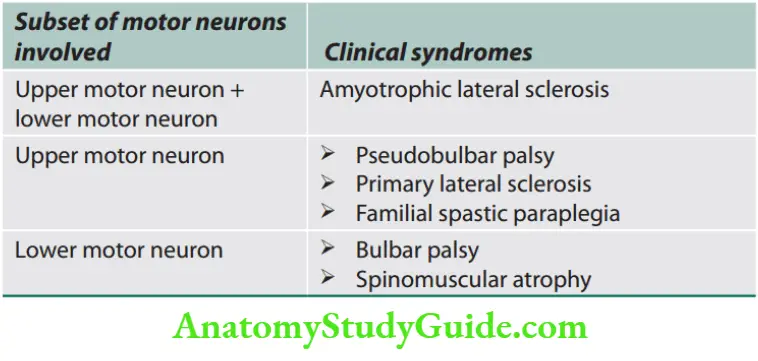
Clinical Features
Question 71. Write a short answer on three neurological signs of upper and lower motor neuron disease.
Answer:
There are four main clinical patterns. There is no involvement of the sensory system. Hence, sensory symptoms (e.g., numbness, tingling, and pain) are not present.
Amyotrophic Lateral Sclerosis
Question 72. Write a short essay/note on amyotrophic lateral sclerosis.
Answer:
- Named by Jean-Martin Charcot in the 19th century. Also known as Lou Gehrig’s disease after the famous baseball player diagnosed with ALS in 1930.
- Degeneration of the motor neuron (UMN and LMN) in motor cortex, brainstem, and spinal cord.
- Amyotrophy: Atrophy of muscle fibers consequent to denervation due to anterior horn cell degeneration
- Lateral sclerosis: Sclerosis of the anterior and lateral corticospinal tracts which are replaced by progressive gliosis.
- Epidemiology: Incidence is 1 to 2.7/lakh, prevalence is 2.7 to 7.4/lakh
- Sex predisposition: M > F (2:1 to 7:1), (F > M in bulbar onset ALS).
- Age: Risk increases with age up to 74 years. Peak onset—6th to the 7th decade (one to two decades earlier in India).
- Prognosis: 20% of patients survive for 5 years and 10%
survive for 10 years.

Etiopathogenesis
- Undetermined etiology. Complex genetic and environmental interaction for neuronal degeneration.
- MND is usually (90–95%) sporadic.
- Molecular pathway: Pathological hallmarks observed in axons of MND is the ubiquitinated cytoplasmic inclusions containing the RNA processing proteins TDP43 and FUS.
This indicates that protein aggregation may be involved in its pathogenesis similar to other neurodegenerative disorders.
Other mechanisms involved in pathogenesis may be oxidative neuronal damage, glutamate-mediated excitotoxicity, mitochondrial dysfunction, and impaired axonal transport.
Clinical presentation
- Typical/spinal form of ALS constitutes 2/3rd of cases. They present with simultaneous involvement of upper and lower motor neurons.
- Usually in one limb, spreading gradually. Often present with focal motor weakness of distal or proximal upper or lower limbs.
- The focal motor weakness spreads to contiguous muscles in the same region before the involvement of another region. Then involves other limbs and trunk muscles.
- Pseudoneurotic pattern: Resembles neuropathy, i.e., involvement of muscles in the apparent distribution of a peripheral nerve. Fasciculations are present on wasted muscles.
- Monomeric: Involvement of one limb (as wasted leg syndrome, monomeric amyotrophy, Chopra’s MND)
- Pseudopolyneuritic: Weakness in both distal lower limbs.
Mills hemiplegic variant: Weakness restricted to one half of the body. - Madras MND: Associated sensorineural deafness, bifacial and bulbar weakness, bilateral optic atrophy in age <15 years.
Bulbar/Pseudobulbar Palsy (20%)
- Initially involves the lower cranial nerve nuclei and their supranuclear connections.
- Presents with weakness of the respiratory group of muscles (e.g., dysarthria, dysphagia, nasal regurgitation of fluids, and choking).
- About 10% present with bilateral upper limb weakness and wasting, flail arm of flail person in a barrel syndrome.
- Head drop.
- Wasting and fasciculations of the tongue are seen almost in all patients
- Cramps in the thighs, abdomen, back, or even tongue.
- Nonmotor symptoms: Sleep disturbance, subtle cognitive dysfunction, and mood changes.
- Rarely involved: Bladder, bowels, autonomic, extraocular movements, and sensory.
Progressive Muscular Atrophy
- It presents as a pure lower motor neuron disease with symptoms of weakness, muscle wasting, and fasciculation.
- Usually starts in one limb and gradually spreads to involve other adjacent spinal segments.
Primary Lateral Sclerosis (Rare 1–2%)
The least common form of MND. It involves upper motor neurons and presents with a slowly progressive tetraparesis and pseudobulbar palsy.
Diagnosis of Amyotrophic Lateral Sclerosis
- No biological marker identified so far.
- Diagnosis is largely by a series of clinical and neurological examinations.
- MRI: Coronal T2WI shows bilateral symmetrical hyperintensity along the corticospinal tract (thin white arrows)
- forming a “wine glass appearance” or “garland sign”.
- Myelogram of the cervical spine (an X-ray analysis): Detects lesions in selected areas of the spinal cord.
- Muscle and/or nerve biopsy.
- Electromyography and nerve conduction velocity (NCV) to measure muscle response to nervous stimulation.
- Split hand phenomenon: In cases of severe changes in the thenar eminence and the relative sparing of hypothenar eminence, observed in the EMG study.
Differential Diagnosis of Amyotrophic Lateral Sclerosis
As amyotrophic lateral sclerosis is untreatable, all secondary causes should be excluded.
Differential diagnosis of amyotrophic lateral sclerosis.
Other motor neuron diseases:
- Primary lateral sclerosis (UMN only), progressive muscular atrophy (LMN only), progressive bulbar palsy
- Structural lesions: Cervical spondylosis, parasagittal/foramen magnum tumor, spinal cord AV malformation
- Neuropathies: Chronic inflammatory demyelinating polyneuropathy (CIDP)
- Myopathies: Polymyositis, inclusion body myositis
- Neuromuscular junction disorder: Myasthenia gravis
- Neurodegenerative diseases:
- Parkinson’s, progressive supranuclear palsy, multiple sclerosis.
- Malignancy: Primary/ metastasis to CNS, motor neuron syndromes with
multiple myeloma, lymphoma, lung, breast - Toxic exposure: Alcohol, heavy metals
- Endocrine: Hyperthyroidism, hyperparathyroidism
- Infectious: HIV, CMV
Treatment
No treatment to arrest degeneration.
Drug therapy
Riluzole: Dose 100 mg/day. It is a sodium channel blocker that reduces glutamate-induced excitotoxicity.
Adverse drug reactions include asthenia, nausea, alterations in liver function tests, headache, abdominal pain, and tachycardia.
Trial drugs: IGF-1, ceftriaxone, edaravone, tamoxifen.
Spasticity
-
- Baclofen 5–10 mg twice daily to three times daily.
- Tizanidine in the dose of 2–4 mg by mouth twice daily up to a total dose of 24 mg daily.
- Memantine starts at 5 mg daily, increasing by 5 mg a week to a maximum of 20 mg twice a day.
- Tetrazepam 50 mg at bedtime, increasing by 25 mg a day to a maximum dose of 150 mg taken two to three times a day.
Rehabilitation
- Foot drop splint
- Finger extension splint
- Respiratory support: Tracheostomy, positive pressure ventilation, cough-assisted device.
- Bulbar involvement: Gastrostomy (for feeding), speech therapy.
- Physiotherapy, speech, and occupational therapy.
Prognosis
The patient usually does not survive for >3 years, though rarely patients may survive for a decade or longer.
Neurogenic Bladder
Nerve Supply of Urinary Bladder
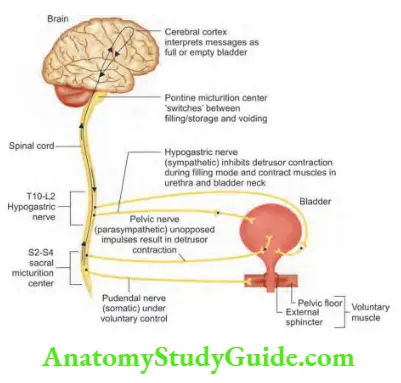
Question 73. Discuss the types of neurogenic bladder.
Answer:
Dysfunction of the urinary bladder due to neurological disorders is known as neurogenic bladder.
Various causes of neurogenic bladder
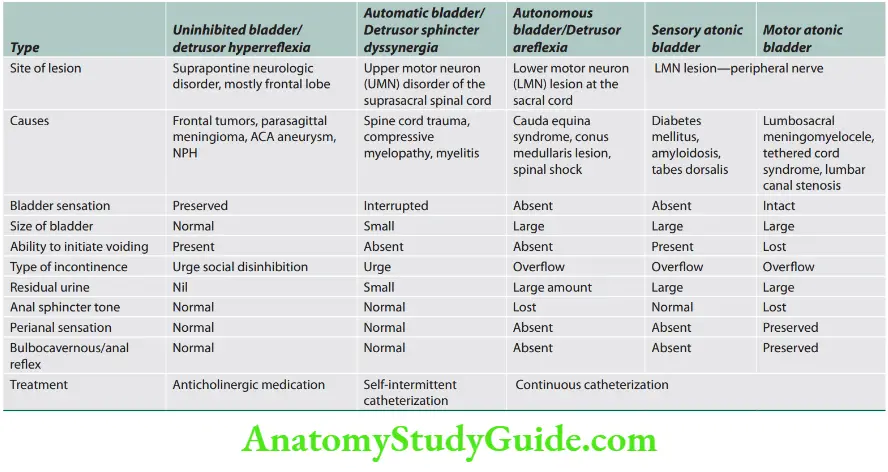
Transverse Myelitis
Question 74. Write a short note on transverse myelitis.
Answer:
Transverse myelitis (TM) is an acute, usually monophasic, demyelinating inflammatory disorder affecting the spinal cord.
It is characterized by acute or subacute motor, sensory, and autonomic (bladder, bowel, and sexual) spinal cord dysfunction.
- Myelitis refers to inflammation of the spinal cord and transverse signifies the involvement across one spinal cord level.
- Usually, 1 or 2 spinal segments are affected with part or all of the cord area at that level involved.
- Varying degrees of motor, sensory, and autonomic disturbances are
produced.
Causes of Transverse Myelitis
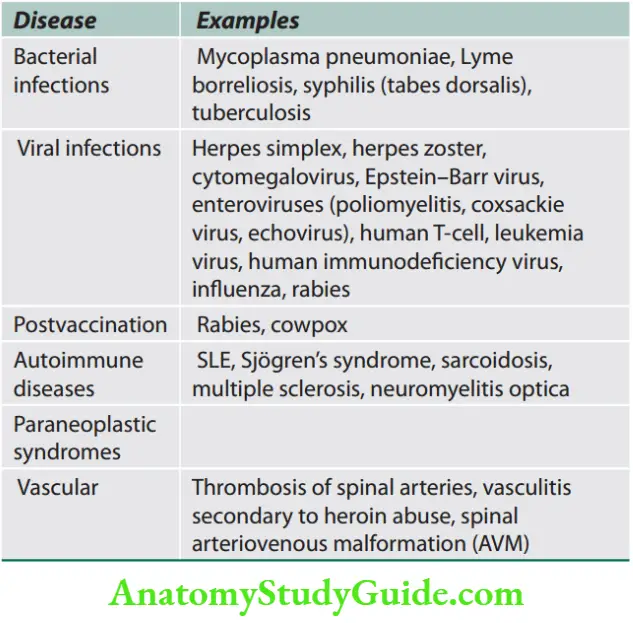
Question 75. Write a short note on the clinical features of transverse myelitis.
Answer:
It is usually thought to be postinfectious in origin and up to half of idiopathic cases will have a preceding respiratory or gastrointestinal illness.
It is one of the causes of a noncompressive spinal cord syndrome in which an immune-mediated process may be responsible for neural injury of the spinal cord.
Clinical Features
Age: It can occur at any age.
Symptoms develop rapidly over several hours to several weeks which may worsen maximally within 24 hours.
Symptoms
- Lower limb weakness.
- Sensory: Sensation is diminished or absent below the level of involved spinal cord.
- Few patients experience tingling or numbness in the legs (subacute paraparesis). Pain and temperature sensation are diminished.
- Bladder and bowel dysfunction: It results in disturbed sphincter control.
- Lhermitte’s sign: It is the radiation of paresthesias down the spine or limbs with neck flexion. It may be positive. It suggests intrinsic cervical spinal cord lesion.
Diagnosis
- First, exclude any mass-occupying lesion compressing the spinal cord.
- Early surgical decompression results in complete recovery. MRI and CSF analysis may be useful in the diagnosis.
MRI: MRI is sensitive and shows spinal cord swelling and edema with gadolinium-enhancing lesions (single or multiple) at the affected level(s). MRI is also useful for excluding other treatable causes of spinal cord dysfunction (e.g., spinal cord compression).
- CSF examination: Shows cellular pleocytosis (usually monocytes/lymphocytes) often with polymorphs at the onset.
- Protein is slightly increased and IgG index is elevated. IgG index is a measure of intrathecal synthesis of immunoglobulin (Ig) and is calculated using the following formula: (CSF IgG/serum IgG)/(CSF albumin/serum albumin).
Normal value is = 0.85 oligoclonal bands are usually absent.
Tests for exclusion of other treatable causes: These include chest X-ray, tuberculin test, ESR, serologic tests (e.g., for mycoplasma, Lyme disease and HIV), vitamin B12 and folate levels, antinuclear antibodies (ANAs) and CSF and blood for VDRL (Venereal Diseases Research Laboratory) tests.
Treatment
- Treat the underlying cause or associated disorder. Otherwise, it is mainly supportive.
- High-dose corticosteroids, intravenous immunoglobulin (IVIg) and plasmapheresis are used in the treatment of idiopathic cases.
- For severe, refractory cases: A course of azathioprine, methotrexate, mycophenolate, or oral cyclophosphamide.
The outcome is variable and evolves over days. There may be no recovery. If present it may be partial or complete (often partial) and follows over weeks or months (1–3 months).
Diseases Of The Peripheral Nervous System
Introduction
Diseases of the PNS are common. They may affect the motor, sensory or autonomic components, either in isolation or in combination.
Cranial nerves III to XII share the same tissue characteristics as peripheral nerves and are prone to the same range of diseases.
Site of lesion: It may be
- Dorsal or ventral nerve root (radiculopathy),
- Brachial/lumbosacral nerve plexus (plexopathy) or
- Cranial nerves (except I and II), and other sensory, motor, autonomic or mixed nerves (neuropathy).
- Neuropathy is a pathological process affecting a peripheral nerve or nerves.
- It may be axonal, demyelinating or neoronopathy.
Classifiation
Peripheral nerve disorders can be broadly classified into three categories.
1. Mononeuropathy simplex: Signifies involvement of a single peripheral nerve [e.g., median nerve in carpal tunnel
syndrome (CTS)].
2. Mononeuropathy multiplex (now called multiple mononeuropathies): Simultaneous or sequential several individual nerves involvement usually at random and noncontiguous (e.g., vasculitis, HIV leprosy).
3. Polyneuropathy: Function of numerous peripheral nerves is affected at the same time.
This leads to a predominantly diffuse, distal and symmetric deficit usually commencing peripherally.
It may be acute, chronic, static, progressive, relapsing or toward recovery.
They are motor, sensory, sensorimotor and autonomic. They are classified into demyelinating and axonal types, depending on principal predominant pathological process.
Typically, there is widespread loss of tendon reflexes with distal weakness and distal sensory loss.
Damage to peripheral nerve may affect the nerve cell body (axon) or the myelin sheath (Schwann cell), leading to axonal or demyelinating neuropathies.
Clinical Features
- Motor nerve involvement: It produces features of a lower motor neuron lesion.
- Sensory nerve involvement: It features depend on the type of sensory nerve involved; small fiber neuropathies are usually painful, present with paresthesias. Large fiber neuropathies cause sensory ataxia.
- Autonomic involvement: It may cause postural hypotension, disturbance of sweating, cardiac rhythm, and gastrointestinal, bladder and sexual functions.
Commonly autonomic involvement complicates other neuropathies.
Diagnosis: By clinical pattern, nerve conduction/EMG, nerve biopsy, usually sural or radial, and detection of systemic or genetic disease.
Mononeuropathies
Focal involvement of a single nerve and implies a local process and may be due to direct trauma, compression or entrapment, leprosy, vascular lesions, neoplastic compression or infiltration, etc.
Peripheral Nerve Compression and Entrapment
- Nerves are susceptible to mechanical compression at a certain location (e.g., ulnar nerve at the elbow, common peroneal nerve at the head of the fibula, radial nerve at spiral groove of humerus, lateral cutaneous nerve of thigh at inguinal ligament, posterior tibial at tarsal tunnel). Focal compression or entrapment is the common cause of a mononeuropathy.
- Entrapment occurs in relatively tight anatomical passages (e.g., the carpal tunnel).
- At the site of compression, focal demyelination and mild degeneration of distal axonal develops.
- Predisposing causes for entrapment neuropathies include diabetes, excess alcohol or toxins, or genetic syndromes.
- These are recognized mainly by clinical features and diagnosis is confirmed by nerve conduction studies.
- Usually recover once the primary cause is removed, either by avoiding the precipitation of activity or by surgical decompression.
Carpal Tunnel Syndrome
Question 76. Write a short essay/note on carpal tunnel syndrome and its causes.
Answer:
Common mononeuropathy due to entrapment of median nerve at the wrist.
Carpal tunnel syndrome is usually not associated with any underlying disease.
However, it may be seen in: hypothyroidism, 3rd trimester of pregnancy, rheumatoid disease, amyloidosis in dialysis patients, and acromegaly.
Clinical features: Nocturnal pain, paresthesia/tingling on palmar aspect of hand and/or forearm and fingers.
It is usually poorly localized and not confined to the anatomical sensory territory of the nerve. At later stages, weakness and wasting of thenar muscles develop.
Tinel’s sign (elicited by tapping the flexor aspect of the wrist: this causes tingling and pain) and Phalen’s test positive. (In Phalen’s, the symptoms are reproduced on passive maximal flexion of wrist).
Treatment: Wrist splint at night or a local steroid injection in mild cases.
In pregnancy, CTS is self-limiting and subsides during postpartum. Definitive treatment is surgical decompression of the carpal tunnel.
Mononeuropathy Multiplex
- Simultaneous/sequential damage to multiple noncontiguous nerves (peripheral or cranial nerves).
- Causes: Ischemia caused by vasculitis (e.g., Churg–Strauss), microangiopathy in diabetes mellitus.
- Less common causes include HIV and hepatitis C infection, granulomatous, amyloidosis, leukemic, or neoplastic infiltration, neurofibromatosis, Hansen’s disease (leprosy), toxins, paraneoplastic, and sarcoidosis.
- Clinical features: Symptoms will depend on the specific nerves involved.
- Several nerves may be affected sequentially or simultaneously, e.g., ulnar, median, radial and lateral popliteal nerves.
- When multifocal neuropathy is symmetrical, it is difficulty distinguishing it from polyneuropathy.
- Treatment: Glucocorticoids, cytotoxic agents.
The causes of peripheral nerve disorders

Polyneuropathies (Peripheral Neuropathy)
Question 77. Write a short essay/note on peripheral neuropathy and its causes/etiology.
Answer:
Polyneuropathy is characterized by a “length dependent” pattern, occurring first in the longest peripheral nerves and affecting the distal lower limbs before the upper limbs.
Sensory symptoms and signs occur in an ascending “glove and stocking” pattern.
Types of peripheral neuropathy.
Causes: Many diseases can produce polyneuropathy
However, the etiology is unknown in about 50% of cases.
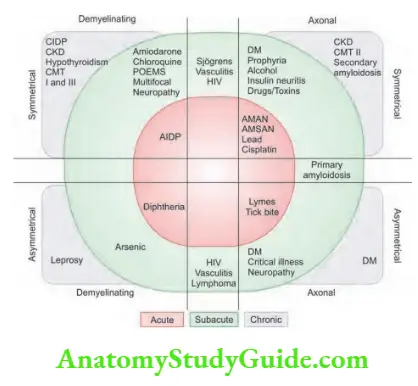
Common causes of axonal and demyelinating chronic polyneuropathies

Question 78. What are causes of peripheral nerve thickening?
Answer:
Causes of peripheral nerve thickening
- Leprosy
- Chronic inflammatory demyelinating
- polyneuropathy (CIDP)
- Amyloidosis
- Charcot–Marie–Tooth disease
- Neurofibromatosis
- Diabetes
- Acromegaly
- Refsum disease
- Idiopathic
- Dejerine–Sottas disease
- Relapsing Guillain–Barré syndrome
Approach to Neuropathy




Pattens Of Neuropathy
- Pattern 1: Symmetric proximal and distal weakness with sensory loss
- Inflammatory demyelinating polyneuropathy (GBS and CIDP)
- Pattern 2: Symmetric distal weakness with sensory loss
- Metabolic disorders, hereditary toxins, drugs
- Pattern 3: Asymmetric distal weakness with sensory loss
- Multiple nerves—vasculitis
- Single nerves/regions—compressive mononeuropathy and radiculopathy
- Pattern 4: Asymmetric distal weakness without sensory loss
- Motor neuron disease—with upper motor neuron findings
- Multifocal motor neuropathy—without upper motor neuron findings
- Pattern 5: Asymmetric proximal and distal weakness with sensory loss
- Polyradiculopathy or plexopathy due to diabetes mellitus
- Meningeal carcinomatosis
- Pattern 6: Symmetric sensory loss without weakness
- Cryptogenic sensory polyneuropathy (CSPN), metabolic (diabetes and others) drugs, toxins
- Pattern 7: Symmetric sensory loss and distal areflexia with upper motor neuron findings
- Vitamin B12 deficiency, HIV, hepatic disease
- Pattern 8: A symmetric proprioceptive sensory loss without weakness
- Sensory neuronopathy (ganglionopathy)
- Pattern 9: Autonomic symptoms and signs
- Neuropathies associated with autonomic dysfunction
- Pattern 10: Syndrome of A/C ascending motor paralysis
- Guillain–Barré syndrome/acute idiopathic polyneuritis
- Diphtheria
- Porphyria
- Triorthocresyl phosphate (TOCP) poisoning
- Paraneoplastic
- Post-vaccinial
- Pattern 11: Syndrome of subacute sensory motor neuropathy
- Deficiency—alcoholic beriberi, pellagra, vitamin B12
- Toxins—arsenic, lead (Pb), mercury (Hg)
- Drugs—nitrofurantoin, isoniazid (INH), dapsone, disulfiram, clioquinol
- Uremic
- Diabetes mellitus
- Polyarteritis nodosa (PAN), sarcoidosis
Approach to a Patient with Neuropathy
Investigation of Peripheral Neuropathy

Approach to a patient with neuropathy.
- Look for 6Ds
- Distribution of the deficits
- Duration
- Deficits (which fibers are involved?)
- Disease pathology (axonal or demyelinating or mixed)
- Developmental or inherited neuropathy
- Drug/toxin exposure
Treatment
- Depends on the underlying cause.
- Symptomatic treatment (more details refer diabetic neuropathy.
- Paresthesia: Carbamazepine (300–1200 mg/day), amitriptyline (25–50 mg/day) or aspirin (350–1200 mg/day) pregabalin, gabapentin, duloxetine.
- Weakness: Physiotherapy.
Guillain–Barré Syndrome and other Immune-mediated Neuropathies
Question 79. Write a short essay/note on Guillain–Barré syndrome and the diagnostic criteria for Guillain–Barré syndrome.
Answer:
Guillain–Barré syndrome is a heterogeneous group of immune-mediated conditions.
Guillain–Barré syndrome is the most common acute, severe fulminant polyradiculopathy/
polyneuropathy.
- Usually demyelinating or rarely axonal.
- Often postinfectious, postvaccinal basis
- Monophasic does not recur.
- Subtypes of Guillain–Barré Syndrome
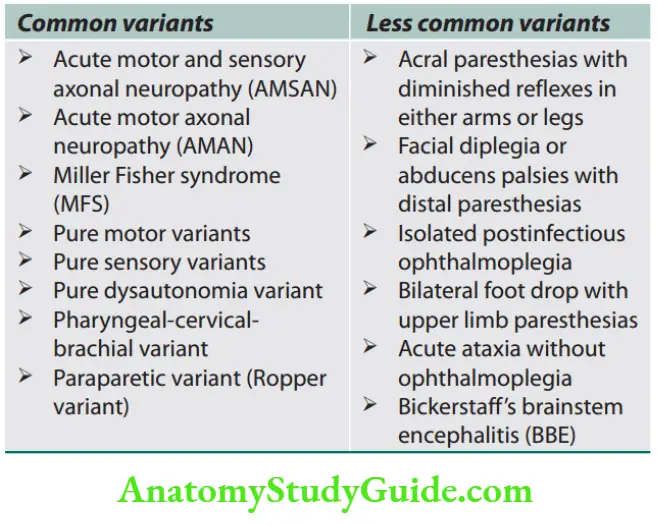
Question 80. What are the variants of Guillain–Barré syndrome?
Answer:
Etiology
Antecedent causes
- Majority of patients (70%) have preceding acute, influenza-like illness or GI infection.
- Infections preceding GBS may be due to Campylobacter jejuni, cytomegalovirus, EBV, herpes virus, CMVand Mycoplasma pneumoniae, or prior recent vaccination (e.g., for swine flu, influenza, rabies (old types).
- Frequently develop in patients with lymphoma, HIV seropositive, and SLE.
Immunopathogenesis
- Guillain–Barré syndrome is an acute-onset immune-mediated demyelinating neuropathy. The autoimmune basis for AIDP (GBS) and other subtypes.
- Both cellular and humoral immune mechanism causes damage. Immune response to self-antigen, infection (mentioned above under “Antecedent causes”), or vaccine induce antibody responses.
- Molecular mimicry, i.e., sharing of homologous epitopes between microorganism oligosaccharides and nerve gangliosides (e.g., GM1), misdirect the antibodies against host peripheral nerves.
- Antiganglioside antibodies mostly to GM1 common in GBS (20–50%) and anti-GQ1b IgG antibodies are seen in Miller Fisher syndrome (MFS) (>90%).
Clinical Features
- Hallmark is an acute/rapid onset of paralysis (predominantly motor paralysis with/without sensory) evolving over days
or weeks with loss of deep tendon reflexes/jerk (areflexia). - Motor paralysis predominant. Weakness beginning in the distal limbs (from lower to upper limbs) that rapidly ascends to affect proximal muscle function (“ascending paralysis”).
- It is more marked in legs than arms and proximally than distally.
- Pain in the low back, neck, and shoulder, in 50% and is often occur early.
- The weakness progresses proximally over few days to a maximum of 4 weeks.
- Sensory involvement is minimal and may precede muscle weakness. It presents as distal paresthesia or loss of pain sensation.
- Facial and respiratory muscle develops in 20–30% of cases requiring ventilatory support.
- No fever or constitutional symptoms at the onset of weakness.
Bladder dysfunction in late and severe cases. - Clinical worsening in 4 weeks reaching a plateau and no further progression.
- Autonomic disturbances common like fluctuation of BP, postural hypotension, cardiac dysrhythmias.
- Pain—common symptom—acute/deep aching pain in weak muscles. It is self-limited.
- Physical examination shows diffuse weakness with loss of reflexes.
- Miller Fisher syndrome: It presents with ophthalmoplegia, ataxia and areflexia.
- Often preceded by diarrhea due to Campylobacter jejuni infection. Bickerstaff’s brainstem encephalitis (BBE) is characterized by alteration in consciousness, paradoxical hyperreflexia, ataxia, and ophthalmoparesis.
- Asbury and Cornblath criteria for GBS.
Investigations/Diagnosis
- CSF findings: Develop after 1 week of illness:
- Raised protein (100–1,000 mg/dL), normal sugar, little or no
pleocytosis. Cell count generally <10 cells/mm3; rarely may be up to 50 cells/mm3, but never above that. This is called as albumincytological dissociation. - CSF pleocytosis (up to 50 cells) is common in patients who have GBS
and concurrent HIV infection.
- Raised protein (100–1,000 mg/dL), normal sugar, little or no
- Electrodiagnostic study: Nerve conduction studies:
- In mild/early stage: Normal
- In demyelination: Prolonged distal latencies, slow conduction, velocity, conduction block, prolonged F wave latencies. Absent H-reflex.
- In primary axonal: Reduced amplitude of compound action potential
without slow conduction.
- Antibodies:
- Antibodies against GQ1b, a ganglioside component of nerve are found in about 25%, usually the motor axonal form.
- Miller Fisher syndrome: Antibodies against GQ1b (ganglioside) have
90% sensitivity.
Diffrential Diagnosis
Exclusion of other causes of an acute neuromuscular paralysis: For example, poliomyelitis, botulism, diphtheria, localized spinal cord syndromes or myasthenia gravis, vasculitis, toxins (organophosphates, lead), diphtheria, porphyria, spinal cord or cauda equina syndrome.
Question 81. Write a short essay/note on principles of management of Guillain– Barré syndrome.
Answer:
Treatment
- Early treatment, each day counts, >2 weeks of 1st motor symptoms immunotherapy not effective.
- Initiative with high dose IVIg/plasmapheresis/combination.
- Intravenous immunoglobulin administration has fewer side effects (0.4 g/ kg daily infusion) for 5 days.
- GBS autoantibodies are neutralized by antibodies in IVIg.
- Patients should be screened for IgA deficiency because severe allergic reactions due to IgG antibodies may develop in patient with congenital IgA deficiency.
- Plasma exchange: 50 mL/kg, on 5 separate occasions over 1–2 weeks.
Glucocorticoid no role. - Full recovery in 55–69% by 1 year.
- Worsening case: Monitoring in intensive care unit (ICU)—blood pressure, cardiac, and nutrition.
- Maintenance of airway and breathing. Ventilatory support
may be needed. - 20-30-40 rule: vital capacity <20 mL/kg, negative inspiratory
pressure < −30 cm H 2O, maximum expiratory pressure <40 cm H 2O is an indication for intubation. - Supportive measures: Deep vein thrombosis prophylaxis, tracheostomy, chest physiotherapy, skin care, bed sore, joint physiotherapy daily reassurance.
Plasma Exchange (Plasmapheresis)
Question 82. Write a short note on plasma exchange (PE) therapy.
Answer:
Plasma exchange (also called plasmapheresis): Plasmapheresis involves the removal of small amounts of plasma (<15% of the patient’s total blood volume).
It can reduce the amount of abnormal protein in the blood. Indications for PE therapy.
Neuromuscular Junction Disorders
Myasthenia Gravis
Question 83. Discuss the etiology, clinical features, investigations, diagnosis, and management/treatment of myasthenia gravis (MG).
Answer:
Myasthenia gravis is an autoimmune neuromuscular junction disorder. Weakness and fatigue of skeletal (preferentially ocular, facial and bulbar) muscles.
Asbury and Cornblath criteria for Guillain–Barré syndrome (GBS).
Required features
- Progressive weakness in both arms and legs
- Areflexia (or hyporeflexia)
Features supportive of diagnosis
- Progression of symptoms over days to 4 weeks
- Relative symmetry
- Mild sensory signs or symptoms
- Cranial nerve involvement, especially bilateral facial weakness
- Recovery beginning 2–4 weeks after progression ceases
- Autonomic dysfunction
- Absence of fever at onset
- Typical CSF (albuminocytologic dissociation)
- EMG/nerve conduction studies (characteristic signs of a demyelinating process in the peripheral nerves)
Features casting doubt on the diagnosis
- Asymmetrical weakness
- Persistent bladder and bowel dysfunction
- Bladder or bowel dysfunction at onset >50 mononuclear leukocytes/mm 3 or presence of polymorphonuclear leukocytes in CSF
- Distinct sensory level
Features that rule out the diagnosis
- Hexacarbon abuse
- Abnormal porphyrin metabolism
- Recent diphtheria infection
- Lead intoxication
- Other similar conditions: poliomyelitis,
- botulism, hysterical paralysis, toxic neuropathy
Indications for plasma exchange therapy.
- Glomerulonephritis—anti-GBM disease
- Guillain–Barré syndrome
- Myasthenia gravis
- Autoimmune encephalitis
- Prerenal transplant
- Systemic vasculitis not responding adequately to immunosuppressive therapy
- Thrombotic thrombocytopenic purpura
Pathophysiology
Pathogenesis of Myasthenia Gravis
Myasthenia gravis is an autoimmune disease.
- Antibodies:
- Anti-AChR antibodies: It is detected in about 80 to 85% of patients.
- Anti-MuSK antibodies: A second group of autoantibodies against muscle-specific receptor tyrosine kinase (anti-MuSK) antibodies have been found in anti-AChR antibody negative
(about 15–20%) patients.
- Changes in thymus: Thymus is abnormal in ~75% of patients with MG. In ~65% the thymus is “hyperplastic”, with active germinal centers. In 10% of patients have thymic tumors (neoplastic). Muscle like cells (myoid cell) within thymus, bearing AChRs on their surface may serve as autoantigen and trigger immune response.
Coexisting Autoimmune Diseases
- Hashimoto’s thyroiditis/thyrotoxicosis (in 5–10%)
- Rheumatoid arthritis
- Pernicious anemia
- Scleroderma
- Lupus erythematosus
Clinical Features
- Age and gender: MG is common in women than in men (2:1). The disease presents with two peaks of incidence, below or above the age of 50, termed early-onset MG (EOMG) and late-onset MG (LOMG), respectively.
- Cardinal feature: Fluctuating weakness (that worsens with exertion and decrease with rest/sleep) and fatigability of muscles. Increase with exercise.
- Muscle affected: Diplopia and ptosis due to involvement of extraocular muscles.
- Show more focal muscle involvement (neck, shoulder, facial, jaw, respiratory, and bulbar muscles).
- Respiratory weakness: Respiratory muscles involvement may become so severe as to require respiratory assistance.
- Patient is said to be in due to diaphragmatic and intercostal muscle weakness. Aspiration may occur if the cough is ineffectual.
- Ocular MG when weakness is exclusive to the eyelids and extraocular muscles, and generalized MG when weakness extends beyond these ocular muscles.
- No sensory signs or signs of involvement of the CNS.
- Aggravating factors: Exertion, hot climate, infection, emotion, pregnancy, menstruation, drugs (amino glycosiden phenytoin).
- A temporary increase in weakness may follow vaccination, menstruation and exposure to extremes of temperature.
- Course: Variable, exacerbation and remission in early years. Remission is incomplete and temporary.
Most cases have a protracted, lifelong course. Exacerbations are usually unpredictable and unprovoked.
Myasthenic Crisis
- A rapid and severe deterioration of myasthenia is called “myasthenic crisis” can bring patient to the brink of respiratory failure and quadriparesis in hours.
- A respiratory infection or a sedative medication with NM (neuromuscular) block may be the reason.
- It can develop at any time after the diagnosis of myasthenia.
Anticipate if patient is restless, anxious with diaphoresis and develops tremor. - Require respiratory support.
Diagnosis
Diagnosis is based on the basis of clinical history, physical findings and two-positive tests (demonstration of autoantibodies and electrophysiological studies).
Serological tests: Antibodies against AChR or MuSK.
Pharmacological tests:
- Anticholinesterase test: Drugs inhibiting AChE allow ACh to interact repeatedly with limited number of AChRs producing strength improvement in myasthenic muscles.
- Edrophonium test (Tensilon test): Edrophonium is a rapidly acting acetylcholinesterase inhibitor.
- Onset of action is rapid (30 seconds) and lasts for short duration (5 minutes). It reverses of muscular weakness dramatically in myasthenia. Test dose (2 mg IV) is given to check for reactions.
- If there is definite improvement drop further test. Pathogenesis of myasthenia gravis. Antireceptor antibodies may inhibit/disturb the normal function of receptors.
- Autoantibodies to the acetylcholine receptor on skeletal muscle cells produce disease by blocking neuromuscular transmission and causing progressive muscle weakness.
- if negative further 8 mg IV is given.
- When the test is positive, it produces substantial improvement in weakness, ptosis, diplopia, nasal voice, etc., within 30 seconds and lasts for up to 5 minutes. Positive test is highly suggestive of MG and the sensitivity of the test is 80%.
- Adverse effect include nausea, diarrhea, salivation, bradycardia, syncope, abdominal cramps, fasciculation (treated with atropine—0.6 mg IV) bronchospasm and syncope.
- Neostigmine/Prostigmine test: 15 mg oral neostigmine, long acting and better evaluation.
- Ice on eyes/ice pack test: Apply an ice pack on eye for 3 to 5 minutes.
- The response is positive when there is increase in at least 2 mm of the palpebral fissure from before to after the test.
Electrodiagnostic:
Repeated nerve stimulation (RNS) test: Electric shock at 2–3 second given to appropriate nerve and corresponding muscle action potential measured.
In normal individuals the amplitude of the evoked muscle action potentials does not change at these rates of stimulation.
However, in myasthenic patients there is a rapid reduction of >10–15% in the amplitude of the evoked responses during repetitive stimulation.
Single fiber electromyogram: Most (95%) sensitive.
Other tests:
- CT, MRI, and X-ray: to exclude thymoma.
- CBC, ESR, RA factor, thyroid function test.
- Pulmonary function test, fasting blood sugar, Mantoux test, ANA, etc., to exclude other diseases.
Screening for associated autoimmune disorders.
- Lambert–Eaton myasthenic syndrome (LEMS) differs from MG in that in LEMS antibodies are against presynaptic calcium channels, deep tendon reflexes are absent and autonomic dysfunction in present and the weakness improves with activity in LEMS.
- It is most commonly associated with small cell carcinoma of lung and the treatment is with aminopyridines.
Question 84. Write a short note on drugs used in myasthenia gravis.
Answer:
Treatment
Goals: To increase the activity of acetylcholine on the remaining receptors at the NMJ. Stop the antibody mediated damage at the NMJ.
1. Oral anticholinesterases: Help weakness but do not change the natural history of myasthenia.
Pyridostigmine: Prolongs acetylcholine action by inhibiting cholinesterase.
Onset of action is within 15–30 minutes and lasts for 3–4 hours.
Dose 30–60 mg/3–4 times daily. Cholinergic crisis characterized by pallor, perspiration, pupillary constriction, paralysis, fasciculation and excessive salivation can be seen with drug over dosage.
Muscarinic effects (e.g., colic and diarrhea) are treated with oral atropine/propantheline (antimuscarinic).
2. Thymectomy
Indications: It is an expert surgical procedure that should be carried out in
All patients with generalized MG between the ages of puberty and at least 55 years. It may be required for thymoma to prevent spread and treat MG.
Advantages: Long-term benefit, improves prognosis, negligible medication.
3. Immunosuppression
- Effective in all. The choice is guided by benefit, risk and urgency. These are used in patients who do not respond to pyridostigmine or who develop relapse on treatment.
- For immediate: IVIg or plasmapheresis.
- For intermediate: Glucocorticoids, cyclosporine, tacrolimus for 1–3 months.
- For long-term: Azathioprine, mycophenolate mofetil (less commonly used) for months/years.
- For refractory: High dose of cyclophosphamide.
The differences between myasthenic crisis and cholinergic crisis
Treatment of myasthenic crisis
- Exacerbation of weakness due to diaphragm and intercostal involvement.
- Treat in ICU setup.
- Cause: Infection (most common), cholinergic crisis due to excess anticholinesterase dose.
- Respiratory assistance (preferably noninvasive, using BIPAP).
- Pulmonary physiotherapy.
- Plasmapheresis or IVIg.
- Stop anticholinesterase if using.

Diseases Of Muscle
- Muscle disease is rare and may be hereditary or acquired.
- Hereditary muscle diseases include muscular dystrophies, muscle channelopathies, metabolic myopathies (including mitochondrial diseases), and congenital myopathies.
- Skeletal muscle disease or myopathies are disorders with structural changes or functional impairment of muscle.
Muscular Dystrophies
Muscular dystrophies (hereditary myopathies) refer to a group of inherited myopathies/disorders characterized by progressive muscle weakness and wasting (due to the destruction of muscle) and may be associated with cardiac and/or respiratory involvement.
Question 85. Write a short note on muscular dystrophies or hereditary myopathies.
Answer:
Muscular dystrophy is subdivided by its mode of inheritance, age at onset, distribution of involved muscles, rate of progression, and prognosis
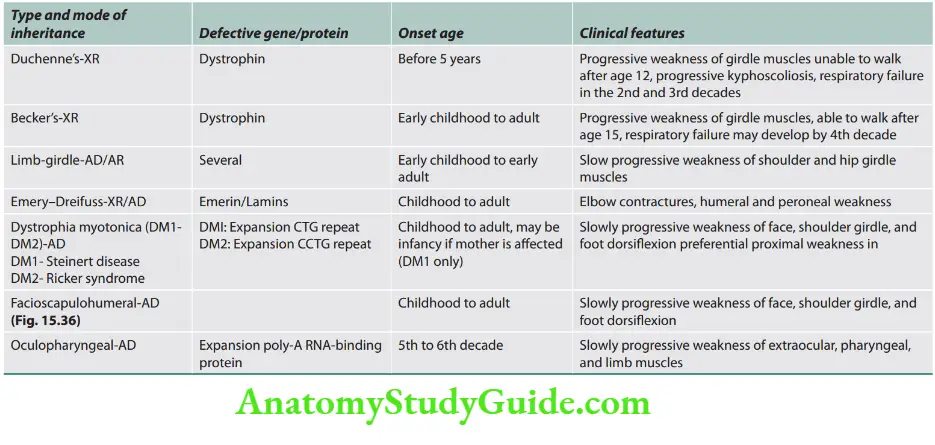
Duchenne Muscular Dystrophy
Question 86. Write a short essay on Duchenne muscular dystrophy.
Answer:
- A most common form of muscular dystrophy.
- Caused by a mutation of the gene responsible for producing dystrophin.
- Shows X-linked recessive inheritance and affects predominantly males.
- Duchenne dystrophy is present at birth, but the disorder usually becomes apparent between ages 3 and 5, by age 12, most patients are wheelchair dependent.
- Muscle selectively involved in order of appearance of weakness is iliopsoas, Quadriceps, Gluteus-Pretibial muscles, muscles of pectoral girdle, muscles of leg and forearm (at the end).
Clinical features: Use of Gower’s maneuver while getting up for the floor, toe walking due to foot drop with associated lordotic posture and pseudo hypertrophy of the calves.
Cardiomyopathy [dialated cardiomyopathy (DCM)] and mental impairment are associated features. But CHF is uncommon. Mental retardation is seen.
Investigations
- Creatine phosphokinase (CPK) levels are invariably elevated to between 20 and 100 times normal.
- Electromyography: Features typical of myopathy.
- Muscle biopsy: Definitive diagnosis can be established on the basis of dystrophin deficiency I in biopsied muscle tissue.
- Gold standard: It is genetic testing.
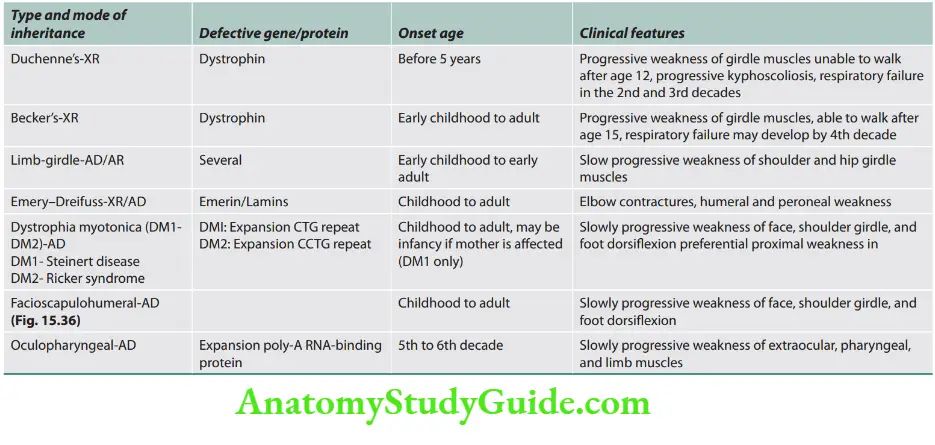
Becker Muscular Dystrophy
- X-linked disorder, results from defects of the same gene responsible for Duchenne dystrophy.
- Pattern of muscle wasting closely resembles that seen in Duchenne.
It is a less severe form of disease and much less frequent than Duchenne. - Clinical features: Becker patients ambulate beyond age 15, while patients with Duchenne dystrophy are typically in a wheelchair by the age of 12; allowing for a clinical distinction between Becker and Duchenne dystrophy.
- Cardiac involvement occurs in Becker dystrophy and may result in heart failure. Mental impairment is uncommon.
- Investigations: Serum CK measurement, EMG and muscle biopsy closely resemble those in Duchenne dystrophy.
Limb-Girdle Muscular Dystrophy
- Most are progressive and affect primarily the pelvic and shoulder girdle muscles.
- Inheritances can AR or AD and it is due to defect of sarcoglycan.
- Clinical features: Respiratory insufficiency from diaphragm weakness may occur.
- In some patients, cardiac involvement results in CHF or arrhythmias, occasional patients present with a cardiomyopathy. Intellectual function remains normal.
- Investigations: An elevated serum CK level, myopathic EMG findings, and muscle biopsy features indicative of myopathy.
Myotonic Dystrophy
- It is an autosomal dominant disorder and is the most common adult muscular dystrophy.
- Clinical features: Frontal baldness, cataracts (Christmas tree cataract), mental impairment, insulin resistance and gonadal atrophy. Hatchet like face, swan neck deformity is seen.
- Involvement of distal more than proximal, facial muscles and handgrip myotonia are pathognomonic features.
- Cardiacdisturbances occur in most patients with myotonic dystrophy. Disturbances in conduction system are common.
- Complete heart block and sudden death may occur. Mitral valve prolapse also occurs commonly in myotonic dystrophy patients.
- Myotonia may improve with phenytoin, mexiletine, and quinidine.
Endocrine Myopathies
Causes of endocrine myopathies are listed.
- Thyroid disorders: Hypothyroidism, hyperthyroidism
- Parathyroid disorders: Hyperparathyroidism,
- hypoparathyroid—overt myopathy
- Adrenal disorders: Glucocorticoid myopathy
- (endogenous/exogenous) most common, Cushingoid,
- adrenal insufficiency, primary hyperaldosteronism (Conn’s disease)
- Vitamin deficiency: Vitamin E, vitamin D
Question 87. Write a short note on causes of endocrine myopathies.
Answer:
Causes of high creatinine phosphokinase
- Brain injury or stroke
- Convulsions
- Delirium tremens
- Dermatomyositis or polymyositis
- Myocarditis
- Muscular dystrophies
- Myopathy
- Rhabdomyolysis
- Pulmonary and myocardial infarction
- Electric shock.
Question 88. List the causes of high creatinine phosphokinase.
Answer:
Disorders Of Cerebellar Function
- Cerebellum is an infratentorial structure in the posterior cranial fossa, attached to the brainstem by the superior, middle and inferior peduncles. The cerebellum is made up of two hemispheres separated by a midline vermis. There are three lobes anterior, posterior, and flocculonodular lobe.
- Midline lesions can produce severe gait and truncal ataxia.
- Cerebellar hemisphere lesions can produce classic ipsilateral limb ataxia (intention tremor, past pointing, and mild hypotonia).
Ataxia
Ataxia may be the result of cerebellar lesion or due to a combination of cerebellar and extracerebellar lesions.
Classifiation of Ataxia
Acquired or sporadic ataxias
- Vascular: Stroke (infarction, hemorrhage), AV malformations
- Infectious/postinfectious diseases: Acute cerebellitis, cerebellar abscess, postinfectious encephalomyelitis, HIV, chickenpox
- Toxin-induced ataxias: Alcohol, drugs (antiepileptic agents, lithium, antineoplastics, cyclosporine, metronidazole), heavy metals (mercury), 5-fluorouracil, cytosine arabinoside
- Structural and neoplastic causes: Gliomas, ependymoma, cerebellar tumor, meningioma, metastatic disease.
- Immune-mediated: Multiple sclerosis, cerebellar ataxia with anti-glutamic acid decarboxylase (GAD) antibodies, paraneoplastic syndrome (small cell lung cancer, breast or ovarian cancer, and lymphoma), gluten ataxia.
- Deficiency: Hypothyroidism, vitamin B1, vitamin B12, and vitamin E Genetic causes
- Autosomal recessive: Friedreich’s ataxia
- Autosomal dominant: Spinocerebellar ataxia
Symmetrical Cerebellar Ataxias
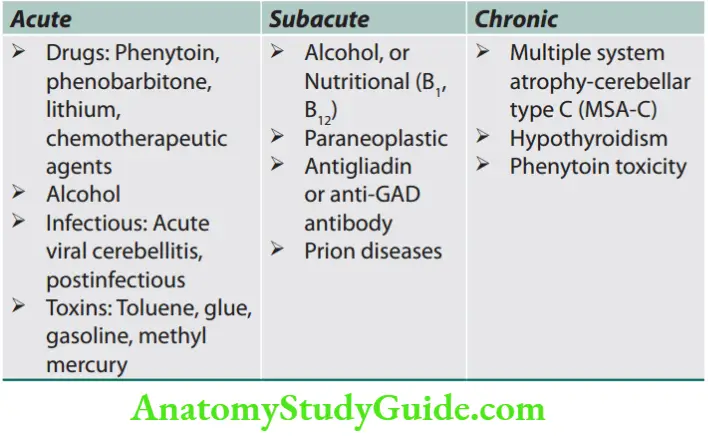
Asymmetrical Cerebellar Ataxias

Treatable Causes of Ataxia
- Hypothyroidism
- Ataxia with vitamin E deficiency (AVED)
- Vitamin B12 deficiency
- Wilson’s disease
- Ataxia with anti-gliadin antibodies and gluten senstive
- enteropathy
- Ataxia due to malabsorption syndromes
- Lyme disease
- Mitochondrial encephalomyopathies, aminoacidopathies,
- leukodystrophies, and urea cycle abnormalities
Clinical Features of Cerebellar Lesions
Cerebellar Ataxia
Question 89. Write a short note on cerebellar ataxia. (or) Write a short note on cerebellar signs.
Answer:
Lesions of the midline vermis of the cerebellum cause truncal ataxia, while lesions of the cerebellar hemispheres cause limb ataxia of the ipsilateral side.
1. Gait ataxia
- Patients will tend to stand with feet well apart and are often frightened to stand.
- Patients tend to reel to the side unilateral lesion or from side to side if central or bilateral (drunken gait) (even if supported).
- Walking along a line of the floor (tandem gait) demonstrates minor degrees of gait ataxia.
- Instability may increase if eyes are closed but patients do not fall.
- This is not a true positive Romberg’s test (true positive Romberg’s test is present when there is impaired joint proprioception/posterior column involvement is known as sensory ataxia).
Question 90. Write a short note on Romberg’s sign/test
Answer:
Romberg’s test is a test for sensory ataxia.
Romberg’s test/sign: Patient stands upright with the feet together (touching each other) and eyes closed.
When there is proprioceptive or vestibular deficit, balance is impaired only when eyes are closed, and the patient may fall if not caught.
Minimal lesions can be demonstrated by asking the patient to stand on his toes with eyes closed.
Principle of Romberg’s test: It is based on the principle that an individual requires at least two of the three following senses to maintain balance while standing:
- Proprioception (the ability to know one’s body in space)
- Vestibular function (the ability to know one’s head position in space); and
- Vision [which can be used to monitor (and adjust for) changes in body position].
2. Truncal ataxia
- Patients cannot sit or stand unsupported and tend to fall backward.
- It is caused by a midline cerebellar lesion, or may be a feature of post-chickenpox cerebellar syndrome.
- Truncal tremor may be evident—constant jerking of trunk and head (titubation).
- Lesions of the cerebellar hemisphere cause ipsilateral limb signs.
- The outstretched arm tends to be held hyper pronated at rest and at a slightly higher level than unaffected side (Riddoch’s sign) and rebounds upward if gently pressed downward and then suddenly released by the examiner.
- Finger-nose and heel-knee-shin tests will demonstrate even mild limb ataxia, with terminal intention tremor and dysmetria (past pointing).
- Limb rebound can be demonstrated by gently pushing down on outstretched arms and then suddenly releasing, causing the arm on the affected side suddenly to fly upward.
- Pendular knee jerks and hypotonia present.
3. Other signs produced by cerebellar lesions/diseases
- Cerebellar dysarthria: Spluttering staccato speech.
- Scanning dysarthria is a jerky and explosive speech with separated syllables may be demonstrated by asking the patient to repeat baby hippopotamus!
- Dyssynergia: It is difficulty in carrying out complex movements.
This results in breaking of an act into its components. - Macrographia: Writing may be larger than normal (contrast with micrographia of Parkinson’s disease).
Rapid alternating movements: Inaccuracies in rapidly repeated movements (dysdiadochokinesia).
This is demonstrated by getting the patient to tap back of their own hand repeatedly with the other hand, or to tap their foot on the floor.
Tremor: Unilateral or bilateral intention tremor, or a truncal tremor.
Nystagmus: Coarse nystagmus, worse, on looking to the side of lesion with fast component toward the affected side.
Pendular knee jerk: The reflexes are less brisk and slower in rise and fall. This produces pendular jerk at the knee.
It produces three on more swings at the knee when the knee reflex is elicited with patient in sitting posture and the legs hanging from bedside.
Nausea and vomiting: Sudden vomiting (without warning) after a positional change, without preceding nausea, is suggestive of a posterior fossa lesion.
Mnemonic for cerebellar signs—DANISH.
- Dysdiadochokinesia and Dysmetria
- Ataxia
- Nystagmus
- Intention tremor
- Slurred speech
- Hypotonia
Hereditary Cerebellar Ataxia—Friedreich’s Ataxia
- Most common form of inherited ataxia. The most common form of degenerative ataxia caused by unstable trinucleotide GAA expansion in the chromosome located on Question The protein involved is frataxin.
- It affects spinal cord—dorsal column, spinocerebellar tract, and pyramidal tract.
- There is also loss of dorsal root ganglion cells with depletion of large myelinated fibers in peripheral nerves.
- Age of onset is usually between 8 and 16 years.
Clinical Features
- Progressive ataxia of gait (due to spinocerebellar tract involvement).
- Lower limb areflexia and later generalized areflexia.
- Leg weakness and extensor plantar responses (due to pyramidal tract involvement).
- Reduced vibration and joint position sense in lower limbs. It is due to involvement of dorsal column.
- Sensory axonal neuropathy detected on nerve conduction test.
Other features: Dysarthria, scoliosis, nystagmus, optic atrophy, deafness, diabetes, pes cavus and cardiomyopathy.
Causes of extensor plantar response with absent ankle jerk.
- Friedreich’s ataxia
- Motor neuron disease (ALS)
- Taboparesis in neurosyphilis
- HIV myeloneuropathy
- Subacute combined degeneration of cord
- (vitamin B 12 deficiency)
- Hypocupremic myelopathy
- Conus-cauda lesion
Investigations
Nerve conduction study (NCS): Normal conduction velocity but small or absent sensory nerve action potential (SNAP) CT and MRI normal cerebellar anatomy, cervical atrophy.
DNA polymerase reaction to detect GAA expansion.
Treatment
No definite treatment, physiotherapy and occupational therapy, spasticity is treated with baclofen, orthopedic advice and surgery.
Spinocerebellar Ataxia (Late-Onset Hereditary Ataxia)
Question 91. Write a short note on the clinical features of spinocerebellar ataxias.
Answer:
Spinocerebellar ataxia (autosomal dominant cerebellar ataxia/ADCA) is an autosomal dominant disease that presents with slowly progressive cerebellar symptoms.
Sometimes with associated optic atrophy, ophthalmoplegia, pyramidal, and extrapyramidal structure.
MRI mostly shows olivopontocerebellar atrophy.
Sensory ataxia
It is due to severe sensory neuropathy, ganglionopathy, or lesions of the posterior column of the spinal cord, e.g., Sjogren’s syndrome, cisplatin, CIDP, paraneoplastic disorders, SACD, tabes dorsalis, Miller Fischer syndrome, celiac disease
Ataxia more at night or while walking through narrow passages (coffee A history of falling into the sink or imbalance when splashing water on the face (wash-basin sign), passing a towel over the face or pulling a shirt over the head should also be sought
Pseudoathetosis—“piano-playing” movements—when the patient has his arms outstretched and eyes closed, the affected arm will wander from its original position.
Vibration and position sense are usually lost together.
Positive Romberg’s test is a hallmark of sensory ataxia.
Vestibular ataxia is due to lesions of vestibular pathways resulting in impairment and imbalance of vestibular inputs, e.g., vestibular, neuronitis, and streptomycin toxicity.
Vertigo and associated tinnitus and hearing loss
The direction of the nystagmus is away from the lesion.
Optic ataxia was first described in a man with lesions of the posterior parietal lobe on both sides of the brain, later became known as Balint’s syndrome.
Among the symptoms that characterize the syndrome are a restriction of visual attention to single objects and a paucity of spontaneous eye movements.
Patients have difficulty completing visually guided reaching tasks in the absence of other sensory cues.
Frontal lobe ataxia (Bruns ataxia) is due to involvement of subcortical small vessels, Binswanger’s disease, multi-infarct state or NPH.
- The gait may appear to be a combination of awkward, magnetic (stuck to the floor), cautious, slow, and shuffling.
This is also known as a frontal gait disorder, referring to the frontal lobe conditions which often cause gait apraxia.
The characteristic features of different types of ataxia.
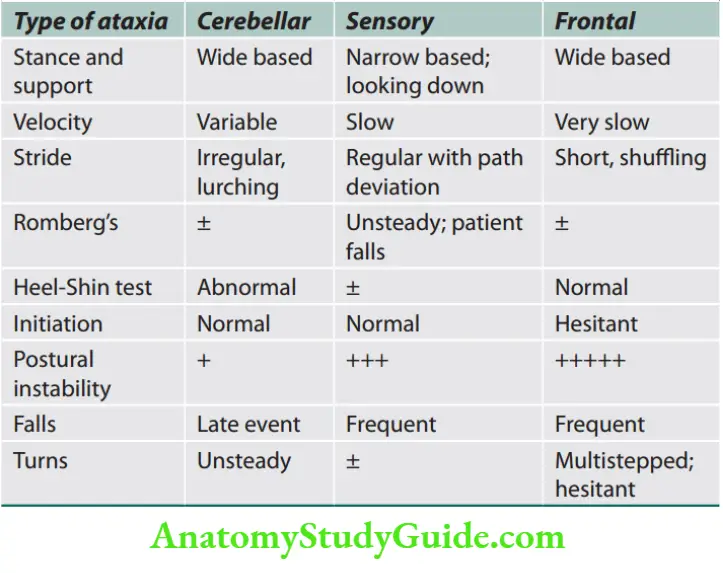
Movement Disorders
Types of Movements
Question 92. Write a short note on CNS disorders characterized by involuntary movements.
Answer:
Akathisia: A subjective feeling of inner restlessness that is relieved by movements (e.g., crossing and uncrossing legs, rocking back and forth and pacing).
Asterixis: Sudden periods of cessation of muscle contraction best seen when the patient’s arms are extended in front. It is a negative myoclonus.
Athetosis: Slow, sinuous, writhing movements, usually of the distal parts of the limbs.
Ballismus: Wild flinging, movements that represent large amplitude proximal choreiform movements. Ballismus is often unilateral (hemiballismus).
Chorea: Semipurposeful flowing movements that flit from one part of the body to another in a continuous and random pattern.
Chorea can be defined as involuntary movements that are abrupt, unpredictable and nonrhythmic, resulting from a continuous random flow of muscle contractions.
Dyskinesia: A general term for any excessive movement. The term dyskinesia is often used as an abbreviation for “tardive dyskinesia” (repetitive oral movements often seen in patients taking certain psychiatric medications).
Dystonia: Twisting movements that are often sustained for variable periods of time with a directional preponderance resulting in posturing.
Question 93. Write a short note on myoclonus and its causes.
Answer:
Myoclonus: Myoclonic movements are sudden, brief, shock-like involuntary movements, which are usually positive (caused by muscle contraction), but can sometimes be negative [due to brief loss or inhibition of muscular tonus, as in asterixis—for example, when caused by hepatic encephalopathy (liver flap) or in uremic encephalopathy]
Tics: Tics are sudden and jerky movements, but in this case the keyword for recognition is the “stereotyped” character of the recurrent movements. Repetitive, stereotypic movements or sounds that are suppressible and that relieve a feeling of inner tension.
Tremor: Regular, oscillatory movements that may be present at rest or with action.
Tremor is characterized by involuntary, rhythmic and sinusoidal alternating movements of one or more body parts.
Question 94. Write a short note on hemiballismus.
Answer:
Hemiballismus: Violent flinging movements of the limbs. It is usually unilateral, and hence called hemiballismus.
Causes of some important movement disorders
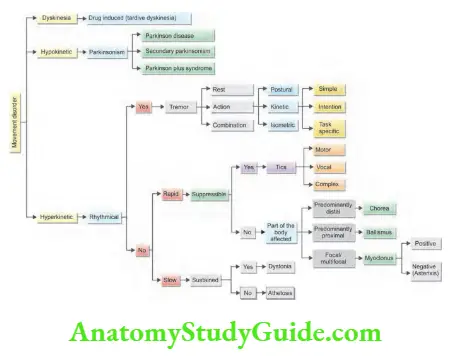
Parkinson’s Disease
Question 95. Describe the etiology, clinical features/ manifestations, diagnosis, and management of Parkinson’s disease.
(or)
What is Parkinsonism? How would you classify Parkinsonism?
(or)
Discuss the management of idiopathic parkinsonism.
Answer: Parkinsonism is a syndrome consisting of a variable combination of tremors, rigidity, bradykinesia, and a characteristic disturbance of gait and posture.
Classification of Parkinsonian disorders

There are many causes for Parkinsonism but the most common cause is Parkinson’s disease (PD).
Idiopathic Parkinson’s Disease (Paralysis Agitans)
- It is a chronic, progressive disorder in which idiopathic parkinsonism occurs without evidence of more widespread neurologic involvement.
- Parkinson’s disease (PD) is distinct from other parkinsonian syndromes both clinically and pathologically.
- Age and gender: Its incidence increases sharply with age. PD generally commences in middle or late life and average age of onset is about 60 years.
Prevalence is higher in men than women (M:F = 1.5:1). It leads to progressive disability with time.
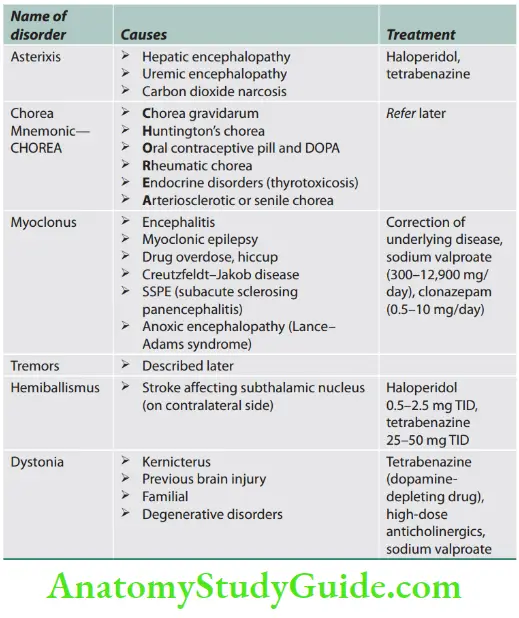
Etiology
Cause of idiopathic Parkinson’s disease is not known. Probably multiple interacting environmental risk factors and genetic susceptibility plays a role.
Environmental factors:
- Small increased risk with rural living and drinking well water.
- Pesticide exposure.
- Oxidative stress: Chemical compound 1-methy l-4-pheny l-1,2,3,6-tetrahydropyridine (MPTP) is a potent mitochondrial toxin. It causes severe Parkinsonism in young drug users of MPTP by producing oxidative stress leading to death of neuronal cell.
- Nonsmokers have a higher risk of PD than smokers.
Genetic factors: Genetic factors may play a role and several single genes causing parkinsonism have been identified.
- Sporadic: Idiopathic PD is not usually familial, but there is a significant genetic component in early onset PD (onset before 40).
- Mutations in many genes has been found in familial cases. Several genetic loci for Mendelian inherited monogenic forms of PD have been identified, designated as PARK 1 11. They are rare but cause early onset and familial PD.
Pathological features
- Hallmark of PD: Degeneration and depletion of the pigmented dopaminergic neurons in the substantia nigra pars compacta (SNc), reduced striatal dopamine, and the presence of α-synuclein and intracytoplasmic proteinaceous eosinophilic inclusions in nigral cells known as Lewy bodies.
- Probably environmental or genetic factors alter the α-synuclein protein, rendering it toxic. This leads to formation of Lewy body within the nigral cells. Lewy bodies are also seen in the basal ganglia, brainstem and cortex.
- Lewy bodies contain tangles of α-synuclein and ubiquitin. They become gradually more widespread and increase as the disease progresses.
- The loss of dopaminergic neurotransmission in the nerve cells (>80%) in the substantia nigra and other nuclei in the midbrain responsible for the symptoms of Parkinson’s disease.
Clinical manifestations
Motor symptoms: Always asymmetrical in onset and become bilateral within a year.
- Tremor is an early and presenting symptom in 70% of patients.
- Frequency is 4–6 Htremor and is typically most prominent at rest and worsens with emotional stress.
- Typically tremor starts with the fingers and hands at rest.
- Often described as pill rolling of finger and wrist, because the patient appears to be rolling something between thumb and forefinger.
- It often begins with rhythmic flexion-extension of the fingers, hand, or foot, or with rhythmic pronationsupination of the forearm.
- Initially, it may be confined to one limb or to the two limbs on one side before becoming more generalized.
- It also affects jaw and chin, but not the head.
- Disappear on voluntary movement and sleep.
Rigidity
- It is a sign rather than a symptom. Increased resistance to passive movement is characteristic clinical feature that accounts for the flexed posture of many patients.
- Rigidity causes stiffness and a flexed posture.
- Stiffness on passive limb movement is described as “lead pipe” rigidity because the increase in muscle tone is present throughout the range of movement.
- Unlike spasticity, it is not dependent on speed of movement.
- When tremor is superimposed on the rigidity, a ratchet like jerkiness is felt, described as “cogwheel” rigidity.
- Akinesia or bradykinesia
- Poverty/slowing of movement are the hallmark of PD.
- Slowness/difficulty of initiating voluntary movement and an
associated reduction in automatic movements, such as swinging of the arms when walking. - There is fixity of facial expression (facial immobility—mask-like face), with widened palpebral fissures and infrequent blinking.
- Repetitive tapping (at about 2 Hz) over the glabella (Glabellar tap) produces a sustained blink response (Myerson’s sign), in contrast to the response of normal subject. Frequency of spontaneous blinking decreases producing a serpentine stare.
- The combination of tremor, rigidity, and bradykinesia results in small, tremulous, and often illegible/difficult handwriting (micrographia).
- It results in difficulty in activities such as tying shoelaces or buttoning, and difficulty rolling over in bed.
Postural changes: A stooped posture is characteristic feature.
- Gait changes: Slow shuffling, freezing and reduced arm swing, small stride length, slow turns, festinating gait (tendency to advance rapid short steps) and catching center of gravity.
- Feet may be glued to floor. Postural instability and freezing may result in fall forward.
- Reduced eye blink.
Speech and swallowing: Speech becomes softer (soft voice—hypophonia), quiet, indistinct, flat/monotonous and stuttering.
Increased salivation/drooling, and dysphagia (swallowing difficulty is a late feature) which may lead to aspiration pneumonia as a terminal event.
Cognitive and psychiatric changes: Cognitive impairment/dementia, depression, sleep disturbances may be present.
Parkinson’s disease timeline
Nonmotor features
- Some nonmotor symptoms (NMSs) may precede the onset of more typical motor symptoms.
- Investigation/Diagnosis
- Diagnosis is made on clinical grounds.
- Structural imaging (CT or MRI) is usually normal.
Functional dopaminergic imaging: By single-photon emission computed tomography (SPECT) or positron emission tomography (PET) is abnormal and shows reduced uptake of striatal dopaminergic markers, particularly in the posterior putamen.
However, it is not specific to PD.
Dopamine transporter (DaT) imaging: It is performed by the use of a radiolabeled ligand binding to dopaminergic terminals to know the extent of nigrostriatal cell loss.
It may be rarely required to distinguish PD from other causes of tremor, or drug-induced parkinsonism.
Stages of Parkinson’s disease.
Question 96. Discuss the staging of Parkinson’s disease.
Answer:
Diffrential diagnosis
1. Secondary parkinsonism
Nonmotor symptoms of Parkinsons’s disease.
Autonomic dysfunction
- Orthostatic
- hypotension
- Urinary incontinence
- Constipation
- Sexual problems
Neuropsychiatric
- Anxiety
- Depression
- Apathy
- Psychosis
- Dementia
Sensory problems
- Reduces sense of smell (hyposmia)
- Pain
Sleep disorders
- Restless legs
- Insomnia
- Daytime somnolence
Rheumatological
- Frozen shoulder
- Periarthritis
- Swan neck deformity
Other
- Seborrhea
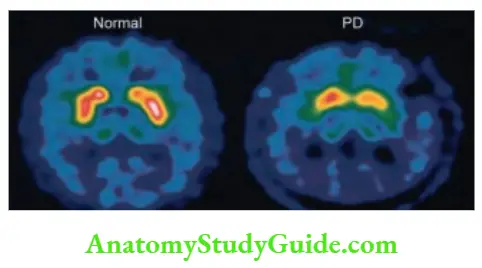
Causes of secondary parkinsonism.
Toxin: Manganese, 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine (MPTP), carbon monoxide, mercury,carbon disulfide, cyanide, methanol Viral: Encephalitis lethargica, Creutzfeldt–Jakob disease
Metabolic: Wilson’s disease Head injury: Punch drunk syndrome
Infectious: Postencephalitic, HIV, SSPE, Prion diseases
Drugs: Dopamine receptor blocking drugs, reserpine, tetrabenazine, alpha-methyldopa, lithium, flunarizine, cinnarizine
Vascular: Multi-infarct, Binswanger’s disease
Trauma: Pugilistic encephalopathy
Others: Parathyroid abnormalities, hypothyroidism, brain tumors, paraneoplastic, normal pressure hydrocephalus (NPH), psychogenic
Question 97. What are the “Parkinson plus” syndromes?
Answer :
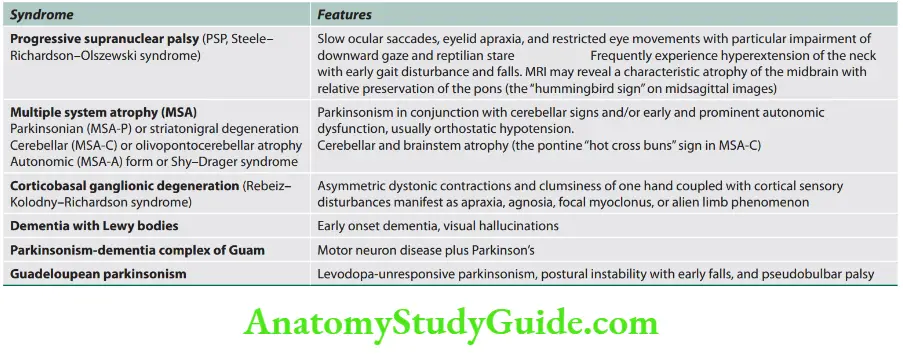
Question 98. Write a short essay/note on drugs used in Parkinson’s disease.
Answer:
Treatment
Symptomatic pharmacologic treatment: Drug treatment in PD is symptomatic rather than curative. None of the currently available drugs are neuroprotective.
Anticholinergic drugs
Nonselective muscarinic antagonists are helpful, especially in relieving tremors. For example, trihexyphenidyl, benztropine, and orphenadrine.
Treatment is started with a small dose (2 mg), which is gradually built up until benefit occurs or side effects limit further increments.
Adverse effects: Urinary retention, dry mouth, blurred vision, worsening of glaucoma, constipation, confusion and hallucinosis in the elderly.
Hence, rarely used as first-line drugs unless the patient has severe tremors.
They should be avoided in patients above 65 years of age.
Levodopa
Levodopa, the metabolic precursor of dopamine. It is the single most effective drug available for the treatment.
It provides symptomatic benefit in most patients with parkinsonism and is often particularly helpful in relieving bradykinesia.
Resolve hypokinesia and rigidity first and tremor later.
Levodopa is metabolized by MAO (monoamine oxidase) and COMT (catecholO-methyl-transferase).
Its plasma half-life is around 2 hours. Early use lowers mortality rate.
Combined with a dopa decarboxylase inhibitor—benserazide (co-beneldopa) or carbidopa (co-careldopa) to reduce the adverse effects (e.g., nausea and hypotension).
Adverse drug reactions:
Postural hypotension, fluctuations in response.
Mydriasis, brownish discoloration of the urine, abnormal smell, transient elevations of transaminases and BUN.
GIT effects: Nausea and vomiting.
Cardiovascular: Tachycardia, ventricular extrasystoles, atrial fibrillation.
Dyskinesias, behavioral disturbances.
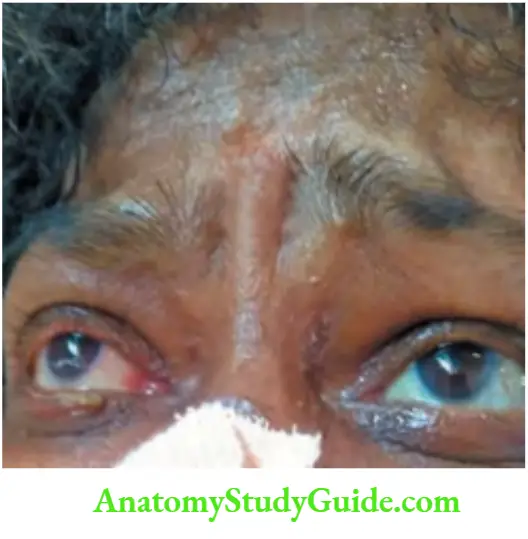

Stage Disease state
Unilateral involvement only, minimal or no functional impairment
Bilateral or midline involvement, without
impairment of balance
First sign of impaired righting reflx, mild to moderate disability
Fully developed, severely disabling disease; patient
still able to walk and stand unassisted
Confiement to bed or wheelchair unless aided
“On-off” effect: Important late complications of levodopa therapy. It is like a light switch; without warning, all of a sudden, person
goes from full control to complete reversion back to bradykinesia, tremor, etc.
It lasts from 30 minutes to several hours and then get control again.
The on-off phenomenon can be controlled in part by reducing dosing, intervals, administering levodopa 1 hour before meals and restricting dietary protein intake or treatment with dopamine agonists.
MAO-B Inhibitors
- Monoamine oxidase type B facilitates breakdown of excess dopamine in the synapse.
- They produce asymptomatic motor benefit when used as a monotherapy and enhance the efficacy of carbidopa levodopa formulations when used as adjuncts voided, e.g., selegiline, rasagiline.
- The addition of selegiline, a monoamine oxidase B inhibitor, reduces the metabolic breakdown of dopamine and may slow down the degeneration in the substantia nigra.
Dopamine receptor agonists
- Dopamine receptor agonists are classified as ergot derived (bromocriptine, pergolide and cabergoline) or nonergot-derived (pramipexole, ropinirole, rotigotine and apomorphine).
- Side effects: Produce impulse control disorders (e.g., pathological gambling, binge eating and hypersexuality) and daytime somnolence.
- Dopamine agonists are contraindicated in patients with psychotic disorders and are best avoided in those with recent myocardial infarction, severe peripheral vascular disease, or active peptic ulceration.
- Ergot-derived agonists are no longer recommended because of rare but serious fibrotic side effects including cardiac valvular fibrosis.
- Nonergot dopamine agonists are preferable to ergot-derived dopamine agonists.
- They are used as an alternative or an addition to levodopa therapy.
Question 99. Write a short note on COMT (Catechol-O-methyltransferase) inhibitors.
Answer:
COMT inhibitors
- Catechol-O-methyl-transferase produces a peripheral breakdown of levodopa (e.g., entacapone and tolcapone).
- Entacapone prolongs the duration of levodopa by decreasing its peripheral metabolism. The more potent tolcapone is less preferred because of rare but serious hepatotoxicity.
Dopamine facilitator
- Amantadine: It is an antiviral agent that potentiates dopaminergic function by influencing the synthesis, release, reuptake of dopamine.
- It has a mild antiparkinsonian effect and short-lived effect on bradykinesia. Hence, it is rarely used and are reserved for patients who are unable to tolerate other drugs.
- Amantadine-either alone or combined with an anticholinergic agent, helpful for mild parkinsonism.
- It acts by potentiating the release of endogenous dopamine.
Adverse effects: Livedo reticularis, peripheral edema, confusion and other anticholinergic effects.
-
- Peripheral dopamine decarboxylase inhibitors (PDI)
- It does not penetrate the BBB; reduce the peripheral metabolism of levodopa.
- Increase plasma levels of levodopa, prolongs the plasma half-life of levodopa, increase available amounts of dopa for entry into the brain and reduce the daily requirement of levodopa by 75%.
- For example, carbidopa, benserazide.
Newer modalities
- Transdermal delivery of Rotigotine improves motor functions.
- Subcutaneous apomorphine infusion
- L-dopa infusion into the duodenum
- Neuroprotective agents that alter pathogenesis
- MAO inhibitors: Selegiline and rasagiline
- Antiexcitotoxicity drugs: Riluzole
- Bioenergetic antioxidant agent, coenzyme Q10
- Antiapoptotic kinase inhibitors (e.g., CEP-1347)
Adenosine A2A receptor antagonists (e.g., istradefylline).
Surgical Treatment
- Indications: Most common indications for surgery in PD are intractable tremor and drug-induced motor fluctuations or dyskinesias.
Different surgeries are:
- Stereotactic surgery (ventrolateral thalamotomy) tried in unilateral cases.
- Pallidotomy improves tremor and dyskinesia
- Deep brain stimulation (DBS): Stereotactic insertion of electrodes into the brain is most often performed bilaterally and simultaneously.
Best site is subthalamic nucleus. DBS in these areas alleviates Parkinsonian motor signs particularly during the off periods and reduces troublesome dyskinesias, dystonia and motor fluctuations that result from drug administration.
DBS is usually reserved for patients with medically refractory tremor or motor fluctuations.
Newer Therapies for PD
Focused ultrasound
Unilateral focused ultrasound lesioning of the STN or thalamus (in tremor-dominant forms of PD) has been found to be beneficial in some patients, particularly if the symptoms are markedly asymmetric.
Cell replacement therapies
Fetal tissue-derived cell transplants—generation dopaminergic neurons from somatic human cells and improvement in the efficacy of differentiation protocols have led to a resurgence of cell transplantation
Surgical delivery of gene therapy
MRI-guided delivery of adeno-associated viral vector serotype-2 encoding the complementary DNA for the enzyme, aromatic L-amino acid decarboxylase (VY-AADC01) into the putamen.
Characteristic Features of Extrapyramidal Lesion
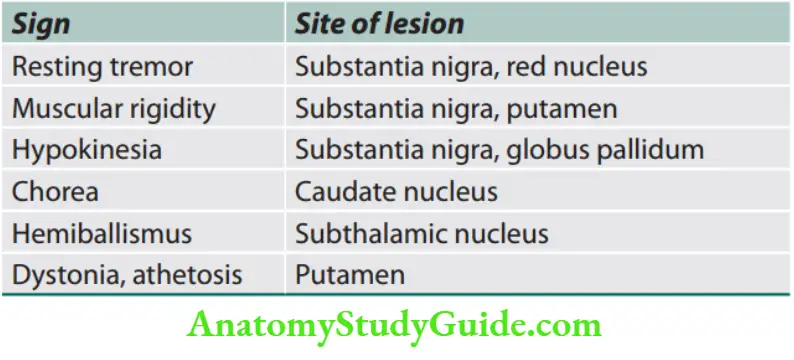
Chorea
Question 100. Write a short note on chorea and mention the disease which causes chorea.
Answer:
General Features
Irregular, semi-purposeful, abrupt, rapid, brief, jerky, unsustained movements that flow randomly from one part of the body to another. These movements disappear during sleep.
When choreic movements are more severe, assuming a flinging, sometimes violent, character, they are called ballism.
Signs in Chorea
- Involuntary protrusion and retraction of the tongue (jack in the box).
- Inability to hold the hands above the head with palms facing each other as it results in pronation of arms so that palms face outward (pronator sign).
- Milking action of patient’s fingers if asked to grasp the physician’s fingers (milk-maid sign).
Sydenham’s Chorea (Saint Vitus Dance)
- The most common cause of nonsuppurative complication of group A β-hemolytic streptococcal pharyngitis.
- It follows acute rheumatic fever by 4–6 months. Severity varies and the disorder may continue for a few months.
- It is due to molecular mimicry between streptococcal and CNS antigens.
- Infection by group A β-hemolytic streptococci in genetically predisposed individuals leads to the formation of cross-reactive antibodies.
- These antibodies disrupt the basal ganglia function. Inflammation is seen in the caudate nucleus.
Clinical features: It is a neuropsychiatric disorder.
- Clinical features include both neurological abnormalities (chorea, weakness, and hypotonia) and psychiatric disorders (such as emotional liability, hyperactivity, distractibility, obsessions, and compulsions).
- These abnormalities lead to an inability to perform normal activities of daily living (ADL) including eating, talking, dressing, writing, walking, learning, and socializing, and thus impact negatively on the child’s quality of life.
- Common sign is motor persistence. It can be demonstrated by an inability to sustain eye closure or tongue protrusion.
- Evaluation for valvular heart disease is a must. Antistreptolysin-O (ASLO) titers and ESR are often normal.
Treatment
Symptomatic
- Sodium valproate 200–600 mg TID is the first-line drug.
- If valproate is not effective, risperidone, a potent dopamine D2 receptor blocker, may be given to control chorea. The dose is 1–2 mg BID.
- Haloperidol 0.5–1.5 mg BD or TID is used occasionally.
- Other drugs include pimozide, carbamazepine, clonidine, and phenobarbital.
- Penicillin prophylaxis is necessary to reduce the risk of cardiac involvement due to future streptococcal infections.
Causes of chorea.
- Rheumatic (Sydenham’s chorea)
- Huntington’s chorea
- Encephalitis, e.g., Japanese encephalitis, measles, mumps
- Vascular, e.g., HIV-related (toxoplasmosis, progressive
- multifocal leukoencephalopathy, HIV encephalitis)
- Immunologic, e.g., systemic lupus erythematosus, antiphospholipid antibody syndrome, paraneoplastic syndromes, acute disseminated encephalomyelopathy, celiac disease
- Drugs, e.g., L-dopa, oral contraceptive, phenytoin
- Degenerative disorders of the brain Benign hereditary
- Pregnancy (Chorea gravidarum)
- Endocrine-metabolic dysfunction, e.g., adrenal insufficiency, hyper/hypocalcemia, hyper/ hypoglycemia, hypernatremia, liver failure
- Miscellaneous, e.g., anoxic encephalopathy, cerebral palsy, kernicterus, multiple sclerosis, post-traumatic
Huntington’s Disease/Chorea
Question 101. Write a short essay on the transmission and clinical features of Huntington’s disease (HD).
Answer: Huntington’s disease is a progressive, fatal, highly penetrant autosomal dominant disorder characterized by motor, behavioral, and cognitive dysfunction. Onset is typically between the ages of 25 and 45 years.
Etiology
- Huntington’s disease is caused by an increase in the number of polyglutamine (CAG) repeats (>40) in the coding sequence of the huntingtin gene located on the short arm of chromosome 4.
- The disease manifests earlier if the number of repeats is larger.
- The gene encodes the highly conserved cytoplasmic protein huntingtin, which is widely distributed in neurons throughout the CNS, but its function is not known.
Manifestations
- Early stages: Chorea tends to be focal or segmental, but progresses over time to involve multiple body regions. Dysarthria, gait disturbance, and oculomotor abnormalities are common features.
- Advancing disease: There may be a reduction in chorea and the emergence of dystonia, rigidity, bradykinesia, myoclonus, and spasticity.
- HD patients eventually develop behavioral and cognitive disturbances and the majority progress to dementia. Depression with suicidal tendencies, aggressive behavior, and psychosis can be prominent features.
Treatment
A multidisciplinary approach, with medical, neuropsychiatric, social, and genetic counseling for patients and their families.
- Medical:
- HD chorea is self-limited and is usually not disabling.
- Psychosis can be treated with atypical neuroleptics. Depression and anxiety are treated with appropriate antidepressants and antianxiety drugs.
- No adequate treatment for cognitive or motor decline.
- Promitochondrial agents such as ubiquinone and creatine are being tested as possible disease-modifying therapies.
Tremor
Question102. Write a short note on the definition of tremor and mention its types with examples.
Answer:
A tremor is an unintentional, rhythmic muscle movement involving to-and-fro movements (oscillations) of one or more parts of the body produced by alternating or synchronous contractions of antagonist muscles.
It is the most common of all involuntary movements and can affect the hands, arms, head, face, voice, trunk, and legs.
Causes of Tremor
- Types/Nature of Tremor
- Resting tremor occurs when the muscle is relaxed. For example, tremors occur when the hands are lying on the lap or hanging next to the trunk while standing or walking. It may be shaking off the limb, even when the individual is at rest. It is usually observed only in the hand or fingers and is seen in Parkinson’s disease.
- Action tremor is detected during any type of movement of an affected body part. There are several subtypes of action tremor.
- Postural tremor occurs when an individual maintains a position against gravity. For example, holding the arms outstretched.
- Kinetic tremor occurs during movement of a part of the body. For example, moving the wrists up and down.
- Intention tremor is present during a purposeful movement toward a target. For example, touching a finger to one’s nose during a medical examination.
- Task-specific tremor occurs when performing highly skilled, goal-oriented tasks. For example, as handwriting or speaking.
- An isometric tremor occurs during a voluntary muscle contraction that is not accompanied by any movement.
Classification
Tremor is most commonly classified by its appearance and cause or origin.
- Essential tremor (benign essential tremor): It is the most common the form of abnormal tremor.
- It may be mild and nonprogressive or slowly progressive. Most commonly observed in the hands but may involve the head, voice, tongue, legs, and trunk. cThe tremor of the hands is typically present as an action tremor.
- Triggers: Heightened emotion, stress, fever, physical exhaustion, or low blood sugar may trigger tremors and/or increase their severity. It decreases alcohol consumption.
Causes of tremor.
- Physiological: Anxiety
- Drugs: Beta-agonists, alcohol
- Intention tremor:
- Cerebellar lesion
- Thyrotoxicosis
- Drug-induced: Amphetamine, steroids
- Alcohol withdrawal, liver failure
- Mercury poisoning
- Wilson’s disease (wing-beating tremor)
Classification
- Parkinsonian tremor is characteristically a resting tremor. Dystonic tremor occurs in individuals affected by dystonia. It may affect any muscle. Dystonic tremors occur irregularly and are often relieved by complete rest.
- A cerebellar tremor is a slow tremor of the extremities that occurs at the end of a purposeful movement (intention tremor). For example, trying to press a button or touching a finger to the tip of one’s nose.
- Caused by lesions in or damage to the cerebellum, e.g., stroke, tumor, or MS. Cerebellar damage can also result in a “wing-beating” type of tremor which is a combination of rest, action, and postural tremors.
- Cerebellar tremors may be associated with dysarthria (speech problems), nystagmus (rapid involuntary movements of the eyes), gait problems, and postural tremors of the trunk and neck.
- Psychogenic tremor (functional tremor) can appear as any form of tremor movement.
- The characteristics may vary but generally include sudden onset and remission, and increased incidence of stress.
- Many individuals have a conversion disorder or psychiatric disease.
- Orthostatic tremors are detected as rhythmic muscle contractions in the legs and trunk immediately after standing.
- Physiologic tremor occurs in every normal individual. It may be exaggerated by strong emotions (such as anxiety or fear), physical exhaustion, etc.
- It is generally not caused by a neurological disease.
- Rubral tremor/Holme’s tremor—seen in midbrain lesions.
Organic Brain Syndrome
Question 103. Write a short note on organic brain syndrome (OBS).
Answer:
It is an abnormal mental state characterized by changes in orientation, memory, judgment, and affect. It is due to diffuse impairment of brain tissue.
The two most important types of OBS are delirium
Coma
Question 104. How would you investigate and manage a 50-year-old patient presenting with a coma?
Answer:
Definitions
Consciousness
- It means the state of the patient’s awareness of self and environment and his responsiveness to external stimulation and inner need.
- Consciousness is maintained by two separate anatomical and physiological systems:
Ascending reticular activating system (ARAS) projecting from the brainstem to the thalamus determines arousal (the level of consciousness).
The cerebral cortex determines the content of consciousness.
Confusion
- Traditionally referred to as “clouding of sensorium”.
- It denotes the inability to think with customary speed, clarity, and coherence, accompanied by some degree of inattentiveness and disorientation.
- Confusion results most often from processes that influence the brain globally, such as toxic or metabolic disturbance or dementia.
Drowsiness
- The inability to sustain a wakeful state without the application of stimuli externally is called drowsiness.
- Slow arousal is elicited by speaking to the patient or applying a tactile stimulus.
Stupor
- A state in which the person can be aroused only by repeated vigorous stimuli is called as stupor.
- There is either absence or slow and inadequate response to spoken commands.
- It is common to find restless or stereotyped motor activity. In these patients there is a reduction in the natural shifting of positions.
- When left unstimulated, these patients quickly return to a sleep-like state.
Lethargic/Drowsiness
The patient can usually be aroused or awakened and may then appear to be in complete possession of her senses but promptly falls asleep when left alone.
It resembles normal sleepiness.
Example: High brainstem disturbances.
Obtundation
It refers to a moderate reduction in the patient’s level of awareness such that stimuli of mild to moderate intensity fail to arouse; when arousal does occur, the patient is slow to respond.
Coma
Coma is a condition characterized by a deep sleep-like stage from which the patient cannot be aroused even with vigorous, continuous stimulation.
The patient does not make any localized responses. However, the patient may grimace or show withdrawal responses to painful stimuli.
Question 105. Write a short essay/note on the causes of coma.
Answer:
Approach to a Patient with Coma
Question 106. How do you proceed to investigate and manage a case of coma?
Answer:
History
Inquire about a history of diabetes, hypertension, head injury, convulsions, alcohol or drug use, circumstances in which patient was found, medications in hospitalized patient like anesthetics, sedatives, antiepileptics, opiates, antidepressants, and antipsychotics.
Onset of Coma
The sudden onset of coma would suggest a vascular cause possibly SAH or brainstem involvement.
Rapid progression from hemispheric signs to coma:
Intracerebral hemorrhage.
Protracted course: Tumor, abscess, chronic SDH.
Altered sensorium (delirium) preceding coma, with no focal neurological deficits, indicates metabolic encephalopathy.
- General Examination
Signs of trauma:
- Raccoon eyes,
- Battle’s sign, and
- CSF rhinorrhea or logorrhea
Blood pressure:
- Hypertension suggests—
- hypertensive encephalopathy or
- intracerebral hemorrhage
- Hypotension suggests—
- myocardial infarction,
- septicemia,
- Addison’s disease, and
- alcohol or barbiturate
- intoxication,
- internal hemorrhage.
Pulse: Bradycardia with periodic breathing and hypertension (Cushing reflex) suggests raised ICP.
Temperature:
- Hypothermia suggests—
- alcohol or barbiturate intoxication,
- myxedema,
- advanced tubercular meningitis, and
- peripheral circulatory failure
- Hyperthermia suggests—
- systemic infection,
- meningoencephalitis,
- heatstroke, and
- anticholinergic
- drugs abuse
- Signs of meningeal irritation:
- Meningitis and
- SAH
Fundus: Raised ICP (papilledema), subhyaloid hemorrhages (SAH), and hypertensive encephalopathy.
The smell of breath: For ketones, alcohol, and hepatic fetor.
Skin inspection:
The rash suggests meningococcemia, staphylococcal endocarditis, typhus, Rocky Mountain spotted fever (RMSF)
Excessive sweating suggests hypoglycemia or shock
Diffuse petechiae suggest thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulopathy (DIC), and fat embolism.
Classification of coma.
Coma without focal signs or meningism
- Exogenous intoxicants: Alcohol, barbiturate, opiates
- Endogenous metabolic disturbances: Anoxia, hypoglycemia, diabetic ketoacidosis (DKA), hyperosmolar nonketotic state (HONK), uremia, hepatic failure, hyponatremia or hypernatremia, Addisonian crisis, carbon monoxide poisoning, myxedema
- Severe systemic infections: Septicemia, typhoid fever, cerebral malaria, pneumonia, peritonitis, Waterhouse–Friderichsen syndrome
- Circulatory collapse (shock) from any cause
- Post seizure states
- Hypertensive encephalopathy
- Hyperthermia and hypothermia
- Concussion
- Acute hydrocephalus
Coma with meningism
- Subarachnoid hemorrhage
- Acute bacterial meningitis
- Viral meningoencephalitis
- Neoplastic meningitis
- Parasitic meningitis
- Pituitary apoplexy
Coma with focal signs
- Hemispheral hemorrhage or massive cerebral infarction
- Brainstem infarction
- Brain abscess, subdural empyema, herpes encephalitis
- Epidural and subdural hemorrhage, brain contusion
- Brain tumor
- Miscellaneous: Thrombotic thrombocytopenic purpura (TTP), fat embolism, ADEM, cortical vein thrombosis, focal infarction caused by bacterial endocarditis
Neurological Assessment
- Neurological assessment is done to determine the depth of coma [Glasgow coma scale (GCS)], brainstem function, and lateralization of pathology.
- Observation first without examiner intervention.
- Observe the posture of the limb, the position of the eyes and head, spontaneous movement, and the pattern of respiration.
- Yawning and spontaneous shifting of body position indicate a minimal degree of unresponsiveness.
- Multifocal myoclonus almost always indicates a metabolic disorder.
- Assess responsiveness by noting the patient’s reaction to calling his name, or to noxious stimuli such as supraorbital or sternal pressure.
Evaluation of Severity of Coma

Question 107. Write a short essay/note on Edinburgh’s classification of coma. Write a short essay/note on Glasgow Coma Scale.
Answer:
Newer Scales for Prognosis of Coma
FOUR (Full Outline of UnResponsiveness) scale:
The new Coma Scale is devised in 2005, with four components (eye, motor, brainstem, and respiration).
Each component has a maximum score of four.
AVPU scale: Alertness, response to Verbal stimuli, response to Painful stimuli, or Unresponsive.
ACDU scale: Alertness, Confusion, Drowsiness, and Unresponsiveness
Grady scale: Scale of I–V along a scale of confusion, stupor, deep stupor, abnormal posturing, and coma.
Stages of Coma
- Grade 1: Individuals who respond with recognition when their name is called and do not lapse into sleep when left undisturbed.
- Grade 2: The person lapses into sleep when undisturbed and is aroused only when a pin is tapped gently over the chest wall.
- Grade 3: Patient who winces in response to deep pain stimulus.
- Deep pain stimulus may result in abnormal postural reflexes either unilateral or bilateral.
- Grade 4: Deep pain stimulus may result in decorticate or decerebrate posturing.
- Grade 5: The patient who maintains a state of flaccid unresponsiveness in spite of deep pain stimulation.
Posture in Comatose Patient
Decerebrate rigidity: It consists of opisthotonus, clenching of jaws, stiff extension of limbs with internal rotation of arms, and plantar flexion of feet.
Extensor posturing arises in a variety of settings: Midbrain compression, cerebellar lesions, metabolic, drug intoxication, etc.
Decorticate rigidity: Arms in flexion and adduction and legs extended signify lesion rostral to midbrain.
Extensor posture of arms with weak flexor responses of legs is seen with lesions at the level of vestibular nuclei.
Brainstem Reflexes (Brainstem Function)
- Pupillary size and reactivity
- Ocular movements
- Corneal responses
- Oculovestibular reflexes
- The pattern of breathing (respiratory pattern)
Question 108. Write a short note on brainstem reflexes.
Answer:
As a rule, when these brainstem activities are preserved, particularly pupil reactions and eye movements, coma must be ascribed to bilateral hemispheral disease.
However, a mass in the hemispheres may cause coma and also produce
herniation.
Pupillary Reactions
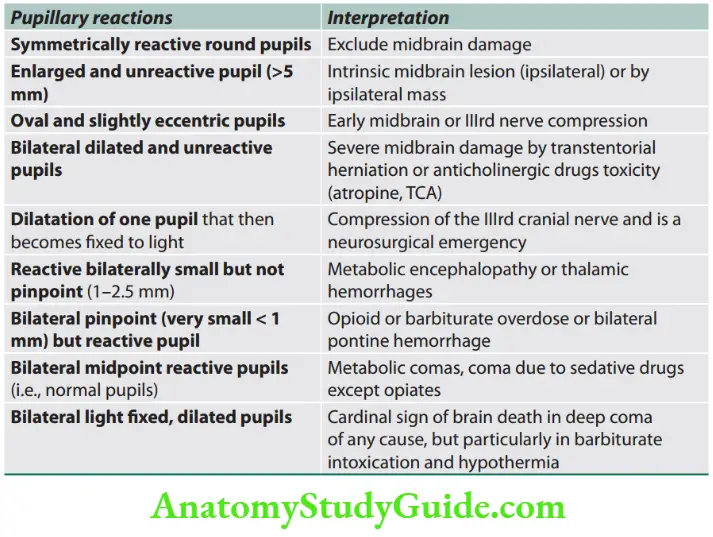
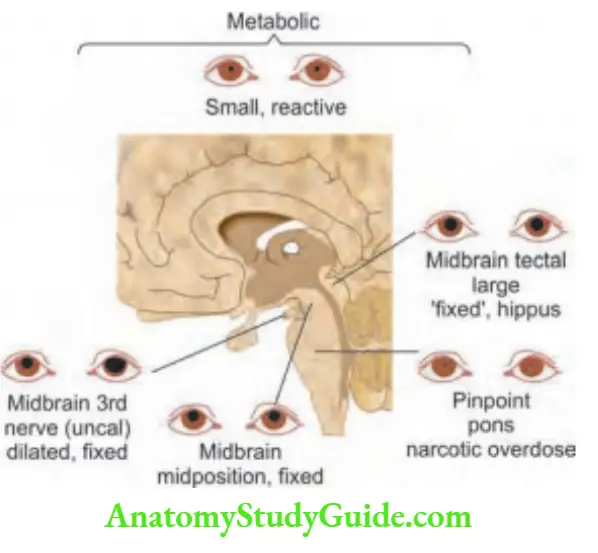
Edinburgh classification of coma.
- Grade 0: Fully conscious
- Grade 1: Drowsy, but responds to verbal commands
- Grade 2: Unconscious, but responds to minimal pain stimulus
- Grade 3: Unconscious, but responsive to strong pain stimulus
- Grade 4: Unconscious, with no response to pain
Eye Movements and Position
- In a light coma of metabolic origin, eyes wander conjugately from side to side in a random fashion. These movements disappear as the coma deepens.
- Adducted eye at rest: VIth nerve palsy. If it is bilateral, it is due to raised ICT.
- Abducted eye at rest: IIIrd nerve palsy.
- Conjugate deviation of eyes toward the hemispheric lesion and away from the unilateral pontine lesion.
- Downward and inward deviation of eyes: Lesions of the thalamus and upper midbrain.
- Eyes turn toward the convulsing side in focal seizures.
- Ocular bobbing: It is characterized by brisk downward and slow upward movements of the eyes associated with loss of horizontal eye movements. It is diagnostic of lesions in the midbrain and pons.
- Ocular dipping: It is characterized by slow downward followed by faster upward movement in patients with normal horizontal gaze.
- It indicates diffuse cortical anoxic damage and drug intoxication.
- Disconjugate eyes (divergent ocular axes): Brainstem lesion, e.g., skew deviation (one eye up, one eye down)
- Oculocephalic reflex: Also called doll’s eye movement.
- Elicited by briskly turning or tilting the head.
- In a coma of metabolic origin or due to lesions of bihemispheric structures, the response consists of conjugate movements of eyes in the opposite direction.
- Positive response indicates Intact oculomotor, abducent, midbrain, and pons.
- Absent reflex indicates Damage within the brainstem. It can be also due to a profound overdose of sedatives or anticonvulsants.
Oculovestibular or Caloric Test
- Method: Irrigate the external auditory canal with cold water.
- Normal response: Causes slow conjugate deviation of eyes toward irrigated ear followed in few seconds by compensatory nystagmus (i.e., fast component away from irrigated ear).
Interpretation:
- Loss of conjugate ocular movements in brainstem damage.
- Loss of fast corrective nystagmus in metabolic or bilateral hemispheral damage.
- The eyes are tonically deflected to the side irrigated with cold water and this position may be held for 2–3 minutes.
Respiratory Patterns
- Slow, shallow, regular breathing: Metabolic or drug depression.
- Cheyne-Stokes respiration (alternating hyperpnea and periods of apnea): Massive supratentorial lesions, bilateral cerebral lesions, and mild metabolic disturbance.
- Central neurogenic hyperventilation: Lesions of lower midbrain and upper pons either primary or secondary to transtentorial herniation.
- Apneustic breathing: Lower pontine lesions.
- Biot’s or ataxic breathing: Lesions of the dorsomedial part of the medulla.
- Agonal gasps: Bilateral lower brainstem damage and terminal respiratory pattern.
- Acidotic (Kussmaul) respiration (deep, sighing hyperventilation): In diabetic ketoacidosis and uremia
Laboratory Studies and Imaging
- Complete blood count
- Random blood sugar
- Renal function tests and liver function tests
- Serum electrolytes
- Urine examination for specific gravity, glucose, acetone, and protein content.
- Arterial blood gases analysis: For acidosis or high CO levels
- Chest X-ray
- ECG
- CT or MRI scan: Quick and effective in demonstrating all types of brain hemorrhage and mass lesions.
- Lumbar puncture: It should be performed in a coma only after careful risk assessment.
- It should not be performed when there is a suspicion of an intracranial mass lesion.
- CSF examination may help in the diagnosis of meningoencephalitis or other infection, or in subarachnoid hemorrhage.
- EEG: It may be useful in the diagnosis of metabolic coma, encephalitis, and nonconvulsive status epilepticus.
- Drugs screen: Blood alcohol and salicylates, urine toxicology (e.g., screening for benzodiazepines, narcotics, amphetamines, etc.). Others, e.g., cerebral malaria and porphyria.
Diffrential Diagnosis
- Coma vigil (vegetative state—VS): The patient is comatose, but the eyelids are open giving the appearance of being awake.
- Patients can open their eyes or have random limb and head movements but there is the complete absence of response to commands or to communicate.
- It is usually due to extensive cortical damage. Brainstem function is intact. Hence, breathing is normal without the need for mechanical ventilation. Patients may remain in this vegetative state for years. Even after 12 months if there is no recovery, it is called a permanent vegetative state (PVS).
- Minimally conscious state (MCS): A condition in which patients have some limited/inconsistent (fluctuating) signs of awareness (unlike patients with a vegetative state).
- These include nonreflexive response to sensory stimulation, awareness of the self or the environment, or language comprehension or expression.
- Akinetic mutism: It refers to a state in which the patient is partially or fully awake, but remains entirely silent and immobile. The patient does nothing voluntarily and has sleep/wake cycles and can maintain vital functions.
- It may be seen in bilateral frontal lobe lesions, hydrocephalus, and a mass in the region of the third ventricle.
- Locked-in-state (locked-in syndrome or pseudocoma): The patient is fully aware and alert but unable to communicate except through eye movements. There is complete paralysis except for vertical eye movements and intact lid movements (blinking in ventral). This syndrome is due to extensive transverse lesions in the pontine and midbrain (infarction).
- Conversion reaction: Patients have normal pupils, corneal reflexes, and plantar reflexes. These patients may keep their eyes firmly closed and resist the opening of the eye by examiners. Their eyes roll up when the lids are raised. It may be due to a feigned or hysterical state.
- Psychogenic coma.
- Brainstem death is discussed below.
Treatment
General management: Comatose patients require careful nursing, maintenance of the airway, and breathing frequent monitoring of vital functions.
Specific treatment: Diagnosis of the underlying cause/lesion should be done as early as possible and appropriate specific treatment is to be given.
For example:
Diabetic with hypoglycemic coma: Administer hypertonic glucose without waiting for reports.
However, bedside estimation of blood glucose and administering dextrose only if the blood glucose is below 100 mg/dL.
Alcoholic with coma: Administer glucose and vitamin B1 100 mg intravenously, without waiting for reports.
- Longer-term management
- Skincare: Change the posture every 2–3 hours and keep the skin clean and dry to avoid pressure/bed sores and pressure palsies.
- Eye care: To prevent corneal damage (lid taping, irrigation).
- Oral hygiene: Mouthwashes and suction.
- Fluids: Through nasogastric tube or IV. Have a secure IV line.
- Feeding: Nutrients through a fine-bore nasogastric tube.
- Sphincters: Catheterization of the urinary bladder and rectal evacuation.
- Posture of the patient: Prevent the aspiration of gastric contents by positioning the patient in a prone or lateral position.
Brain Death
Question 109. Write a short note on brain death.
Answer:
Brain death occurs from irreversible brain injury which is sufficient to permanently eliminate all cortical and brainstem function (i.e., loss of all functions of the brain, including the brainstem).
- Because the vital centers in the brainstem maintain cardiovascular and respiratory functions, brain death is incompatible with survival despite mechanical ventilation and cardiovascular and nutritional supportive measures.
- It develops when intracranial pressure (ICP) exceeds cerebral perfusion pressure (CPP) and results in cessation of CBF and oxygen delivery.
Significance of brain death:
- It permits the withdrawal of costly lifesaving equipment and drugs.
- Family can be offered the opportunity for organ donation.
Diagnosis of Brain Death
Brain death is a clinical diagnosis. No other tests are necessary and complete clinical examination including independent brain death determinations by two licensed physicians is conclusive.
Clinical Evaluation (Prerequisites)
- Establish known irreversible causes of coma.
- The first and foremost critical step in establishing the diagnosis of brain death is to establish an irreversible, untreatable cause of the brain injury (e.g., global ischemia due to cardiac arrest, intracranial bleeding, and severe head injury).
- Exclude potentially reversible conditions like hypothermia, drug intoxication, poisoning, and metabolic disorders (e.g., hypoglycemia, acidosis, and electrolyte imbalance).
- Hypothermia should also be excluded—the rectal temperature must exceed 35°C.
- Achieve body temperature >36°C.
- Achieve normal systolic BP (>100 mm Hg)
Clinical Evaluation (Neuroassessment)
- Establish coma
- Establish absence of brainstem reflexes
- Pupillary reflex (absent)
- Eye movements
- Oculocephalic: Absent (doll’s eye movements)
- Oculovestibular: Absent (cold caloric test)
Facial sensation and motor response: No corneal reflex, no jaw reflex, no grimacing to deep pressure on the nail bed, supraorbital ridge, or temporomandibular joint
- Pharyngeal (gag) reflex absent
- Tracheal (cough) reflex absent.
Establish Apnea by Apnea Test
Prerequisites for apnea test: Body temperature > 36°C, systolic blood pressure > 100 mm Hg, normal electrolytes profile, and normal PaCO 2 (35–45 mm Hg).
- The procedure of apnea test:
- Connect a pulse oximeter and disconnect the ventilator.
- Deliver 100% O
- 2 by catheter through an endotracheal tube at 6 L/minute.
- Observe for respiratory movement at least for 8–10 minutes.
- Discontinue testing: If BP drops to <90 mm Hg and PaO2 decreases to 85% by pulse oximetry for 30 seconds.
- If respiratory drive/movement is observed after 8 minutes. Take the next blood sample for blood gas studies.
- This indicates apnea, the test result is negative.
- Absence of respiration drive: If respiratory movements are absent and arterial PaCO2 is 60 mm Hg or 20 mm Hg increase over a baseline, normal PaCO2 indicates apnea, the test result is positive and supports the clinical diagnosis of brain death.
Ancillary Tests/Confirmatory Testing
- EEG: Electrocerebral silence absence of electrical activity for at least 30 minutes.
- Cerebral angiography: Absence of intracranial blood flow.
- PET: Glucose metabolism studies/dynamic nuclear brain scan: “Hollow skull” sign of brain death.
Documentation
Time of death is the time the arterial PaCO2 reached the target value or when the ancillary test was officially interpreted.
Diseases Of Cranial Nerves
1st Cranial Nerve: Olfactory Nerve Common causes of anosmia.
2nd Cranial Nerve: Optic Nerve
Clinical Examinations
The optic nerve is tested by various modalities of vision: Visual acuity, visual fields, color vision, and fundoscopy.
- Visual acuity
- Snellen chart is used to measure visual acuity for distant vision.
- Visual acuity for near vision is tested by Jaeger’s chart.
- Pathway of the optic nerve
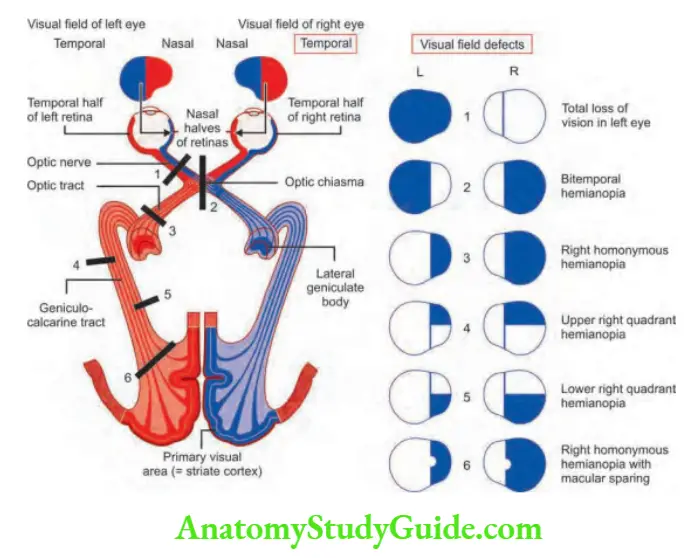
Question110. Discuss the field defects produced and the localization of lesions at various levels.
Answer:
Visual fields
- Visual can be impaired by damage to the visual system anywhere from the eyes to the occipital lobes.
- One can localize the site of the lesion with considerable accuracy by mapping the visual field deficit by finger confrontation
and then correlating it with the topographic anatomy of the visual pathway. - At the optic chiasm, fibers from nasal ganglion cells decussate into the contralateral optic tract.
- Symmetric compression of the optic chiasm by a pituitary adenoma, meningioma, craniopharyngioma, glioma, or aneurysm results in bitemporal hemianopia.
- A unilateral post-chiasmal lesion leaves the visual acuity in each eye unaffected, although the patient may read the letters on only the left or right half of the eye chart.
- Damage to the optic radiations in the temporal lobe (Meyer’s loop) produces a superior quadrantic homonymous hemianopia, whereas injury to the optic radiations in the parietal lobe results in an inferior quadrantic homonymous hemianopia.
- Lesions of the primary visual cortex give rise to dense, congruous hemianopia field defects by the posterior cerebral artery infarct.
- They have macular sparing because collaterals from the middle cerebral artery supply the macular representation at the tip of the occipital lobe.
- Destruction of both occipital lobes produces cortical blindness.
This condition can be distinguished from bilateral pre-chiasmal visual loss by noting that the pupil responses and optic fundi remain normal.
Visual loss
Lesions in any areas between the retina and the visual cortex can produce visual loss.
Patterns of visual field loss.
- Visual symptoms involving one eye: Due to lesions anterior to the optic chiasm.
- Transient visual loss: It is the sudden onset of visual loss lasting < 15 minutes. It is usually due to vascular disease and may be difficult to identify whether the visual loss was monocular (carotid circulation) or binocular (vertebrobasilar circulation).
Color vision
- The retina has three classes of cones, with visual pigments of differing peak spectral sensitivity: red (560 nm), green (350nm), and blue (430 nm).
- The red and green cone pigments are encoded on the X chromosome; the blue cone pigment is on chromosome 7.
Common causes of anosmia.
- Local causes
- Acute local inflammation
- Heavy smoking
- Atrophy of bulb
- Chronic kidney diseases
- Syndromes associated
- Foster Kennedy syndrome (anosmia, optic atrophy of one eye and contralateral eye papilledema due to the tumor in the brain)
- Pseudo – FosterKennedy syndrome (above features in the absence of tumor)
- Systemic causes
- Parkinsonism
- Meningitis
- Head trauma
- Intracranial tumors
- Endocrine diseases
- Diabetes mellitus
- Hypothyroidism
- Kallmanns syndrome
- Turners syndrome
- Vitamin B12 deficiency
- The most common anomalies of color vision are the various types of red-green deficiency inherited as a sex-linked recessive condition.
- Only males are affected (X-linked) and present in 8% of males.
- Ishihara color plates can be used to detect red-green color blindness.
- Acquired defects of color vision occur in macular and optic nerve diseases, and due to certain drugs (e.g., ethambutol and chloroquine).
Papilledema
Question 112. List the causes and eye findings in Argyll Robertson’s pupil.
Answer:
It is the pupillary change where the accommodation reflex is present but the light reflex is impaired.
Site of lesion: Tectum of the midbrain or peripherally in the branch of IIIrd cranial nerve.
- Ophthalmological findings in Argyll Robertson pupil.
- Pupils: Small, irregular, and unequal in size
- Iris: Atrophy and depigmentation Light reflex: Absent for direct (always) and consensual (usually stimulus)
- Accommodation reflex: Intact
- Ciliospinal reflex: Absent
Causes of Argyll Robertson pupil.
- Neurosyphilis (tabes dorsalis) generally bilateral
- Diabetes
- Multiple sclerosis
- Sarcoidosis
- Tumors of the pineal region
Pinpoint Pupil
Question 113. List the causes of the pinpoint pupil.
Answer:
Causes of pinpoint pupil
- Pontine hemorrhage
- Organophosphorus poisoning
- Opium poisoning
- Pilocarpine instillation
- Thalamic hemorrhage (occasionally)
Adie’s Pupil
Question 114. Write a short note on Adie’s pupil.
Answer:
Normally, pupil reaction to light is absent or markedly reduced when tested in the routine examination.
However, Adie’s pupil reacts slowly with prolonged maximal stimulation.
Once Adie’s pupil reacts to accommodation, the pupil remains tonically constricted and dilates very slowly.
Cause: Destruction of ciliary ganglion.
Blindness
Question 115. Write a short note on the causes of blindness.
Answer:
Causes of blindness
- Cataract leading cause (47.9%)
- Glaucoma (12.3%)
- Age-related macular degeneration (AMD) (8.7%)
- Corneal opacities
- Diabetic retinopathy
- Childhood blindness
- Trachoma
Horner’s Syndrome
Question 116. Write a short essay/note on Horner’s syndrome.
Answer:
- Horner’s syndrome is a syndrome complex caused by the involvement of the oculosympathetic tract.
- Sympathetic nervous supply to the eye consists of a three-neuron pathway:
- Fibers through the IIIrd nerve innervate the levator muscle of the eyelid (Muller’s muscles).
- Fibers through the nasociliary nerve supply the blood vessels of the eye.
- Fibers through the long ciliary nerve innervate the pupil.
- Damage to any part of the pathway results in Horner’s syndrome.
- Components of Horner’s Syndrome
Causes of Horner’s Syndrome
- Miosis due to reduced pupillodilator activity
- Partial ptosis of eyelid
- Enophthalmos
- Anhidrosis of ipsilateral half of face
- Absence of ciliospinal reflex
Causes of Horner’s Syndrome
- Cerebral and brainstem lesions: Hemispherectomy, massive cerebral infarction, brainstem demyelination
- Cervical cord lesions: Syringomyelia and cord tumors (e.g., ependymoma, glioma)
- Thoracic root level: Apical lung tumor (Pancoast “tumor”) or TB, cervical rib, trauma to brachial plexus
- Sympathetic chain and carotid artery in the neck: Following thyroid/laryngeal/carotid surgery, carotid artery dissection, neoplastic infiltration, cervical sympathectomy
- Miscellaneous: Congenital Horner’s syndrome, cluster headache (transient), idiopathic.
Third, 4th, and 5th Cranial Nerves: Oculomotor, Trochlear, and Abducens Nerves
- Normal palpebral fissure 9–12 mm.
- The narrowing of the palpebral fissures due to the inability to open an upper eyelid is called ptosis.
- Ptosis may be congenital or acquired, unilateral or bilateral, partial or complete
- Congenital ptosis: It is due to bilateral congenital hypoplasia of the IIIrd nerve nuclei, and results in bilateral ptosis.
- Acquired ptosis: Acquired ptosis may be unilateral or bilateral.
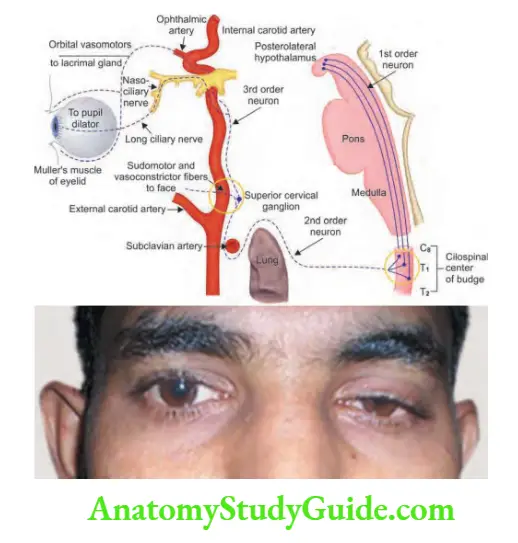
Causes of ptosis
Question 117. Write a short note on the causes of ptosis.
Answer:
Causes for ptosis
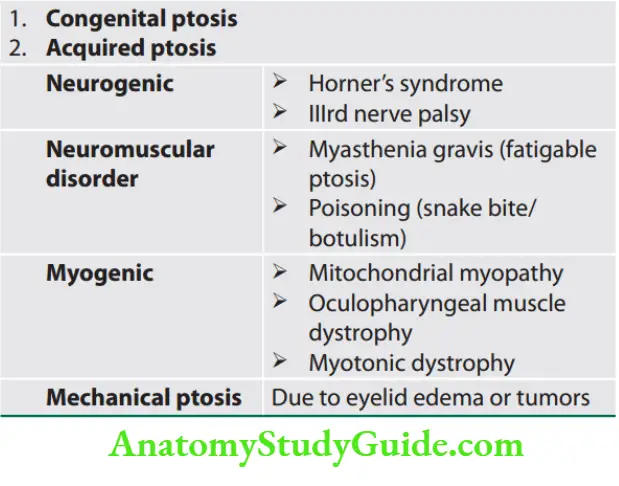
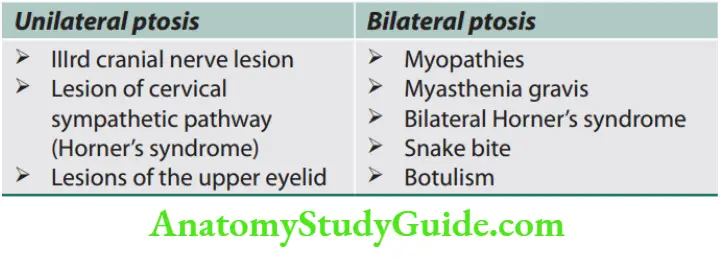
- Partial ptosis: This occurs with a lesion of the cervical sympathetic pathway (Horner’s syndrome) due to weakness of the tarsal muscles, innervated by cervical sympathetic nerves.
- The upper eyelids can however be raised voluntarily.
- Complete ptosis: This occurs with IIIrd nerve lesions due to paralysis of the levator palpebrae superioris, innervated by the IIIrd nerve.
- The patient is not able to voluntarily open the affected eye.
- Ptosis and Pupillary Size
- Ptosis with a small pupil: Horner’s syndrome
- Ptosis with a large pupil: IIIrd nerve palsy (compressive lesions).
- Ptosis with normal papillary size: Infarction of IIIrd nerve, myasthenia gravis, myopathies, or GBS.
Ophthalmoplegia
- Ophthalmoplegia is the paralysis or weakness of the eye muscles.
- Supranuclear ophthalmoplegia—also called gaze palsies.
- It is due to the involvement of corticonuclear fibers of the IIIrd, IVth, and VIth cranial nerves.
- Internuclear ophthalmoplegia—due to the involvement of medial longitudinal fasciculus (MLF) and paramedian pontine reticular formation (PPRF) which connect the IIIrd nerve to the contralateral VIth nerve.
- Nuclear/Intranuclear ophthalmoplegia—involvement of individual cranial nerves. (CN III, IV, and VI).
- Internal ophthalmoplegia—paralysis of constrictor pupillae and ciliary muscle.
- External ophthalmoplegia—paralysis of extraocular muscles.
- Total ophthalmoplegia—a combination of external and internal ophthalmoplegia.
Internuclear Ophthalmoplegia
Question 118. Write a short note on internuclear ophthalmoplegia.
Answer:
- It is caused by a lesion of the MLF, which carries signals from the abducens nucleus to the contralateral medial rectus oculomotor subnucleus.
- The abducens nerve and MLF coordinate conjugate horizontal eye movements with co-contraction of ipsilateral lateral rectus and contralateral medial rectus muscles.
- Classic signs of unilateral internuclear
- ophthalmoplegia include impaired adduction of the ipsilesional eye and abducting nystagmus of the contralateral eye.
- Despite ipsilateral adduction weakness with direct motility testing, adduction is often intact with convergence because
convergence signals to the medial rectus nucleus are distinct from the MLF. - Multiple sclerosis and microvascular brainstem ischemia are the most common causes.
Diplopia
Question 119. Write a short note on diplopia.
Answer:
Diplopia means double vision. Most common subjective complaints elicited by lesions in the oculomotor system.
It occurs more frequently with lesions of the extraocular muscles or oculomotor nerves than with supranuclear lesions which result in gaze palsies.
Monocular diplopia
- The first point to clarify is whether diplopia persists in either eye after covering the fellow eye. If it does, the diagnosis is monocular diplopia.
- The cause is usually intrinsic to the eye. For example, corneal aberrations, uncorrected refractive error, cataracts, or foveal traction may give rise to monocular diplopia.
Binocular diplopia
- Diplopia alleviated by covering one eye is binocular diplopia and is caused by disruption of ocular alignment, which occurs only if both eyes are open.
- Binocular diplopia occurs from a wide range of processes. For example, infectious, neoplastic, metabolic, degenerative, inflammatory, and va scular.
- Here two images, one real and one false are formed. The real image is closer to the eye and distinct; the false image is farther away from the eye and indistinct.
Nystagmus
Question 120. List the causes of nystagmus.
Answer:
Nystagmus is involuntary, conjugate, repetitive, and rhythmic movement of eyeballs. It is a sign of disease of the retina, cerebellum, and/or vestibular systems, and their connections.
Types of Nystagmus


- Other common types of nystagmus
- Description Condition seen
- See-saw nystagmus Upward deflection of one eyeball with a downward deflection on the contralateral eyeball
- Suprasellar region anterior to IIIrd ventricle
- Upbeat nystagmus Fast movement upward Lesions in the vermis of the cerebellum
- Downbeat nystagmus Fast component is down Foramen magnum lesions
- Optokinetic nystagmus Railway track nystagmus Deep parietal lobe lesions
- Convergence-retraction nystagmus Attempted upgaze provokes jerk nystagmus with a fast component in an inward convergent manner
- Lesion at superior colliculus—Parinaud’s syndrome
- Vth Cranial Nerve: Trigeminal Nerve
Question 121. Discuss the clinical features and management of trigeminal neuralgia.
Answer:
- Trigeminal Neuralgia (Tic Douloureux)
- Trigeminal neuralgia is also known as prosopalgia or Fothergill’s disease.
- Tic Douloureux means painful jerking.
- It is a neuropathic disorder. It is defined as sudden, episodes of usually unilateral, severe, brief, stabbing, intense, lancinating, and recurring pain in the distribution of one or more branches of the Vth cranial nerve (trigeminal nerve).
- Middle age and later. Usually, it starts in the 6th and 7th decades and the major risk factor is hypertension.
- Etiology
- Usually produced due to compression of the trigeminal nerve at or near the pons by an ecstatic vascular loop.
- Pain like trigeminal neuralgia can be seen in other conditions.
Clinical features
Pain
- Characteristics of pain: Sudden, unilateral, intermittent (paroxysmal), sharp, shooting/knife-like/lancinating/electric shock-like.
- Pain rarely crosses the midline. In extreme cases, the patient will have a motionless face known as the “frozen or mask-like face”.
- In 10–12% of cases it is bilateral and usually due to intrinsic brainstem pathology (e.g., MS) or expanding cranial tumor (acoustic schwannomas, meningiomas, epidermoids).
- Duration of pain: Pain is of short duration (lasting seconds), but may recur with variable frequency (maybe many times a day).
- Attacks do not occur during sleep.
- Pain occurs along the cutaneous distribution of the Vth nerve (full or branches).
- The pain usually commences in the mandibular division but may spread to involve the maxillary and occasionally the ophthalmic divisions.
- Pain is precipitated by minor trauma to the trigger zones (e.g., slight touch, chewing, shaving, rinsing the mouth, exposure to cold wind).
- Common trigger zones can be external/cutaneous (around the ala of nose, corners of lips and check) or internal/intraoral (teeth, gingivae, tongue).
- Trigger areas on the face are so sensitive that touching or even air currents can trigger an episode.
- Neither objective signs of sensory loss nor signs of Vth nerve dysfunction can be demonstrated on examination.
Diffrential diagnosis
- Reactivation of the varicella-zoster virus is seen in older people and has a predilection for ophthalmic division of the trigeminal nerve.
Treatment
- The first line of treatment: Carbamazepine (anticonvulsant) to be started with a dose of 100–200 mg/day, increase in 2–3 weeks to 200–400mg TID.
- Second line of treatment: Baclofen, lamotrigine, oxcarbazepine, phenytoin, gabapentin, pregabalin, sodium valproate.
- Long-acting anesthetic agents: Localized pain is managed by injecting any of the following into a particular branch of the nerve.
Alcohol injection
- Peripheral glycerol injection
- Surgery: Indicated if the drug fails or is not tolerated.
- Peripheral neurectomy (nerve avulsion)
- Open procedures (intracranial procedures)
- Microvascular decompression
- Percutaneous rhizotomies
- Gamma knife radiosurgery: Using stereotactic imaging of the trigeminal nerve root entry zone, radiation to delivered to the trigeminal nerve.
Causes of trigeminal neuralgia.
- Usually idiopathic
- Demyelination
- Multiple sclerosis
- Petrous ridge compression
- Post-traumatic neuralgia
- Intracranial tumors
- Intracranial vascular abnormalities
- Viral etiology
7th Cranial Nerve: Facial Nerve
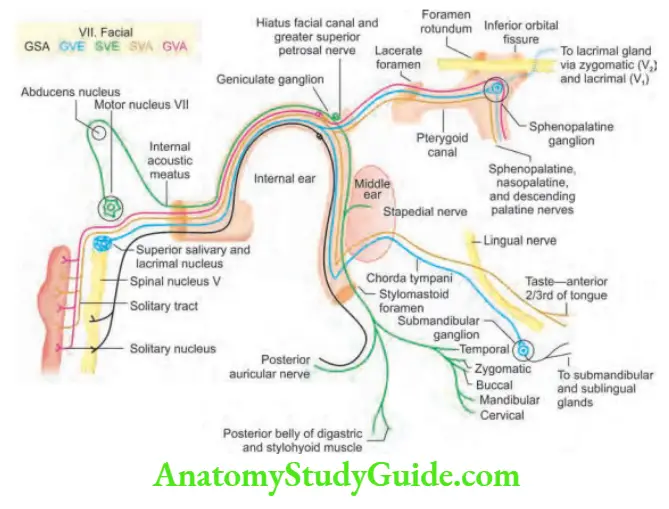
Causes of Facial Nerve Palsy
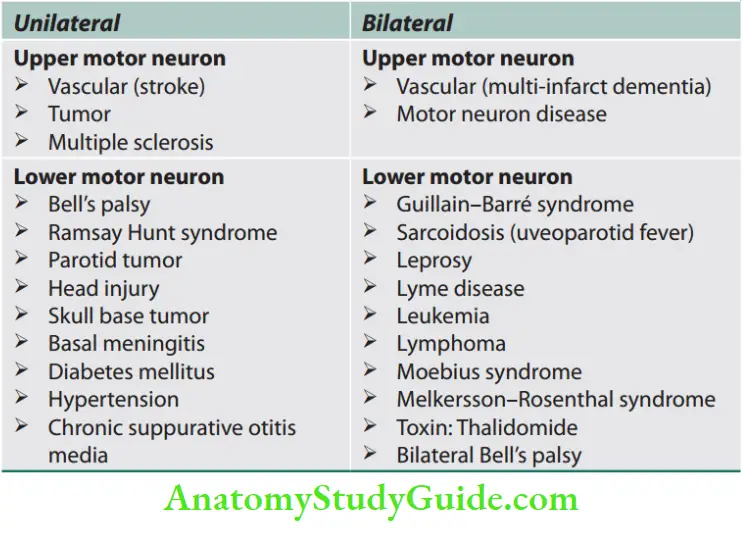
Bell’s Palsy
Leave a Reply