Antibiotics Introduction
Antibiotic is a chemical compound that inhibits or abolishes the growth of microorganisms, such as bacteria, fungi or protozoan. It also includes any agent with biological activity against living organisms; however, the term is commonly used to refer any substances with anti-bacterial, anti-fungal or anti-parasitical activity.
Table of Contents
History: Although potent antibiotic compounds for the treatment of human diseases caused by bacteria such as tuberculosis, plague or leprosy were not isolated and identified until twentieth century. The first known use of antibiotics was by the ancient Chinese over 2,500 years ago.
Many other ancient cultures, including the ancient Egyptians and ancient Greeks already used molds and plants to treat infections, owing to the production of antibiotic substances by these organisms. At that time, however, the compounds having antibiotic activity present in molds or plants were unknown.
The antibiotic properties of Penicillium spp. were first described in France by Ernest Duchense in 1897. However, his work went by without much notice from the scientific community until Alexander Fleming’s discovered Penicillin.
Modern research on antibiotic therapy began in Germany with the development of narrow-spectrum antibiotic Salvarsan by Paul Ehrlich in 1909, for the first time allowing an efficient treatment of the then-widespread problem of syphilis. The drug, which was also effective against other spirochetal infections, is no longer in use in modern medicine.
Antibiotics were further developed in Britain following the re-discovery of Penicillin in 1928 by Alexander Fleming. More than ten years later, Ernst Chain and Howard Florey become interested in his work and came up with the purified form of
Penicillin. The three shared the 1945 Nobel Prize in Medicine. “Antibiotic” was originally used to refer only to substances extracted from a fungus or other microorganism, but has come to include also the many synthetic and semi-synthetic drugs that have antibacterial effects.
Antibiotics Classification
Antibiotics are classified in many ways based on chemical structure, its spectrum of activity, pharmacological activity.
- Based on Chemical Structure
-
- B-lactam antibiotics : eg. Penicillins, Cephalosporins, Monobactams, Carbapenems.
- Aminoglycoside antibiotics: eg. Streptomycin, Neomycin, Paromomycin, Kanamycin, Amikacin, Gentamicin, Tobramycin, Netilmicin, Sisomicin, Spectinomycin.
- Tetracyclines : eg. Tetracycline, Chlortetracycline, Rolitetracycline, Oxytetracycline, Methacycline, Demeclocycline, Meclocycline, Doxycycline, Minocycline.
- Macrolide antibiotics : eg. Erythromycin, Clarithromycin, Azithromycin, Dirithromycin, Troleandomycin.
- Lincomycins : eg. Lincomycin, Clindamycin.
- Polypeptide antibiotics : eg. Vancomycin, Teicoplanin, Bacitracin, Polymyxin B, Colistin, Gramicidin.
- Nitrobenzene derivative : eg. Chloramphenicol.
- Steroidal antibiotic : eg. Fusidic acid.
- Miscellaneous : eg. Novobiocin, Mupirocin, Quinupristin.
- Based on Pharmacological Activity
- Antifungal antibiotics.
- Polyenes : eg. Amphotericin B, Nystatin, Natamycin.
- Others : eg. Griseofulvin.
- Anticancer antibiotics : eg. Dactinomycin, Daunorubicin, Doxorubicin, Bleomycin, Idarubicin, Mitomycin, Plicamycin, Streptozocin, Valrubicin.
- Antityphoid antibiotic : eg. Chloramphenicol.
- Antidiarrheal antibiotic : eg. Colistin.
- Antituberculor antibiotics : eg. Rifampicin, Rifabutin, Cycloserine, Capreomycin.
ẞ- Lactam Antibiotics
B-lactam antibiotics are a broad class of antibiotics which include Penicillin derivatives, Cephalosporins, Monobactams, Carbapenems and B-lactamase inhibitors. Basically they are antibiotic agent which contains a ß-lactam nucleus in its molecular structure. They are the most widely used group of antibiotics available.
Clinical use: ß-lactam antibiotics are indicated for the prophylaxis and treatment of bacterial infections caused by susceptible organisms. Whilst traditionally ß-lactam antibiotics were mainly active against Gram-positive bacteria only. The development of broad-spectrum ß-lactam antibiotics active against various Gram-negative organisms has increased their usefulness.
Mode of Action: ß-lactam antibiotics are bactericidal and act by inhibiting the synthesis of peptidoglycan layer of bacterial cell wall. The peptidoglycan layer is important for cell wall structural integrity, especially in Gram-positive organisms. The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by transpeptidases known as penicillin binding proteins.
Penicillins
Penicillin refers to a group of ß-lactam antibiotics used in the treatment of bacterial infections caused by susceptible, usually Gram-positive organisms.
History: The discovery of penicillin is usually attributed to Scottish scientist Sir Alexander Fleming in 1928, though others had earlier noted the antibacterial effects of Penicillium. The development of penicillin for use as a medicine is attributed to the Adelaide born Nobel Laureate Howard Walter Florey. In March 2000, doctors of the San Juan de Dios Hospital in San Jose published manuscripts belonging to the Costa Rican scientist and medical doctor Clodomiro-Picado-Twight (1887-1944).
The manuscripts explained Picado’s experiences between 1915 and 1927 about the inhibitory actions of the fungi of genera penic. Apparently Clorito-Picado had reported his discovery to Paris Academy of Sciences in Paris, yet did not patent it, even though his investigation had started years before Fleming’s. Fleming, at his laboratory in St. Mary’s Hospital in London, noticed a halo of inhibition of bacterial growth around a contaminant blue-green mold Staphylococcus plate culture.
Fleming concluded that the mold was releasing a substance that was inhibiting bacterial growth and lysing the bacteria. He grew a pure culture of the mold and discovered that it was a Penicillium mold, now known to be Penicillium notatum. Fleming coined the term “Pencicillin” to describe the filtrate of a broth culture of the Penicillium mold. Even in these early stages, penicillin was found to be, most effective against Gram-positive bacteria and ineffective against Gram-negative organisms and fingi.
The chemical structure of penicillin was determined by Dorothy Crowfoot Hodgkin in the early 1940s, enabling synthetic production. A team of Oxford research scientists led by Australian Howard Walter Florey and including Ernst Boris Chain and Norman Heatley discovered a method of mass producing the drug.
Chemist John Sheehan at MIT completed the first total synthesis of penicillin and some of its analogues in the early 1950s, but his methods were not efficient of mass production. Florey and Chain shared the 1945 Nobel Prize in medicine with Fleming for this work. Penicillin has since become the most widely used antibiotic to date and is still used for many Gram-positive bacterial infections.
Development from Penicillin: The narrow spectrum of activity of the penicillins, along with the poor activity of the orally-active Phenoxy methyl penicillin, led to the search for derivatives of penicillin which could treat a wider range of infections.
The first major development was Ampicillin, which offered a broad spectrum of activity than either of the original Penicillins. Further development yielded B-lactamase-resistant penicillins including Flucloxacillin, Dicloxacillin and Methicillin. These are ineffective against Methicillin-resistant Staphylococcus aureus strains that subsequently emerged.
The line of true penicillins was the antipseudomonal penicillins, such as Ticarcillin and Piperacillin, useful for activity against Gram-negative bacteria. However, the usefulness of the B-lactam ring was such that related antibiotics, including Mecillinams, Carbapenems and most importantly, Cephalosporins, have this at the centre of their structures.
Mechanism of Action: B-lactam antibiotics work by inhibiting the formation of peptidoglycan crosses links in the bacterial cell wall. The ß-lactam moiety of penicillin binds to the enzyme (transpeptidase) that links the peptidoglycan molecules in bacteria and this weakens the cell wall of the bacterium. In addition, the build-up of peptidoglycan precursors triggers the activation of bacterial cell wall hydrolysis which further digests the bacteria’s existing peptidoglycan. When the bacteria lose their cell walls they are then called spheroplasts.
Classification
Penicillins are also classified on many ways depending upon the nature and spectrum of activity.
- Natural (Biosynthetic) Penicillins: eg. Benzylpenicillin (Penicillin – G), 2- Pentenyl penicillin (Penicillin F), 3-Pentenyl penicillin, n- Pentyl penicillin, n- Heptyl penicillin (Penicillin – K), p – Hydroxy benzyl penicillin, (Penicillin- X), Phenoxymethyl penicillin. (Penicillin-V) and Zero allergy penicillin (Penicillin- O).
-
- Semi-synthetic Penicillins: eg. Ampicillin, Amoxicillin, Methicillin, Nafàcillin, Oxacillin, Cloxacillin, Dicloxacillin, Flucloxacillin, Carbenicillin, Ticarcillin, Piperacillin, Mezlocillin and Cyclacillin.
- Penicillin Combination: eg. Benzathine penicillin and Procaine penicillin
- Based on Spectrum of Activity
- Effective against Gram-positive bacteria: eg. Benzyl penicillin, Ampicillin, Oxacillin.
- Effective against Gram-negative bacteria: eg. Temocillin.
- Broad spectrum penicillins: eg. Ampicillin, Amoxicillin, Carbenicillin.
- Penicillinase resistant penicillins: eg. Methicillin, Nafacillin, Oxacillin, Cloxacillin, Dicloxacillin, Flucloxacillin.
- Acid resistant penicillins: eg. Benzathine penicillin, Phenoxymethyl penicillin, Phenoxy ethyl penicillin, Phenoxy propyl penicillin.
Chemistry
The penicillin structure composed of fused 4- member ß-lactam ring with thiazolidine nucleus. It contains three chiral carbon atoms (C3, Cs and Co). 6-Amino penicillanic acid (6-APA) is the basic skeleton structure of all penicillins.
Benzylpenicillin potassium
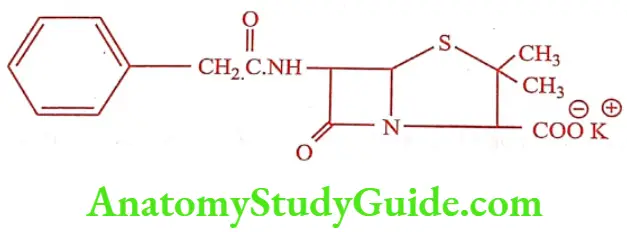
It is commonly known as Penicillin G, is the gold standard type of penicillin. It is given by a parenteral route of administration because of it instability in the stomach PH. It is a white crystalline powder, very soluble in water, practically insoluble in chloroform, ether, fixed oils and liquid paraffin. It contains not less than 96 percent and not more than 100.5 percent of penicillins. It is stored in tightly-closed container in a cool and dry place. It is available as intramuscular or intravenous injection or infusion in divided doses.
Benzathine penicillin (Pencomla)
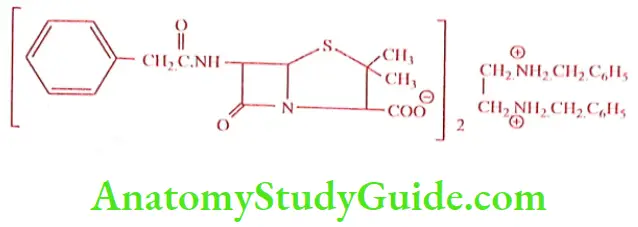
It is a combination of two molecule Benzyl penicillin with one molecule of Benzathine (N, N’ dibenzyl ethylene diamine). It is a white crystalline powder, odorless, freely soluble in formamide and DMF, slightly soluble in ethanol (95%), very slightly soluble in chloroform and water, practically insoluble in ether. In should be stored in a tightly closed container, in a cool and dry place. It is given orally, IM or IV in a divided dose. A single injection is found to be effective for a week.
Procaine penicillin (Fortified procaine penicillin)
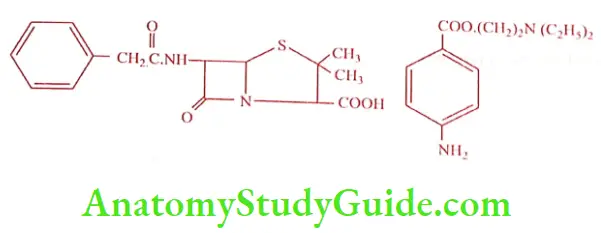
It is a combination of equimolar mixture of one molecule of Benzyl penicillin with one molecule of Procaine. It is a white crystalline powder, slightly soluble in water, sparingly soluble in ethanol (95%). It should be stored in a tightly closed container in a cool and dry place. It is given as IM injection. It is a popular respiratory product which reduces the injection frequency.
Chemical Degradation
The main cause of deterioration of penicillin is the reactivity of the strained lactam ring. The hydrolysis and the nature of degradation products are influenced by the pH of the solution. Thus the B-lactam carbonyl group of penicillin readily undergoes nucleophilic attack by water or hydroxide ion to form an inactive penicilloic acid which on decarboxylation produces penilloic acid. On treatment with strong acid the ultimate product formed is penicillamine and penilloaldehyde. On subject to pH 2.0 and 4.0 gives rise to penillic acid and penicillenic acid respectively.
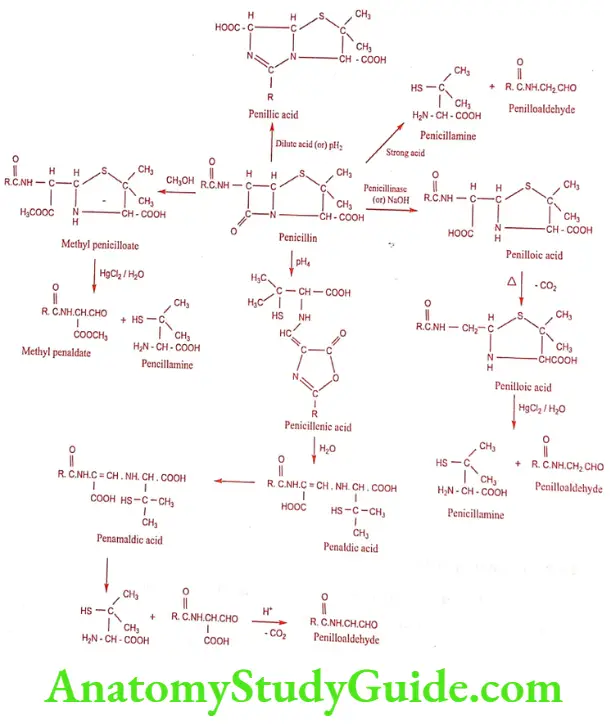
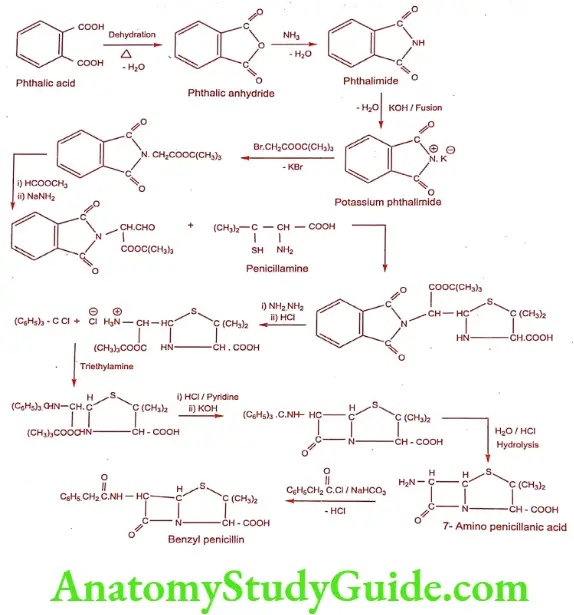
Synthesis of Benzylpenicillin
Cephalosporins
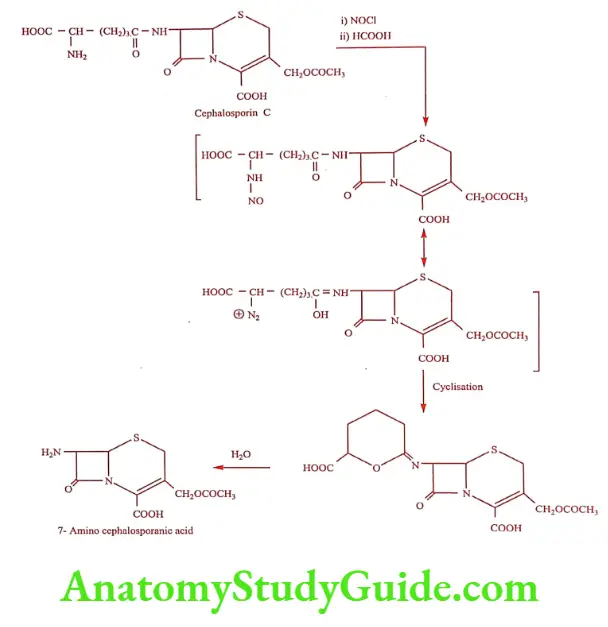
Cephalosporin compounds were first isolated from cultures of Cephalosporium acremonium from a sewer in Sardinia in 1948 by Italian scientist Giuseppe Brotzu. He noticed that these cultures produce substance that was effective against Salmonella typhi, the cause of typhoid fever. Researchers at the Sir William Dunn School of Pathology at the University of Oxford isolated Cephalosporin C, which had stability to B-lactamase but was not sufficiently potent for clinical use. The cephalosporin nucleus, 7-Amino cephalosporanic acid (7-ACA), was derived from Cephalosporin C and proved to be analogous to the penicillin nucleus 6-Amino penicillanic acid. Modification of the 7-ACA side-chains resulted in the development of useful antibiotic agent, and the first agent Cephalothin was launched by Eli Lilly in 1964.
Mode of Action: Cephalosporins are bactericidal and have the same mode of action as other ß-lactam antibiotics (such as Penicillins). Cephalosporins disrupt the synthesis of the peptidoglycan layer of bacterial cell walls. The peptidoglycan layer is important for cell wall structural integrity, especially in Gram-positive organisms. The final transpeptidation step in the synthesis of the peptidoglycan is facilitated by transpeptidases known as penicillin binding proteins.
Clinical use: Cephalosporins are indicated for the prophylaxis and treatment of bacterial infections caused by susceptible organisms. First-generation cephalosporins are predominantly active against Gram-positive bacteria and successive generations have increased activity against Gram-negative bacteria.
Chemistry
The nomenclature of Cephalosporin is more complex than penicillins because of the presence of a double bond in dihydrothiazine ring. Basic nucleus is made up of 7-Amino cephalosporanic acid (7-ACA).
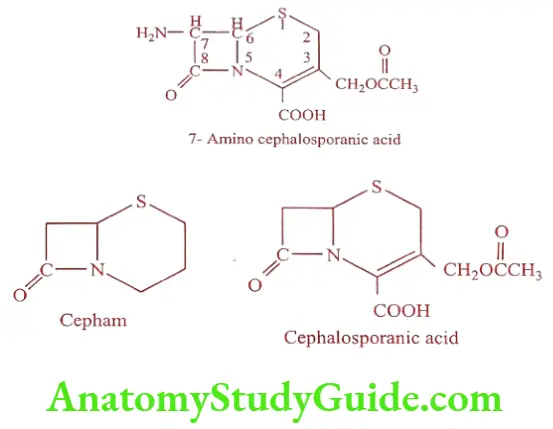
Semi synthetic modification of the basic 7-ACA nucleus has resulted from acylation of the 7-amino group with different acids or nucleophilic substitution or reduction of the acetoxyl group.
The semi synthetic Cephalosporin’s has got the following advantages over natural Cephalosporin’s.
- increased acid stability.
- better oral absorption.
- broad antimicrobial spectrum.
- increased activity against resistant microorganisms.
- decreased allergenicity.
- increased tolerance.
Classification
The classification of Cephalosporin into generations is commonly practiced, although the exact categorization of Cephalosporin’s is often in precise.
- Based on Generations
-
- First Generation: eg. Cefacetrile, Cefalexin, Cefadroxil, Cefaloglycin, Cefalonium, Cefaloridine, Cefalotin, Cefapirin, Cefatrizine, Cefazaflur Cefazedone. Cefazolin, Cefradine, Cefroxadine Ceftezole.
- Second Generation: eg. Cefonicid, Cefprozil, Cefuroxime, Cefuzonam, Cefaclor, Cefamandole, Ceforanide, Cefotiam, Carbacephems, Loracarbef, Cephamycins, Cefbuperazone, Cefmetazole, Cefminox, Cefotetan and cefoxitin.
- Third Generation: eg. Cefcapene, Cefdaloxime, Cefdinir, Cefditoren, Cefetamet, Cefixime, Cefmenoxime, Cefodizime, Cefoperazone, Cefotaxime, Cefpimizole, Cefpodoxime. Cefteram, Ceftibuten, Ceftiofur, Ceftio lene, Ceftizoxime, Ceftriaxone, Ceftazidime, Cefpiramide, Ceftinir, Cefetamet, Cefpodoxime and Cefsulodin.
- Fourth Generation: eg. Cefelidine, Cefepime, Cefluprenam, Cefoselis, Cefozopran, efpirome and Cefquinome.
- Yet to be Classified: eg. Cefaclomezine, Cefaloram, Cefaparole, Cefcanel, Cefedrolor, Cefempidone Cefetrizole, Cefivitril, Cefmatilen, Cefmepidium, Cefovecin, Cefoxazole Cefrotil, Cefsumide, Ceftioxide, Ceftobiprole, Cefuracetime.
- Based on Route of Administration
- Oral Cephalosporins: eg. Cephalexin, Cephadrine, Cefadroxil, Cefachlor, Cefprozil, Loracarbef, Cefuroxime, Cefpodoxime proxetil, Cefixime.
- Parenteral Cephalosporins: eg. Cephalothin, Caphapirin, Cefazolin, Cefamandole, Cefonicid,Cefornaide, Cefuroxime, Cefotaxime, Ceftizoxime, Ceftrixone, Ceftazidime, Cefoperazone.
- Cefalosporin Combinations: eg. Cefoperazone with Sulbactam, Cefotaxime with Sulbactam, Ceftriaxone with Sulbactam,
Synthesis of 7-Amino cephalosporanic acid from Cephalosporin C.
Synthesis of Cefalexin (Cefbact, Keflex)
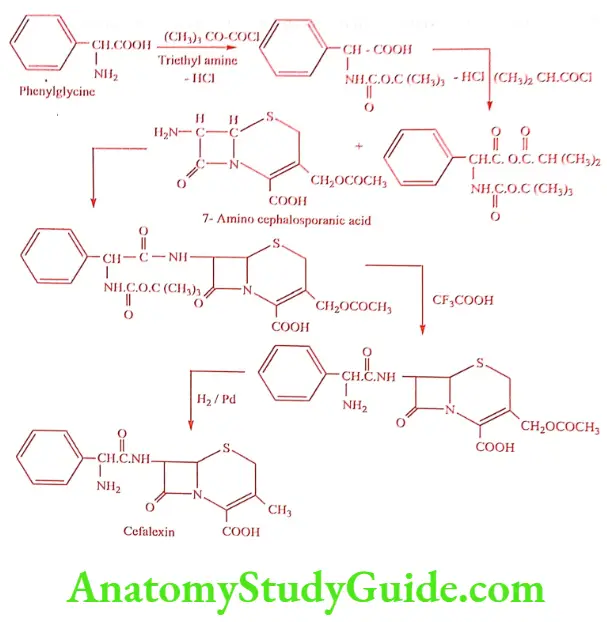
ADR: Pain at injection site, hypersensitivity, GI disturbances and eosinophilia.
Dose: 1to 2gm daily given in divided doses, increased to 6gm in deep seated.
Synthesis of Cefazolin (Reflin, Ciprid)
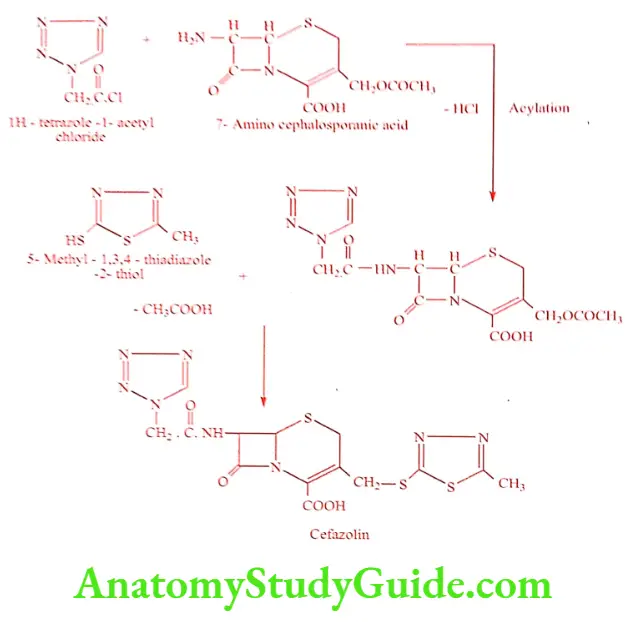
ADR: Super infection, nausea, vomiting and abdominal pain.
Dose: 0.5 to 1.0gm deep i.m, slow i.v injection over 3 to5mts or as i.v infusion every 6 to 12 hrs.
Synthesis of Cefadroxil (Neodrox, Vistadrox)
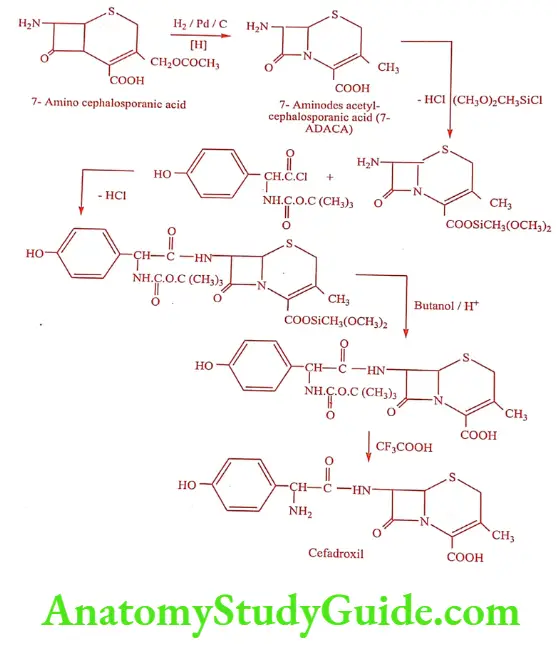
ADR: Allergic reaction, nausea, vomiting and candidiasis.
Dose: 250 to 500mg/8hrs, maximum of 4gm daily.
Degradation of Cephalosporins
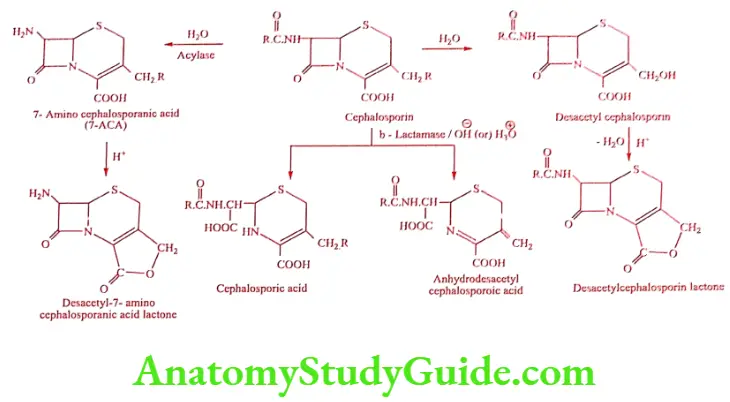
Aminoglycoside Antibiotics
Introduction
Aminoglycoside antibiotics are from the Actinomycetes, particularly from the genus Streptomyces. Among the many antibiotics isolated from the genus, important one is Streptomycin. Several other closely related structures are Kanamycin, Neomycin, Gentamycin, Paromomycin, Tobramycin and Netilmycin.
All the structures consists of amino sugars linked glycosidically. All have one amino hexose and some have a pentose. Additionally each aminoglycosides contains a highly substituted 1, 3 – diamino cyclohexane central ring. They are all basic, water-soluble compounds or mixtures of compounds.
Spectrum of Activity: Aminoglycoside antibiotics are broad-spectrum antibiotics, particularly effective against Gram-negative bacilli, Gram-negative and Gram-positive cocci except Staphylococci, tent to be less sensitive. Streptomycin is particularly active against Mycobacterium tuberculosis. Paromomycin is used in amoebic dysentery.
Mechanism of Action: They act directly on the bacterial ribosome to inhibit the initiation of protein synthesis and to interfere with translation of the genetic message.
They bind to the 30S ribosomal subunit to form a complex that cannot initiate proper amino acid polymerization.
Streptomycin (Ambistryn, Isos)
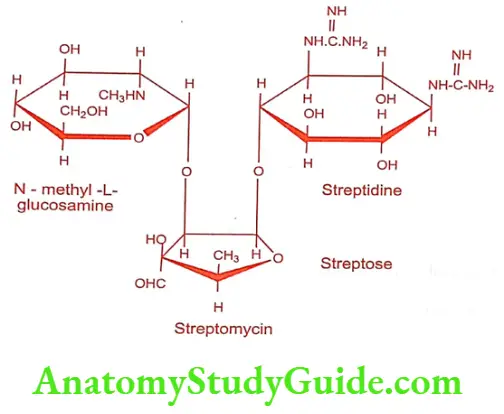
Streptomycin is produced by a culture of Streptomyces griseus. It is official as streptomycin sulfate salt, which is a white, odorless, hygroscopic powder, but stable towards light and air. It is freely soluble in water, slightly soluble in alcohol but insoluble in most of the organic solvents.
Hydrolysis

Streptose cannot be isolated because of its instability.
ADR: Giddiness, vertigo, tinnitus, ataxia and hypersensitivity reactions.
Dose: 15mg/kg, 3 times weekly for first 2 to 3 months for severe diseases.
Use: It is active against various Gram-positive and Gram-negative bacteria. It is effective against Mycobacterium tuberculosis. It is used in the treatment of TB along with other drugs PAS and Rifampicin.
Neomycin B (Clostagen, Neosporin)
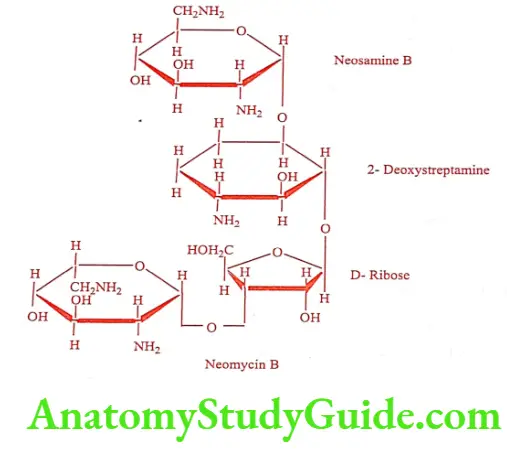
Neomycin is a mixture of three substance isolated from Streptomyces fradiae. The three components A, B and C have been separated by counter – current distribution technique. Neomycin A is a disaccharide, which is a common degradation product of Neomycin B and C.
Neomycin B and C consists of four carbohydrate units and differ only in the configuration of the amino-methyl group in the neosamine ring linked to the ribose unit. Neomycin as a sulfate salt is a white to slightly yellow crystalline hygroscopic and photosensitive powder, very soluble in water.
ADR: Ototoxicity, hypersensitivity, contact dermatitis and local irritation.
Dose: For the treatment of pre or post operative otitis 0.5% drops; 1to 2 drops 3 to 4 times daily.
Use: It is used for treatment of gastrointestinal infections, dermatological infections and acute bacterial peritonitis.
Kanamycin (Efficin, Kancan)
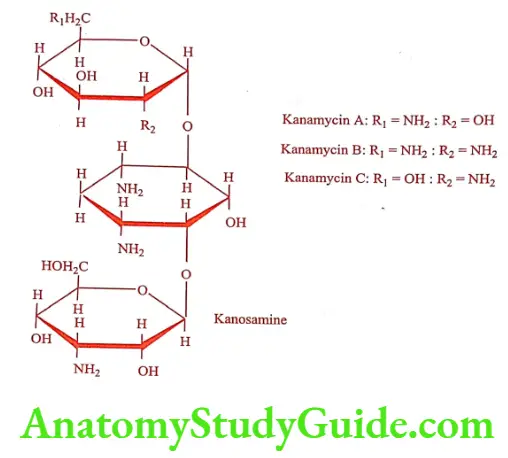
Kanamycin is isolated from a culture of Streptomyces kanamyceticus. Kanamycin is a mixture of component. They differ in the substituted D-glucose attached glycosidically to the 4th position of the desoxystreptamine ring. Kanamycin A contains 6-Amino-6-deoxy-D-glucose; Kanamycin B contains 2, 6-Diamino-2,6- dideoxy-D-glucose; Kanamycin C contains 2-Amino-2-deoxy-D-glucose. It is basic and forms salts with acids through its amino groups. It is water soluble. It is stable to heat and chemicals.
ADR: Pain, inflammation at injection site, GI disturbances and mal-absorption of fat.
Dose: 1gm every hr for 4 hrs, then 1gm every 6 hrs for 3 to 4 days.
Use: It is used in the treatment of infections of the intestinal tract and systemic infections.
Gentamicin (Garamycin, Gensat)
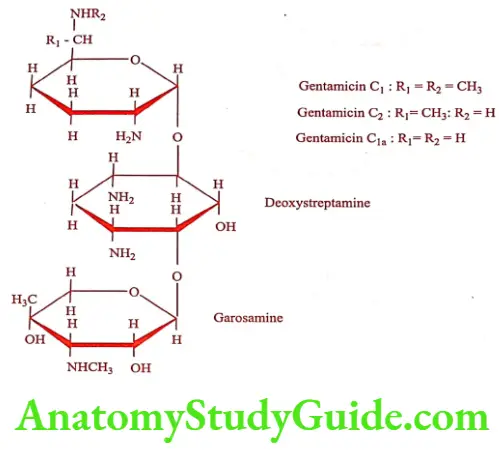
Gentamicin is a group of antibiotics obtained from Micromonospora purpurea. Mixture is identified as Gentamicins C1, C2 and C1a. Gentamicin sulfate is a white to buff substance, soluble in water and insoluble in alcohol, acetone and benzene. It is chemically incompatible with Carbenicillin.
ADR: Dizziness, acute renal failure and acute tubular necrosis.
Dose: 3 to 5mg/kg/day, given in divided doses for every 8 hrs for 7 to 10 days.
Use: It has a broad spectrum of activity against both Gram-positive and Gram-negative organisms. In particular it exhibited strong activity against Pseudomonas aeruginosa and other Gram-negative bacilli. It is also effective in the treatment of a variety of skin infections.
Tobramycin (Tobacin, Tobamist)
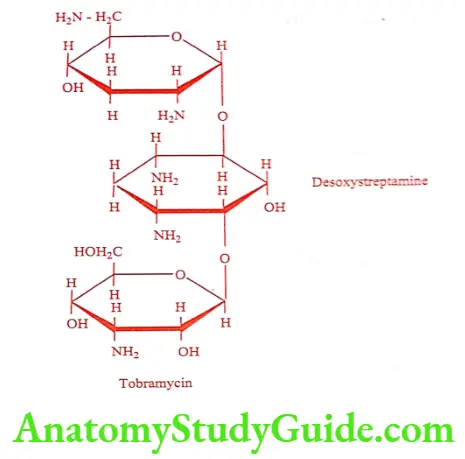
It is obtained from a strain of Streptomyces tenebrarius. Tobramycin more closely resembles Kanamycin B in structure.
ADR: Nausea, vomiting, dizziness and acute renal failure.
Dose: 2 to 3mg/kg once daily or up to 8 to 10mg/kg/day in cystic fibrosis.
Use: It is two to four folds active against Pseudomonas aeruginosa, when compared to Gentamicin.
Amikacin (Amistar, Amitax)
It is a semi synthetic aminoglycoside analogue of Kanamycin A. Chemically it is 1-N-amino-a-hydroxybutyryl kanamycin A.
ADR: Tinnitus, vertigo, ataxia and ototoxicity.
Dose: 15mg/kg daily in equally divided doses injected every 8 or 12 hrs for 7 to 10 days.
Use: It is effective against strains of bacteria that are resistant to other aminoglycoside antibiotics especially Gentamicin and Tobramycin. It is less ototoxic than Gentamicin and Tobramycin.
Netilmicin (Netromycin, Netromax)
It is a semi synthetic derivative of Sisomicin obtained from Micromonospora inyoensis. Chemically it is 1-N-ethyl-sisomicin. Netilmicin structurally resembles Gentamicin la.
ADR: Head ache, malaise and visual disturbances.
Dose: 4 to 6mg/kg once daily or in equally divided doses given every 8 or 12 hrs.
Use: It is active against many Gentamicin-resistant strains, especially E.coli, P.aeruginosa, Enterobacter and Klebsiella.
Tetracyclines
Introduction
Tetracyclines are obtained by fermentation from Streptomyces spp. or by chemical transformation of natural products. They are derivatives of an octahydro- naphthacene, a hydrocarbon system that comprises four annulated six member rings. The group name is derived from this tetracyclic system. The first member of the tetracycline group of antibiotic is Chlortetracycline from a culture of Streptomyces aureofaciens.
Later Oxytetracycline was isolated from Streptomyces rimosus. Tetracycline is a group of antibiotic that include Tetracycline. Volitetracycline, Oxytetracycline, Chlortetracycline, Demeclocycline, Meclocycline, Methacyclin, Doxycycline and Minocycline.
The stereochemistry of the tetracycline is very complex. Carbon atom 4, 4a, 5, 5a, 6 and 12a, are potentially chiral depending on substitution. Oxytetracycline and Doxycycline possess 5a-hydroxy substituent have six chiral carbon and others have only five chiral carbon.
Structure of Tetracyclines: The tetracyclines are amphoteric compounds, forming salts with either acids or bases. It exists mainly as Zwitter ions in neutral solutions.
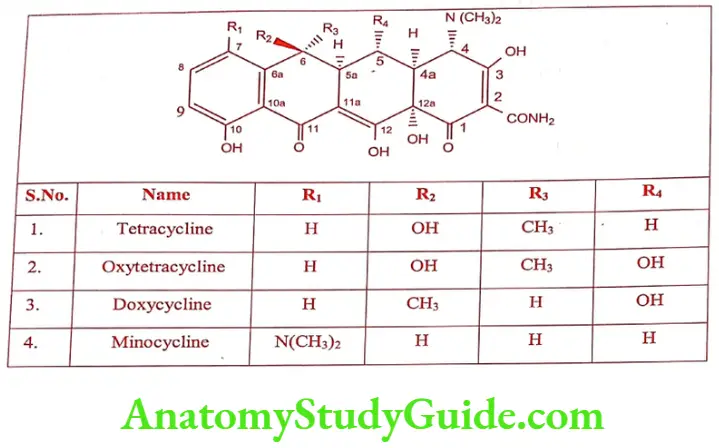
They are yellow in colour. The hydrochloride salts are used for oral administration and usually given is the form of capsule to mask the bitter taste.
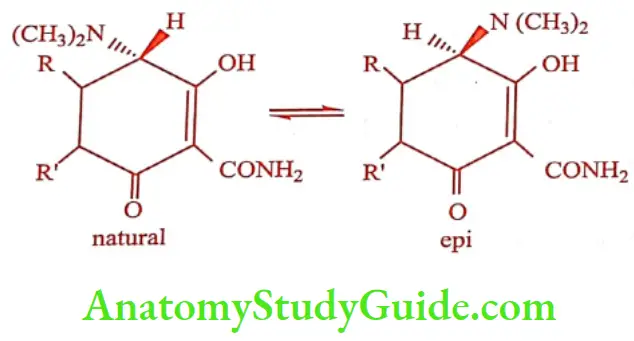
The tetracycline is their ability to undergo epimerization at C4 in solutions of intermediate pH range. These isomers are called epitetracycline. They exhibit less activity than the natural isomers.
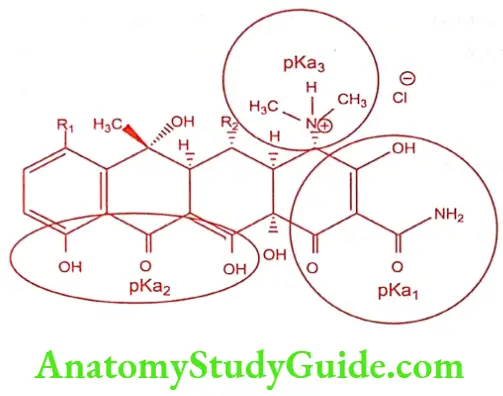
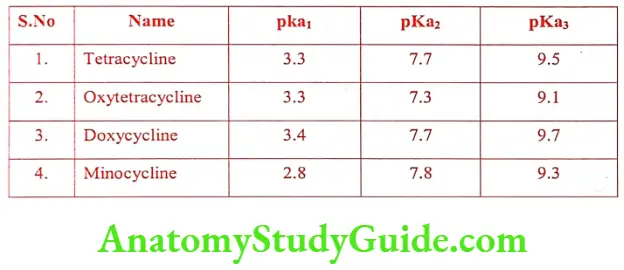
Tetracyclines forms a stable chelate with many metals including calcium, magnesium and iron, such chelates are insoluble in water, leading to impaired absorption of tetracyclines in the presence of milk, calcium, magnesium and aluminium-containing antacids and iron salts.
The affinity of tetracyclines for calcium causes them to be incorporated into newly forming bones and teeth as tetracycline calcium orthophosphate complexes. Deposits of these antibiotics in teeth cause a yellow discoloration that darkens over time. Tetracyclines are distributed into the milk of lactating mothers and will cross the placental barrier into the fetus.
Mechanism of Action: Tetracyclines are specific inhibitors of bacterial protein synthesis. They bind to the 30s ribosomal subunit and thereby prevent the binding of aminoacyl tRNA to the mRNA ribosome complex.
Spectrum of Activity: Tetracyclines are broad spectrum antibiotics, active against wide range of Gram-positive and Gram-negative bacteria, spirochetes, mycoplasma, rickettsiae and chalmydiae. It is used in the treatment of life-threatening infections such as septicemia, endocarditis and meningitis.
Tetracycline (Tetrabact, Lupiterra)
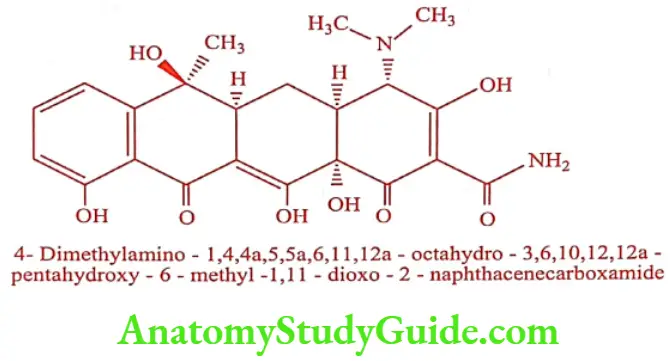
Chlortetracycline on controlled catalytic hydrogenolysis gives Tetracycline. It is also obtained from fermentation of Streptomyces spp. The hydrochloride salt is used most commonly in medicine.
ADR: Oesophageal ulceration, nausea, vomiting and diarrhea.
Dose: 250 to 500mg, every 6 hrs, increased to 4gm daily in severe infections.
Use: It is abroad spectrum antibiotic used in the treatment of bacterial infection of respiratory system, genitor urinary system and gastro-intestinal system.
Oxytetracycline (Terramycin, Oxter)
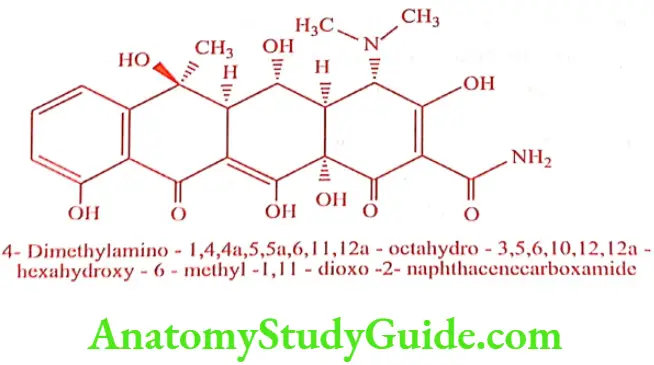
It is isolated from Streptomyces rimosus. It is used for parenteral administration.
ADR: Anorexia, nausea, vomiting and diarrhea.
Dose: 250 to 500mg, 4 times daily.
Use: It is used in the treatment of bacterial infection of respiratory infection, genitorurinary and gastro-intestinal infection and rickettsial infections.
Doxycycline (Lupidox, Doxt)
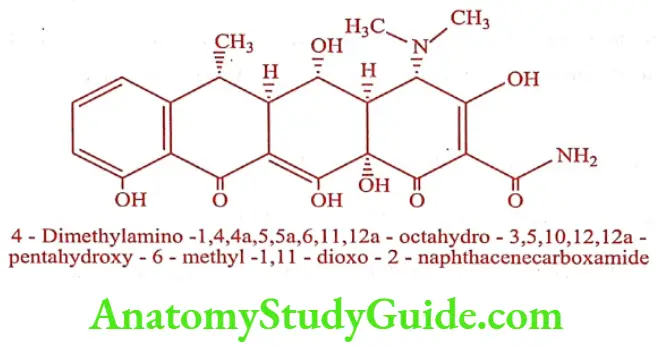
It is produced by catalytic hydrogenation of Methacycline. The 6a-methyl epimer is 3 times as active as its ẞ-epimer.
Synthesis
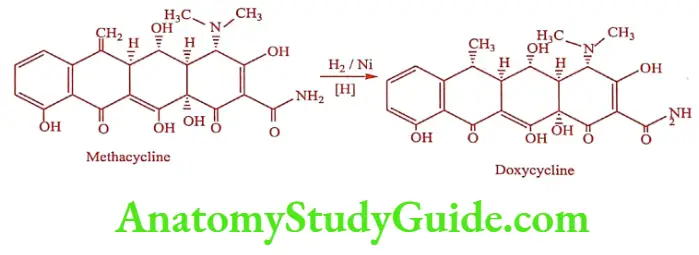
ADR: Permanent staining of teeth, rash, super infection and GI upsets.
Dose: 200mg on day one followed by 100mg once daily.
Use: It is used in the treatment of respiratory tract infection, dental, ophthalmic and ENT infections.
Minocycline (Minolox, Cynomycin)
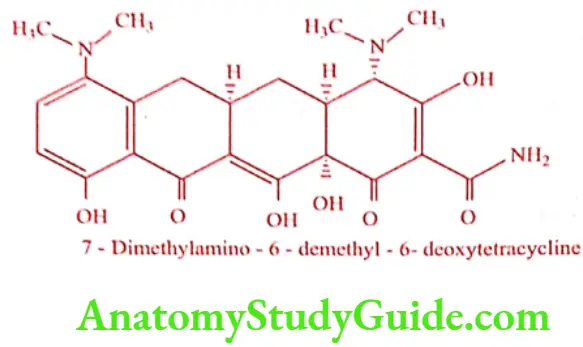
The most potent tetracycline currently used in therapy. It is obtained by reductive methylation of 7-Nitro – 6 – demethyl – 6-deoxytetracycline.
ADR: Oesophageal ulceration, dizziness and decreased hearing.
Dose: Initially, 200mg followed by 100mg every 12 hrs.
Use: It is used in the treatment of pelvic inflammatory diseases, urinary tract infection, gonorrhea and syphilis.
Degradation of Oxytetracycline
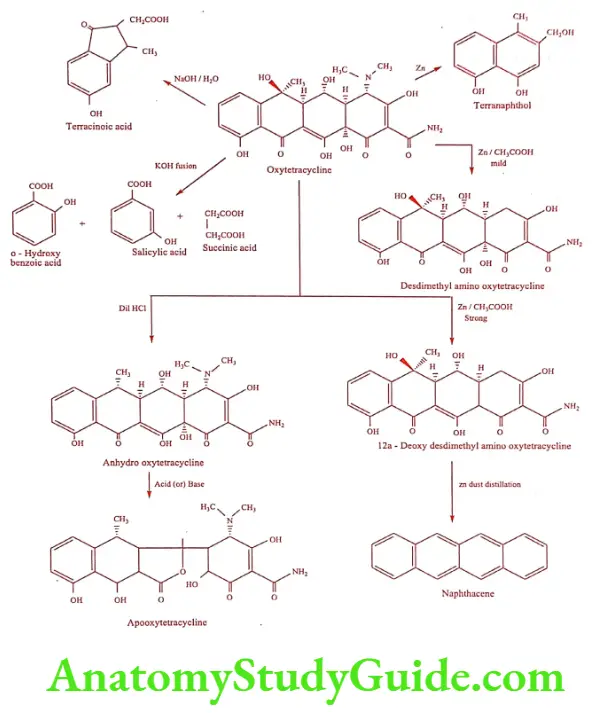
Structure Activity Relationship
- Functional groups at position 5-, 6- and 7- or rings B, C and D in tetracyclines can be removed without drastically altering antimicrobial properties.
- Conversion of carboxamide group to nitriles causes a 20 fold loss of activity though some carboxamide methylamines are highly active.
- Epimerization of Csa adnC4 dehydrogenation (C5a, C11a) results in the loss of activity.
- The monomethylamino and trimethylamino analogues are relatively inactive.
Macrolide Antibiotics
Introduction
Macrolide antibiotics are produced by various strains of Streptomyces. They have three common chemical characteristics
- a large non-planar strain less ring
- a keto group
- a glycosidically linked amino sugar
Usually the lactone ring has 12, 14 or 16 atoms in it, unsaturated with an olefinic group conjugated with the ketone function. They may have a neutral sugar also, in addition to the amino sugar, which is linked glycosidically to the lactone ring.
Mechanism of Action: Macrolide binds to 50s subunit of the bacterial ribosome, to prevent the translocation step of bacterial protein synthesis. It does not bind to mammalian ribosome.
Spectrum of Activity: The spectrum of antibacterial activity of macrolides resembles that of penicillins. They are effective against most species of Gram-positive bacteria, both cocci and bacillai and also Gram-negative cocci especially Neisseria spp. They are also active against Treponema palladium, Mycoplasma, Chalmydia, Compylobacter and Legionella spp.
Erythromycin (Erycin, Erycip)
Erythromycin was isolated form a culture of Streptomyces erythreus. It is a mono – acidic base, which on hydrolysis gives a basic sugar desosamine and a neutral sugar cladinose. It is a very bitter, white or yellow white crystalline powder. It is insoluble in alcohol, but only slightly soluble in water.
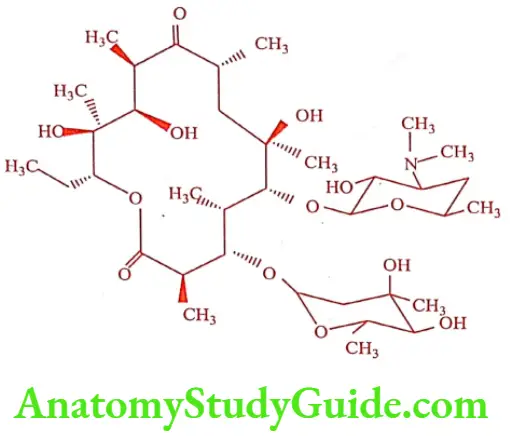
ADR: Rash, urticaria, nausea, vomiting and GI discomfort.
Dose: 1 to 2gm/day, increased up to 4gm/day for severe infection.
Use: The spectrum of activity resembles penicillin. It is the drug of choice in the treatment of infections caused by Campylobacter, Chalmydia, Corynebacterium diptheriae, Bordetella pertussis, Legionella Pneumophila, Mycoplasma pneumoniae and Ureoplasma ureolyticum. It is most effective against Gram-positive cocci, Streptococci, Pneumococci and Staphylococcus aureus, Neisseria meningitidis, Gonorrhoea, Listeria, H. influenzae and Acnes.
Clarithromycin (Clear, Calrithro)
Chemically Clarithromycin is 6-Methyl ether of Erythromycin. It is prepared by simply methylating Erythromycin at 6-hydroxyl group. This semi – synthetic derivative fully retain the antibacterial activity of Erythromycin with increased acid stability and oral bioavailability and reduced gastrointestinal side effects. It inhibits Cytochrome p450 oxidase and thus potentiates the actions of drugs metabolized by these enzymes. It occurs as white crystalline solids, practically insoluble in water, but sparingly soluble in alcohol, freely soluble in acetone.
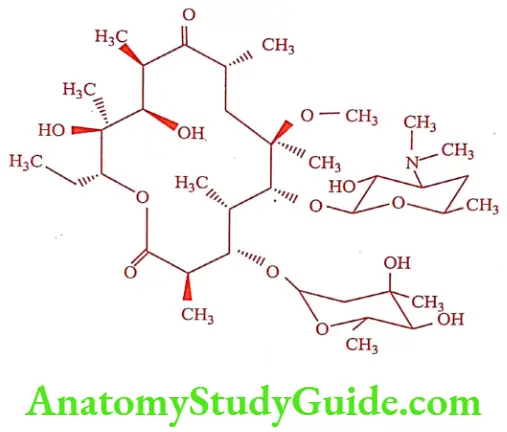
ADR: GI upset, altered taste, head ache and hallucination.
Dose: 250mg bid increased to 500mg bid for severe infection if necessary for 7 to 14 days.
Use: It exhibit greater potency against Chalmydia pneumoniae, H.influenza, M. pneumoniae: Legionella spp and M. catarrhalis. It is significantly more active than Erythromycin against Streptococci and S. pneumoniae, but it is more expensive than Erythromycin.
Azithromycin (Azithro, ATM)
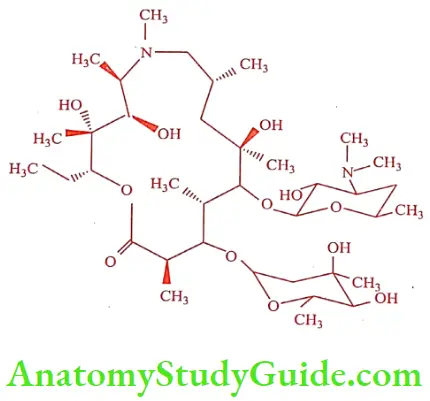
It is a semi-synthetic derivative of Erythromycin. It is a prototype of a series of nitrogen-containing, 15-member ring macrolides known as azalides. It is more stable than other macrolide antibiotics.
ADR: Mild to moderate nausea, vomiting, abdominal pain and flatulence.
Dose: 500mg once daily for 3 days.
Use: Spectrum of antimicrobial activity is similar to Erythromycin and Clarithromycin. It is more active against Gram-negative bacteria and less active against Gram-positive bacteria.
Lincomycins
Introduction
Lincomycins are sulfur-containing antibiotics isolated from Streptomyces lincolensis. Lincomycin is the most active and medicinally useful compounds. Structural modification of Lincomycin resulted in 7- Chloro-7- deoxy derivative called Clindamycin. Clindamycin possess great antibacterial potency, when compare to Lincomycin. Lincomycin resembles macrolide in antibacterial spectrum. They are active against Gram – positive bacteria, particularly cocci and also effective against non-spore-forming anaerobic bacteria, actinomycetes, mycoplasma and Plasmodium spp. Lincomycin binds to 50s ribosomal subunit to inhibit protein synthesis.
Lincomycin hydrochloride (Link, Lincozin)
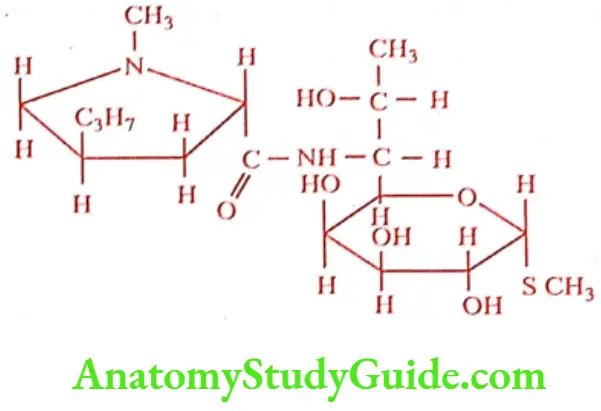
Lincomycin is a basic antibiotic forming a readily water- soluble hydrochloride. It is a white crystalline solid, readily soluble in water and alcohol. It is active against many Gram-positive organisms including penicillinase-producing Staphylococci, B-hemolytic Streptococci and Penumococci.
ADR: Hypotension, vertigo, dermatitis, rash and urticaria.
Dose: 500mg 3 or 4 times daily.
Use: It is used to treat a wide variety of upper respiratory, skin and tissue infection.
Clindamycin hydrochloride (Clincin, Climycin)
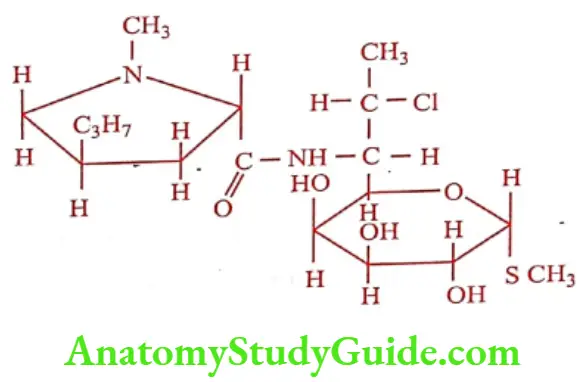
Replacement of 7(R) hydroxy group of Lincomycin with chlorine gives Clindamycin with enhanced antibacterial activity. Chemically it is called 7(S) – Chloro -7- deoxy lincomycin. It is available as crystalline, water soluble hydrochloride hydrate. Clindamycin palmitate hydrochloride salt in oral dosage form as wells as Clindamycin phosphate ester in solution for IM or IV injection is also available.
ADR: Diarrhea, abdominal pain, nausea and vomiting.
Dose: 150 to 300mg every 6 hrs. May be increased to 450mg every 6 hrs in severe infection.
Use: It is used in the treatment of a wide variety of upper respiratory, skin and tissue infection. It is active against Streptococci, Staphylococci and Pneumococci.
Polypeptide Antibiotics
Introduction
Polypeptide antibiotics are composed of amino-acids joined by peptide links into cyclic structures with or without branches from the ring. They contain unusual amino acids such as a,y-Diaminobutyric acid and a,ẞ-Diaminopropionic acid as well as saturated aliphatic acid such as Methyl octanoic acid (MOA). They possess a number of interesting and unique characteristics:
- They frequently consist of several structurally similar but chemically distinct entities isolated from a single source.
- Most of them are cyclic except Gramicidin.
- They contain D-amino acids which are not found in higher plants or animals.
- Many of them contain non-amino acid moieties, such as heterocyclic, fatty acids, sugars etc.,
Polymyxin (Aerosporin, Neosporin)
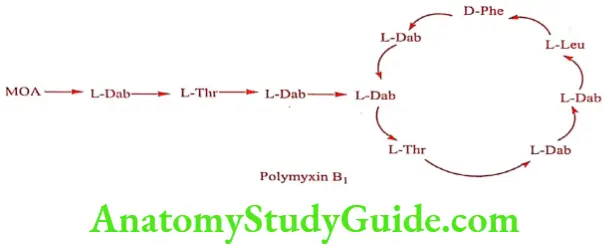
Polymyxin was isolated from Bacillus polymyxa and it produces a mixture of five antibioticis: A, B, C, D and E. The two most important of these are Polymyxin B and Polymyxin E (Colistin) and the other member of the groups are too toxic for clinical use. Polymyxin B itself is a mixture of B1 and B2, which can be separated by countercurrent distribution, of which Polymyxin B1 is used clinically. On hydrolysis both produces L – a,y-diaminobutyric acid, L-threonine, D-phenylalanine and L-leucine. They differ in that B1 contains (+) – 6 methyl octanoic acid, where as B2 contains isooctanoic acid.
ADR: Occasional nausea, vomiting, diarrhea, flushing and paresthesias.
Dose: 1.5 to 2.5mg/kg/day in divided doses.
Use: Polymyxin B is useful against many Gram-negative organisms. It is mainly used in topical application for local infection in wounds and burns.
Colistin (Colistop, Walamycin)
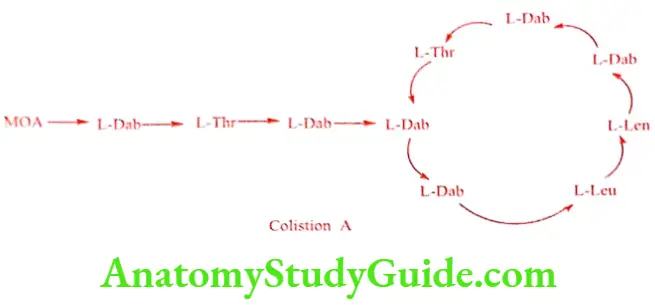
Polymyxin E is also called as Colistin. It is available in the form of sulfate salt. It is a mixture of antibiotics produced by Bacillus polymyxa var. colistinus. On hydrolysis La‚y a,y diaminobutyric acid, L-threonine, DL – leucine and (+) – 6 – methyl octanoic acid are produced.
ADR: Renal damage, neurological disturbances and GI disturbances.
Dose: 25 to 100mg three times a day.
Use: It is used for the treatment of urinary tract infections caused by Gram-negative organisms such as Aerobacter, Bordetella, Escherichia, Klebsiella, Pseudomonas, Salmonella and Shigella spp.
Vancomycin (Vanlid, Vancomate)
Vancomycin is a glycopeptide antibiotics isolated from Streptomyces orientalis. It is freely soluble in water, but insoluble in ether, chloroform.
ADR: Ototoxicity, nephrotoxicity, eosinophilia and red-man syndrome
Dose: 500mg every 6 hrs infused over at least 60mts or 1gm every 12 hrs infused over 100mts.
Use: It is highly active against Gram-positive Cocci, Neisseria and Clostridia. It inhibits synthesis of peptidoglycan in cell-wall formation.
Nitrobenzene Derivative
Chloramphenicol (Chloraxin, Paraxin)
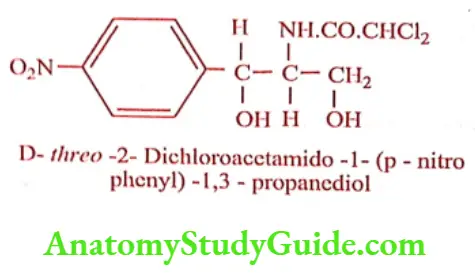
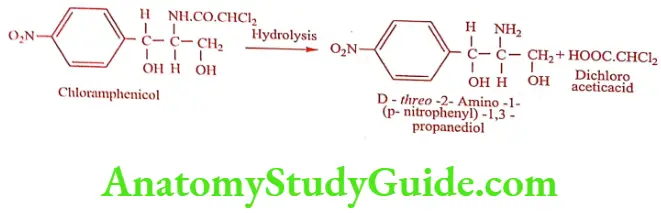
Chloramphenicol was isolated from a culture of Streptomyces venezuelae. Hydrolysis (acid or alkali) yielded dichloro acetic acid and a base called 2-Amino-p- nitro phenyl-1, 3- propanediol. This base contains two chiral carbon give rise to threo and erythro forms. By relating the structure to L-Norephedrine, L-Pseudo norephedrine and D-series, it was assigned to D (-) threo configuration.
It is a yellowish white needle -like crystals, slightly soluble in water, very soluble in alcohol. It has no odor, but has a very bitter taste. It possesses two chiral carbon atoms in the acylamido propanediol chain. Biological activity resides almost exclusively in the D-threo isomer, the L-threo and D and L-erythro isomers are virtually inactive. It is relatively a simple compound possessing aromatic nitro group previously unknown in a natural products. Because of this nitro functional group, it can be reduced, diazotized and coupled with ẞ-naphthol to give a red azo-dye. This reaction is useful for the quantification of Chloramphenicol.
Mode of Action: It inhibits protein synthesis in bacteria. It acts primarily by binding reversibly to the 50s ribosomal subunit and appears to prevent the binding of the amino acid containing end of aminoacyl tRNA to the acceptor site on the 50s ribosomal subunit.
Structure Activity Relationship: In an attempt to establish a relationship between the structure of Chloramphenicol and its biological activity several compounds have been synthesized and following observations made:
- Replacement of – NO2 group with a number of other substituent’s like – CN, CONH2, NH2, OH, N(CH3)2, NHR, NHCH2R, X, Ph or various heterocyclic groups resulted in some reduction or complete loss of antibacterial activity.
- Shifting of the NO2 group from p-position reduces the antibacterial activity.
- When phenyl group is replaced by other aromatic or alicyclic groups like naphthyl, pyridyl, quinolyl, thienyl, furyl, cyclohexyl, etc, they are less potent than Chloramphenicol.
- The primary alcohol group is required since its replacement with H or alkyl group leads to compounds with much less potent activity.
In general, electro negativity, molecular volume and p – quinoid type system are the factors which determine activity. Studies of the metabolic fate of Chloramphenicol have shown that the major portion (80%) of the antibiotic is eliminated in the urine as glucuronic acid conjugate.
Synthesis
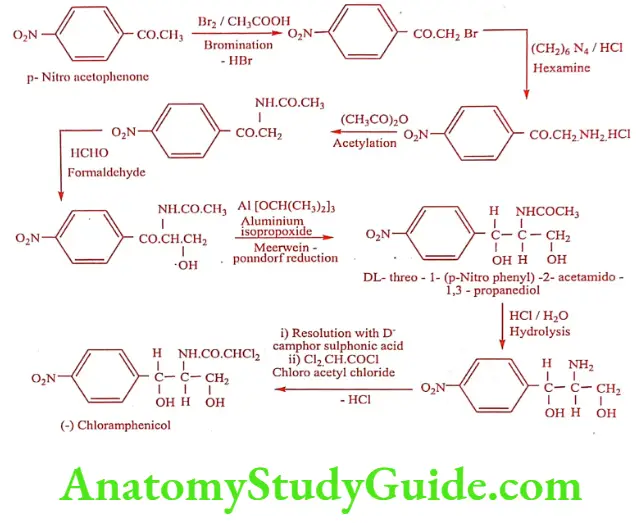
ADR: GI symptoms, bleeding, peripheral and optic neuritis.
Dose: 500mg/kg/day in 4 divided doses increased to 1000mg/kg/day for severe infection.
Use: Chloramphenicol is an important drug of choice for the treatment of typhoid fever caused by Salmonella typhi. It is also effective against most bacteria, Haemophilus influenzae, Neisseria meningitidis.
Steroidal Antibiotic
Fusidic acid (Fucidin,Fusital)
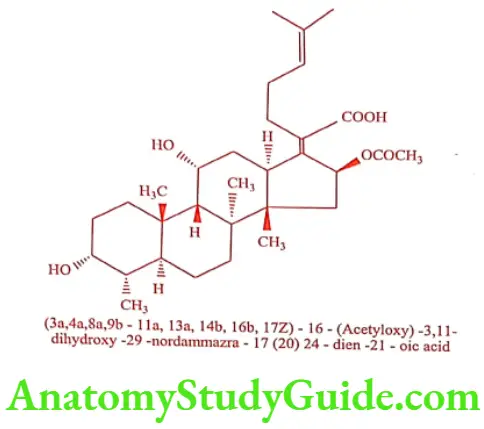
Fusidic acid was isolated from the broth of Fusidium coccineum. It is a member of an unusual class of fused-ring antibiotic with a steroidal structure. It reversibly inhibits the process of translocation at the level of systems containing prokaryotic ribosomes as well as eukaryotic ribosomes. It binds to the 50s ribosomes subunit and to the y-core, which does not contain any more peptidyl transferase.
ADR: Jaundice, liver dysfunction and GI disturbances.
Dose: As 2% ointment/cream/gel apply onto affected area 3to 4 times daily.
Use: It is active against a number of Gram-positive bacteria.
Miscellaneous
Mupirocin (Mupirax, Pirocin)
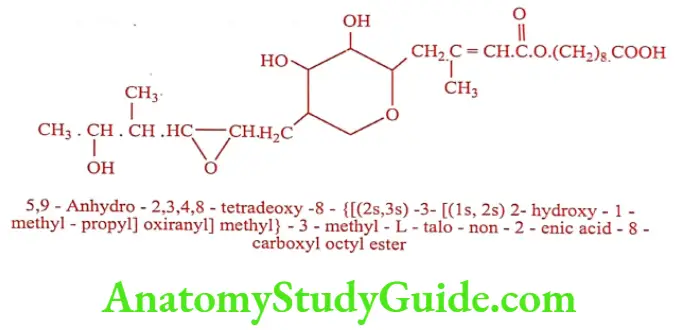
It is produced by the submerged fermentation of Pseudomonas fluorescens. It interferes with RNA synthesis and protein synthesis in susceptible bacteria. It reversibly binds with bacterial isoleucyl tRNA synthase to prevent the incorporation of isoleucine into bacterial protein.
ADR: Burning, stinging, itching, erythema, dermatitis and rash.
Dose: Apply as 2% ointment tid for 10 days.
Use: Its antibacterial activity is confined to Gram-positive and Gram-negative Cocci including Staphylococci, Strephtococci, Neisseria spp. The use of Mupirocin is confined to external application. It is supplied in water – miscible ointment containing 2% of the antibiotics in PEG 400 and 3350.
Leave a Reply