White Blood Cells Disorder and Lymph Nodes
Question 1. Enumerate the Causes of Eosinophilia.
Answer:
Causes of increased Eosinophil count:
- Allergic disorders such as asthma, hay fever
- Parasitic infestations
- Drug reaction
- Malignancies (Hodgkin disease)
- Autoimmune disorders (pemphigus, dermatitis herpetiformis)
- Vasculitis
Read and Learn More Preparatory Manual of Pathology Question and Answers
Question 2. Write a note on the leukemoid reaction.
Answer:
Leukemoid reactions
- Presence of a large number of immature granulocytes in the blood, mimicking myeloid leukemia
- More common in children
- Seen in severe infection, hemolysis, or in malignancies
Question 3. Write a note on agranulocytosis.
Answer:
Agranulocytosis
- Clinically significant reduction in neutrophils
- This leads to an increase in the susceptibility to bacterial and fungal infections
- Serious infections are most likely when the neutrophil count falls below 500 per mm3
- The most common cause is drug toxicity, e.g. alkylating agents, antimetabolites
Question 4. Classify non-Hodgkin lymphoma.
Answer:
Non-Hodgkin lymphoma
1. Peripheral B-cell neoplasms
- Chronic lymphocytic leukemia/Small lymphocytic lymphoma
- Extranodal marginal zone lymphoma
- Mantle cell lymphoma
- Follicular lymphoma
- Marginal zone lymphoma
- Hairy cell leukemia
- Plasmacytoma/plasma cell myeloma
- Diffuse large B-cell lymphoma
- Burkitt lymphoma
2. Peripheral T-cell and NK-cell neoplasms
- Mycosis fungoid’s/Sézary syndrome
- Anaplastic large cell lymphoma
- Angioimmunoblastic T-cell lymphoma
- Enteropathy-associated T-cell lymphoma
- Panniculitis-like T cell lymphoma
- Hepatosplenic γδT cell lymphoma
- Adult T cell leukemia/lymphoma
- Extranodal NK/T cell lymphoma
- NK-cell leukemia
3. Precursor B-cell neoplasms
- B-cell acute lymphoblastic leukemia/lymphoma (B-ALL)
4. Precursor T cell neoplasms
- T cell acute lymphoblastic leukemia/lymphoma (T-ALL)
Question 5. Discuss acute lymphoblastic lymphoma in relation to its pathogenesis, morphology and prognosis.
Answer:
Acute Lymphoblastic Lymphoma (ALL)
Acute Lymphoblastic Lymphoma Pathogenesis
- 90% of ALLs have hyper ploidy (>50 chromosomes)
- T cell ALLs show gain-of-function mutations in NOTCH1
- B-cell ALLs show loss-of-function mutations in PAX5 or balanced t(12;21) translocation
- Mutations lead to the maturation arrest of lymphoid cells with immature lymphoid cell proliferation
Acute Lymphoblastic Lymphoma Morphology
- Bone marrow: Hypercellular and packed with lymphoblasts
- Tumor cells have scant basophilic cytoplasm with nuclei larger than those of small lymphocytes, stippled nuclear chromatin with convolutions in the nuclear membrane
- Lymphoblasts are periodic acid–Schiff (PAS) positive, and myeloperoxidase negative
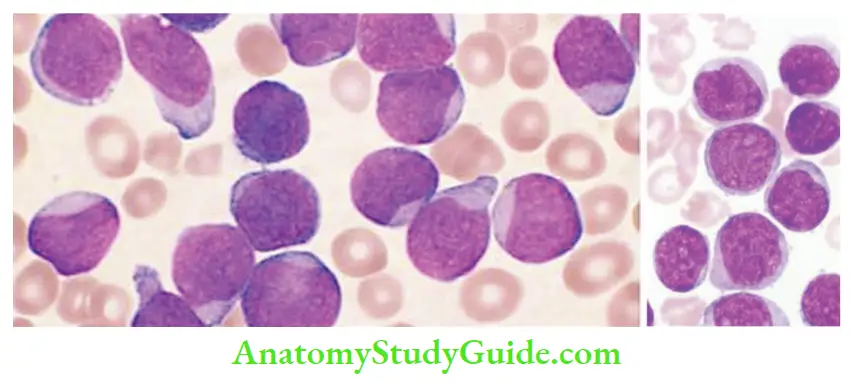
Acute Lymphoblastic Lymphoma Prognosis
Worse prognostic factors
- Age < 2 years
- Presentation in adolescence or adulthood
- High tumor burden (peripheral blood blast count greater than 100,000/mm3)
Favorable prognostic factors
- Age between 2 and 10 years
- Low white cell count
- Hyperdiploidy, trisomy of chromosomes 4, 7, and 10, and the presence of t(12;21)
Question 6. Write a note on Chronic Lymphocytic Leukemia.
Answer:
Chronic lymphocytic leukemia/small lymphocytic lymphoma
- Most common leukemia affecting adults
- In CLL, lymphocytosis is present at the time of making a diagnosis (absolute lymphocyte count) is >5000 per mm3
- CLL leukemic presentation, SLL localized disease
- The median age at diagnosis is 60 years
- M:F:: 2: 1
Chronic Lymphocytic Leukemia Morphology
- Diffuse effacement of lymph nodes by an infiltrate of predominantly small lymphocytes
Chronic Lymphocytic Leukemia Proliferation centers
- Admixed are variable numbers of larger activated lymphocytes that form loose aggregates
- Are pathognomonic for CLL/SLL
Chronic Lymphocytic Leukemia Peripheral Smear
- A large number of small round lymphocytes with scant cytoplasm
- Smudge cells: Disrupted cells, while making smears
Chronic Lymphocytic Leukemia Immunophenotype
- Tumor cells express: CD19, CD20 (Pan B-cell markers), CD5 and CD23 positive
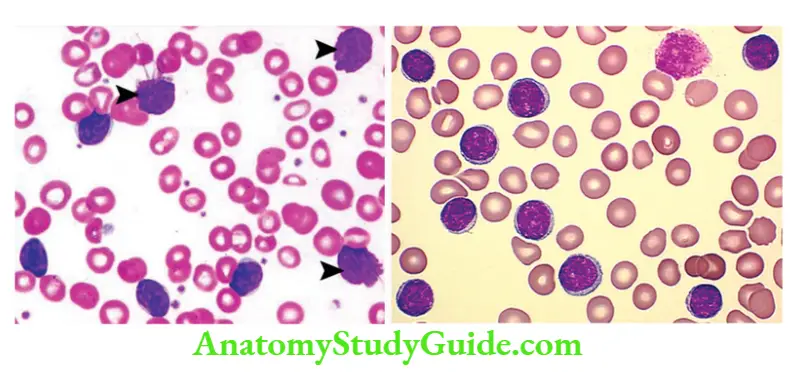
Chronic Lymphocytic Leukemia Clinical features
- Easy fatigability, weight loss, anorexia
- Generalized lymphadenopathy and hepatosplenomegaly
- Hypogammaglobulinemia, which predisposes to infections
- Autoimmune hemolytic anemia (due to auto-antibody production by B-cells)
Question 7. Write a note on Burkitt’s lymphoma.
Answer:
Burkitt’s Lymphoma
Burkitt’s Lymphoma Three types:
1. African (endemic) Burkitt lymphoma:
- Young children most commonly involve—mandible
- Abdominal viscera – kidneys, ovaries, and adrenal glands can be involved
2. Sporadic (non-endemic) Burkitt lymphoma:
- In developing countries, affects children and young adults
- Presents as a mass involving the ileocecum and peritoneum
3. HIV-associated Burkitt lymphoma:
- Involves lymph nodes, brain, bone marrow, liver, and GIT
Burkitt’s Lymphoma Pathogenesis
- All three types of Burkitt’s lymphomas are associated with EBV infection
- Associated with increased MYC expression
Increased MYC expression occurs due to either of the following translocations:
- t(8;14): IgH locus on chromosome 14 and MYC on chromosome 8
- t(2;8): Igκ on chromosome 2 and MYC on chromosome 8
- t(8;22): Igλ on chromosome 22 and MYC on chromosome 8
Burkitt’s Lymphoma Morphology
- Diffuse infiltrate of lymphoid cells with round to ovoid nuclei, coarse chromatin, several nucleoli, and a moderate amount of cytoplasm
- Tumor exhibits a high mitotic index and contains numerous apoptotic cells
- Nuclear remnants of these apoptotic cells are phagocytosed by benign macrophages
- These phagocytes have abundant clear cytoplasm, creating a characteristic “starry sky” pattern
Burkitt’s Lymphoma Immunophenotype
- Tumor cells express CD19, CD20, CD10, BCL6
Burkitt’s Lymphoma Note:
- Burkitt lymphoma is very aggressive but responds well to intensive chemotherapy
- Most children and young adults can be cured
Question 8. Write a note on the diagnostic criteria of symptomatic multiple myeloma.
Answer:
Diagnostic criteria of symptomatic multiple myeloma:
1. M-protein in serum or urine
- No levels of serum proteins are included. However, most cases have IgG >3 g/dl, IgA >2.5 g/dl or >1 gm/24 hr of urine light chain
2. Bone marrow clonal plasma cells or plasmacytoma
- No minimal level is designated; however, monoclonal plasma cells exceed 10% of nucleated cells
3. Related organ or tissue impairment (CRAB: Hypercalcemia, renal insufficiency, anemia, bone lesions)
- Manifestations of end-organ damage are the most important criteria of symptomatic myeloma
Question 9. A 70-year-old woman admitted with worsening anemia and pathological fracture of the humerus had an ESR of 120 mm in 1 hour. Her peripheral smear showed increased rouleaux formation. X-ray of the skull showed multiple punched-out osteolytic lesions.
1. What is the most probable diagnosis? Write briefly on the etiopathogenesis of this disease.
2. Describe the bone marrow changes of this disease.
3. Enumerate the diagnostic criteria of symptomatic multiple myeloma.
Answer:
Diagnosis: Multiple myeloma
Pathogenesis of multiple myeloma:
1. Translocations of Ig heavy-chain (IgH) gene on chromosome 14q32 with
- Cyclin D1 on chromosome 11q13
- Cyclin D3 on chromosome 6p21
2. Deletions of chromosome 17p
3. Mutations involving the NF-ΚB pathway and rearrangements involving MYC
Morphology
- Pathological fractures are seen in the vertebral column followed by ribs, skull, pelvis, femur, clavicle, and scapula (in descending order)
- Bone lesions appear radiographically as punched-out defects, measuring 1 to 4 cm in diameter
- Marrow contains an increased number of plasma cells, comprising >30% of the cellularity
- Plasma cells have an eccentrically placed nucleus with perinuclear clearing (due to the prominent Golgi apparatus)
Other cells which can be seen in the marrow
- Flame cells with fiery red cytoplasm
- Mott cells with multiple grape-like cytoplasmic droplets
- Russell bodies: Intra-cytoplasmic globular inclusions
- Dutcher bodies: Intra-nuclear inclusions
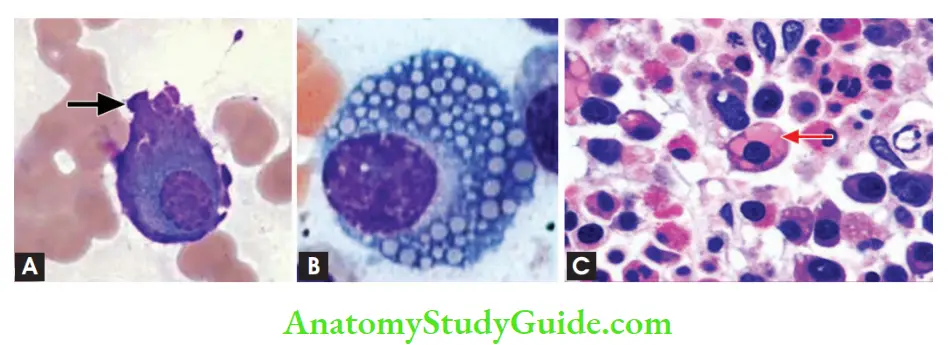
Peripheral Smear
Rouleaux formation can be seen
Plasma cell leukemia: Tumor cells (plasma cells) appear in peripheral blood Diagnostic criteria of symptomatic multiple myeloma: (already discussed above)
1. M-protein in serum or urine:
- No levels of serum proteins are included. However, most cases have IgG >3 g/dl, IgA >2.5 g/dl or >1 gm/24 hr of urine light chain
2. Bone marrow clonal plasma cells or plasmacytoma:
- No minimal level is designated; however, monoclonal plasma cells exceed 10% of nucleated cells
3. Related organ or tissue impairment (CRAB: Hypercalcemia, renal insufficiency, anemia, bone lesions)
- Manifestations of end-organ damage are the most important criteria of symptomatic myeloma
Intracellular Accumulation Of Protein
Question 10. Discuss the pathognomonic features of hairy cell leukemia.
Answer:
Hairy cell leukemia
- Affects middle-aged males, M: F::5:1
Pathogenesis
- Associated with BRAF mutations in >90% of cases
Clinical features
- Massive splenomegaly is the most common finding
- Hepatomegaly and lymphadenopathy
- Pancytopenia due to marrow infiltration and splenic sequestration
- Infections—increased incidence of atypical mycobacterial infections
- Excellent prognosis
Morphology
- Leukemic cells have fine hair-like projections that are best recognized under a phase-contrast microscope
- Peripheral smear: Hairy cells have round, oblong, or reniform nuclei and moderate amounts of pale blue cytoplasm with thread-like or bleb-like extensions
- Bone marrow aspirate: Tumor cells are enmeshed in reticulin fibrils, which results in dry tap
Immunophenotype
- Hairy cell leukemias express pan-B-cell markers CD19 and CD20, and distinctive markers, such as CD11c, CD25, CD103, and annexin A1
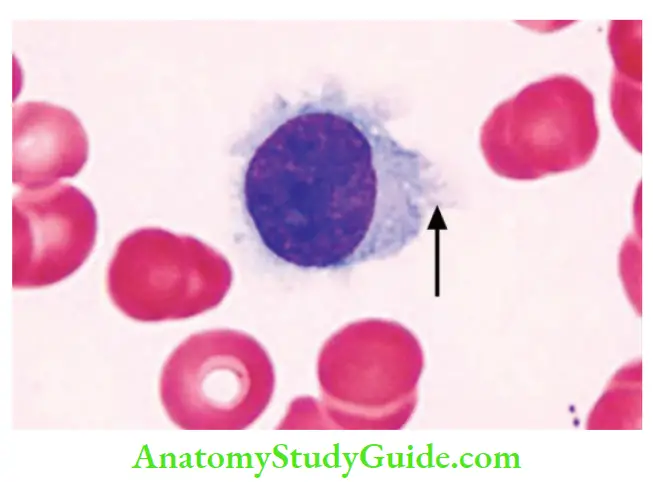
Question 11. Discuss in detail the classification, molecular pathogenesis, and morphology of Hodgkin lymphoma and its types.
Answer:
WHO classification recognizes five subtypes of Hodgkin lymphoma (HL)
1. Hodgkin lymphoma Classical types:
- Nodular sclerosis
- Mixed cellularity
- Lymphocyte-rich
- Lymphocyte depletion
2. Lymphocyte predominance type
Intracellular Accumulation Of Protein
Pathogenesis of Hodgkin Lymphoma
1. NF-ΚB gets activated by one of the following mechanisms:
- Due to EBV infection, which turns on genes that promote lymphocyte survival and proliferation
- EBV-positive tumor cells express latent membrane protein-1 (LMP-1), that upregulates NF-B
- Reed-Sternberg cells show gains in REL proto-oncogene on chromosome 2p, which leads to an increase in NF-B activity
2. Florid accumulation of reactive cells in the tissues involved by classical HL occurs in response to a wide variety of cytokines (for example IL-5, IL-10, and M-CSF), chemokines (for example eotaxin), and other factors (for example immunomodulatory factor galectin-1)
Hodgkin lymphoma Morphology
- Diagnostic Reed-Sternberg (RS) cells are large cells (45 m in diameter) with abundant cytoplasm and single or multiple nuclei
- The Nucleus of the RS cell has a large inclusion-like nucleolus about the size of a small lymphocyte (5 to 7 m in diameter)
- RS cells undergo a peculiar form of cell death in which the cells shrink and become pyknotic, a process described as “mummification”
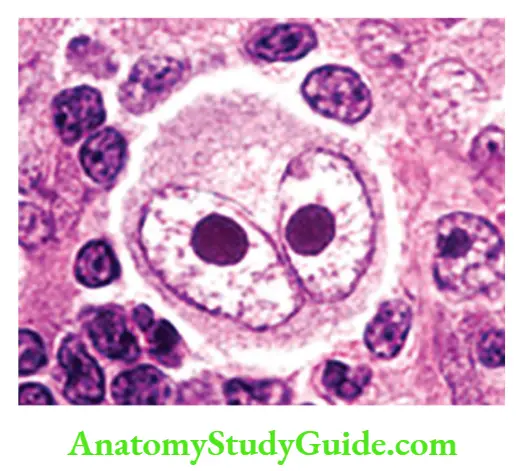
Hodgkin lymphoma Variants
1. Nodular sclerosis type
- The most common form of Hodgkin lymphoma
- Deposition of collagen in bands is seen that divide the involved lymph nodes into circumscribed nodules
- Characterized by the presence of lacunar cell variant of Reed-Sternberg cell
- Lacunar cells: Folded or multilobed nucleus, that lies within an open space, which is an artifact created by disruption of the cytoplasm during tissue sectioning, leaving the nucleus sitting in an empty hole (a lacuna)
- Reed-Sternberg cells are found in a polymorphous background of T cells, eosinophils, plasma cells, and macrophages
- Excellent prognosis
2. Mixed-cellularity type
- Diffuse effacement of lymph nodes, with a cell population comprising of T cells, eosinophils, plasma cells, and macrophages admixed with Reed-Sternberg cells
- Mononuclear variants of RS cells are seen
- Mononuclear RS cell: Contains a single nucleus with a large inclusion-like nucleolus
3. Lymphocyte-rich variant
4. Lymphocyte-depleted variant
Note:
- All four mentioned subtypes are associated with EBV infection
- Immunophenotype of HL cells show PAX-5 (B-cell transcription factor), CD15, and CD30 positivity
5. Lymphocyte predominance variant:
- Effacement of lymph node by a nodular infiltrate of small lymphocytes admixed with L&H (lymphocytic and histiocytic) variants of RS cells (popcorn cells)
- Lymphohistiocytic variants (L&H) cells have infolded nuclear membranes, small nucleoli, fine chromatin, and abundant pale cytoplasm
- L&H variant cells express CD20 and BCL6, and are negative for CD15 and CD30
- Not associated with EBV infection
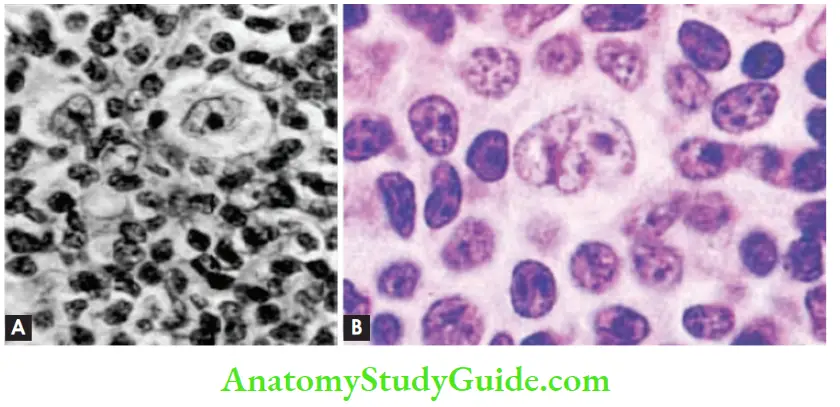
Question 12. Differentiate between Hodgkin lymphoma and non-Hodgkin lymphoma.
Answer:

Question 13. What is mycosis fungoid?
Answer:
Mycosis fungicides
- Tumor comprising of CD4+ helper T cells, which shows infiltration of the epidermis and upper dermis by neoplastic T cells
- Neoplastic T cells appear cerebriform due to marked infolding of the nuclear membranes
- Involvement of the skin is manifested as generalized exfoliative erythroderma
- Sézary syndrome
- When the tumor cells (Sézary cells) invade the peripheral blood
- Leukemia comprising of Sézary cells, with characteristic cerebriform nuclei
- Question 14. Classify acute myeloid leukemia. Describe molecular pathogenesis, lab investigations and clinical features of acute myeloid leukemia.
Answer: - WHO classification acute myeloid leukemia:
1. AML with genetic aberrations:
- AML with t(8;21)
- AML with inv (16)
- AML with t(15;17)
- AML with t(11q23)
2. AML with MDS-like features
3. AML, therapy-related
- 4. AML, not otherwise specified
- AML, minimally differentiated
- AML without maturation
- AML with myelocytic maturation
- AML with myelomonocytic maturation
- AML with monocytic maturation
- AML with erythroid maturation
- AML with megakaryocytic maturation
Intracellular Accumulation Of Protein
Pathogenesis:
- t(8;21) and inv (16) lead to the formation of fusion gene products, which block the maturation of myeloid cells
- AML with t(15;17) (acute promyelocytic leukemia) leads to the formation of PML-RAR (retinoic acid receptor-) fusion gene product, which interferes with the terminal differentiation of granulocytes
- Patients with PLM-RARα mutations can be treated with either all-trans retinoic acid or arsenic trioxide
Hodgkin lymphoma Lab diagnosis and morphology
- The number of leukemic cells in the blood varies from more than 100,000/mm3 to 10,000/ mm3
- Diagnosis of AML is based on the presence of at least 20% myeloid blasts
- Myeloblasts have delicate nuclear chromatin, two to four nucleoli, and more voluminous cytoplasm than lymphoblasts
- Auer rods, distinctive needle-like azurophilic granules, are present in AML with the t(15;17) (acute promyelocytic leukemia)
- Monoblasts have folded or lobulated nuclei, and lack Auer rods
- In AML, if blasts show megakaryocytic differentiation, marrow can show accompanying fibrosis
- Aleukemic leukemia: Blasts are entirely absent from the blood
- Bone marrow examination is essential to exclude acute leukemia in pancytopenia patients
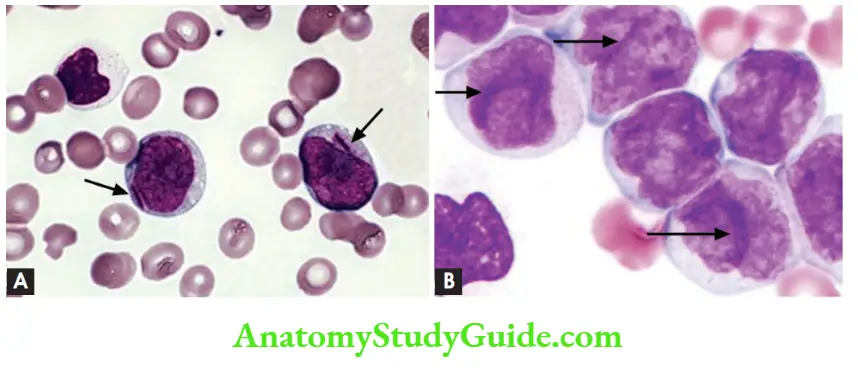
Hodgkin lymphoma Note:
- Myeloid precursors show MPO positivity
- Monoblasts and promonocytes show non-specific esterase (NSE) positivity
- Erythroid precursors show PAS positivity
Hodgkin lymphoma Clinical features
- Anemia, neutropenia, and thrombocytopenia, lead to fatigue, fever, and spontaneous mucosal and cutaneous bleeding
- Procoagulants and fibrinolytic factors released in AML with t(15;17), exacerbate the bleeding tendency
- Tumors with monocytic differentiation often infiltrate the skin (leukemia cutis) and the gingiva
- Occasionally, it presents as a localized soft-tissue mass known as myeloid sarcoma or chloroma
Question 15. Write a note on myelodysplastic syndromes.
Answer:
Myelodysplastic syndromes
- Clonal stem cell disorders characterized by maturation defects
- Associated with ineffective hematopoiesis and a high risk of transformation to AML
- Two types—primary (idiopathic) or secondary to drug or radiation therapy (t-MDS)
Myelodysplastic syndromes Classification of MDS (WHO)
- Refractory anemia
- Refractory anemia with ring sideroblasts
- Refractory cytopenia with multilineage dysplasia (RCMD)
- Refractory anemia with excess blasts-1 (RAEB-1)
- Refractory anemia with excess blasts-2 (RAEB-2)
- MDS-U (unclassified)
- MDS with isolated del (5q) deletion
Morphology of Myelodysplastic syndromes :
Bone marrow examination:
- Cellularity: Hypercellular marrow, can be normocellular or hypocellular
- Characteristic finding: Dyspoises affecting the erythroid, granulocytic, monocytic, and megakaryocytic lineages
1. Erythroid series:
Ring sideroblasts (erythroblasts with iron-laden perinuclear granules seen on Prussian blue stain)
Megaloblastic maturation and nuclear budding abnormalities
2. Myeloid lineage:
- Neutrophils contain decreased numbers of secondary granules, toxic granulations, and/or Döhle bodies
- Pseudo-Pelger-Hüet cells: Neutrophils with only two nuclear lobes
3. Megakaryocytic lineage:
- Pawn ball megakaryocytes: Megakaryocytes with multiple separate nuclei
Myelodysplastic syndromes Note:
Myeloblasts can be increased but comprise <20% of marrow cellularity
Question 16. Describe chronic myeloid leukemia in relation to its peripheral smear, bone marrow findings and clinical outcome.
Answer:
Chronic myelogenous leukemia:
- A characteristic feature is the BCR-ABL gene fusion product derived from portions of the BCR gene on chromosome 22 and the ABL gene on chromosome 9
- BCR-ABL is created by (9;22) translocation (the so-called Philadelphia [Ph] chromosome)
- BCR-ABL gene product leads to increase proliferation of granulocytic and megakaryocytic precursors with their release into the blood
Chronic myelogenous leukemia Morphology
1. Chronic phase Bone marrow
- Markedly hypercellular marrow with increased numbers of granulocytic precursors
- Megakaryocytes are also increased and usually include small, dysplastic forms
- Erythroid progenitors are present in normal or mildly decreased numbers
- Sea-blue histiocytes: Presence of scattered macrophages with abundant wrinkled, green-blue cytoplasm
Peripheral smear in chronic phase
- Leukocytosis >100,000 cells/mm3
- Consists predominantly of band forms and myelocytes admixed with neutrophils, eosinophils, and basophils
- < 10% blasts are seen
- Elevated platelet count
- Splenomegaly, as a result of extramedullary hematopoiesis
2. Accelerated phase
- Persistent or increasing WBC count or increasing splenomegaly
- Persistent thrombocytosis
- Persistent thrombocytopenia
- 20% or more basophils
- 10–19% myeloblasts in the blood or bone marrow
3. Blast phase
- Blasts are equivalent to or greater than 20%
- Extramedullary blast proliferation
- In 70% of cases, blast lineage is myeloid and in 20–30% of cases blasts are of lymphoid lineage
Pawn Ball Megakaryocytes
Chronic myelogenous leukemia Clinical features
- Anemia
- Splenomegaly (producing a dragging sensation in the abdomen or acute onset left upper quadrant pain due to splenic infarction)
- Treatment: BCR-ABL inhibitors
Question 17. Write the diagnostic criteria of polycythemia vera.
Answer:
Diagnostic criteria for polycythemia vera (PV)
1. Major criteria
- Hemoglobin >18.5 gm/dl in men, 16.5 gm/dl in women
- Presence of JAK2 V617F mutation
2. Minor criteria
- Bone marrow biopsy showing hypercellularity for age with an increase in all three cell lines (panmyelosis)
- Serum erythropoietin levels below the reference range for normal
- Endogenous erythroid colony formation in vitro
polycythemia vera Note:
For diagnosis, PV, 2 major, and 1 minor criterion are required.
Question 18. Write a note on primary myelofibrosis.
Answer:
Primary myelofibrosis
WHO criteria for primary myelofibrosis:
Primary myelofibrosis Major:
- Presence of megakaryocytic proliferation and atypia, accompanied by fibrosis
- Not meeting WHO criteria for polycythemia vera, CML, MDS
- Demonstration of JAK2 V617F mutation or another clonal marker
Primary myelofibrosis Minor:
- Leucoerythroblastosis
- Increase in serum lactate dehydrogenase levels
- Anemia
- Splenomegaly
Primary myelofibrosis Note:
For diagnosing, all 3 major and 2 minor criteria should be fulfilled
Primary myelofibrosis Morphology:
- Bone marrow (early/cellular phase):
- Marrow is hypercellular due to an increase in maturing cells of all lineages
- Erythroid and granulocytic precursors appear normal, but megakaryocytes are large, dysplastic, and abnormally clustered
Primary myelofibrosis Bone marrow (late phase):
- Hypocellular and diffusely fibrotic
- Clusters of atypical megakaryocytes with “cloud-like” nuclei are seen
- Hematopoietic elements are found within dilated sinusoids
Pawn Ball Megakaryocytes
Question 19. Discuss Langerhans cell histiocytosis.
Answer:
Langerhans cell histiocytosis (LCH)
- Histiocytosis means the proliferation of dendritic cells or macrophages
- In LCH, there occurs proliferation of immature dendritic cells, Langerhans cell
- Most commonly associated with BRAF mutation
Can present as:
1. Letterer-Siwe disease
- Age <2 years
- Cutaneous lesions (seborrheic eruptions), over the front and back of the trunk and on the scalp
- Hepatosplenomegaly, lymphadenopathy, pulmonary lesions, destructive osteolytic bone lesions
- Anemia, thrombocytopenia, and recurrent infections due to bone marrow infiltration
- Rapidly fatal
2. Eosinophilic granuloma
- Arises within the medullary cavities of bones, most commonly the calvarium, ribs, and femur
- Hand-Schüller-Christian triad: Calvarial bone defects, exophthalmos, and diabetes insipidus (due to involvement of pituitary stalk)
- Eosinophils make a prominent component of the infiltrate
- Spontaneous remission and good prognosis
3. Pulmonary Langerhans cell histiocytosis
- Adult smokers, due to reactive proliferations of Langerhans cells
- May regress spontaneously upon cessation of smoking
Morphology
- Langerhans cells have abundant, vacuolated cytoplasm and vesicular nuclei containing linear grooves or folds
- Tumor cells express S-100 and CD1a
Electron microscopy
- The presence of Birbeck granules in the cytoplasm is characteristic
- Birbeck granules are rod-shaped with dilated terminal ends producing a tennis racket-like appearance and contain the protein langerin
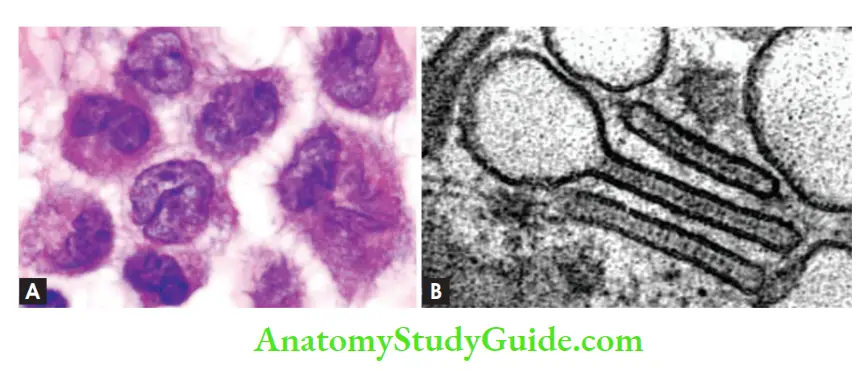
Leave a Reply